

Abstract
The caspase family of proteases play essential roles in apoptosis and innate immunity. Among these, a subgroup known as initiator caspases are the first to be activated in these pathways. This group includes caspase-2, -8, and -9, as well as the inflammatory caspases, caspase-1, -4, and -5. The initiator caspases are all activated by dimerization following recruitment to specific multiprotein complexes called activation platforms. Caspase Bimolecular Fluorescence Complementation (BiFC) is an imaging-based approach where split fluorescent proteins fused to initiator caspases are used to visualize the recruitment of initiator caspases to their activation platforms and the resulting induced proximity. This fluorescence provides a readout of one of the earliest steps required for initiator caspase activation. Using a number of different microscopy-based approaches, this technique can provide quantitative data on the efficiency of caspase activation on a population level as well as the kinetics of caspase activation and the size and number of caspase activating complexes on a per cell basis.
Keywords: Biochemistry, Issue 133, Caspase, Apoptosis, Bimolecular Fluorescence Complementation, Microscopy, Venus, Time-lapse
Introduction
The caspase protease family are known for their critical roles in apoptosis and innate immunity 1. Because of their importance, determining when, where, and how efficiently specific caspases are activated can provide crucial insights into the mechanisms of caspase activation pathways. The imaging based protocol described here enables the visualization of the earliest steps in the caspase activation cascade. This technique takes advantage of the dynamic protein:protein interactions that drive caspase activation.
The caspases can be divided into two groups: the initiator caspases (caspase-1, -2, -4, -5, -8, -9, -10, and -12) and the executioner caspases (caspase-3, -6, and -7). The executioner caspases are present in the cell as preformed dimers and are activated by cleavage between the large and small subunit 2. When activated, they cleave numerous structural and regulatory proteins resulting in apoptosis 3. The initiator caspases are the first caspases to be activated in a pathway and generally trigger the activation of executioner caspases. In contrast to executioner caspases, initiator caspases are activated by dimerization 4,5. This dimerization is facilitated by recruitment of inactive monomers to specific large molecular weight complexes known as activation platforms. Assembly of activation platforms is governed by a series of specific protein:protein interactions. These are mediated by conserved protein interaction motifs present in the proform of the initiator caspase and include the death domain (DD), death effector domain (DED) and caspase recruitment domain (CARD) 6 (Figure 1A). Activation platforms generally include a receptor protein and an adaptor protein. The receptor is usually activated upon binding of a ligand, inducing a conformational change that allows for the oligomerization of numerous molecules. The receptor then either recruits the caspase directly or adaptor molecules that in turn bring the caspase to the complex. Thus, numerous caspase molecules come into close proximity permitting dimerization. This is known as the induced proximity model 7. Once dimerized, the caspase undergoes autoprocessing, which serves to stabilize the active enzyme 4,8. For example, assembly of the Apaf1 apoptosome is triggered by cytochrome c following its release from the mitochondria in a process called mitochondrial outer membrane permeabilization (MOMP). Apaf1 in turn recruits caspase-9 through an interaction that is mediated by a CARD present in both proteins 9. Similar protein interactions result in the assembly of the CD95 death inducing signaling complex (DISC) that leads to caspase-8 activation; the PIDDosome, which can activate caspase-2; and various inflammasome complexes that initiate caspase-1 activation 10,11,12. Thus, initiator caspases are recruited to specific activation platforms by a common mechanism resulting in induced proximity and dimerization, without which activation will not occur.
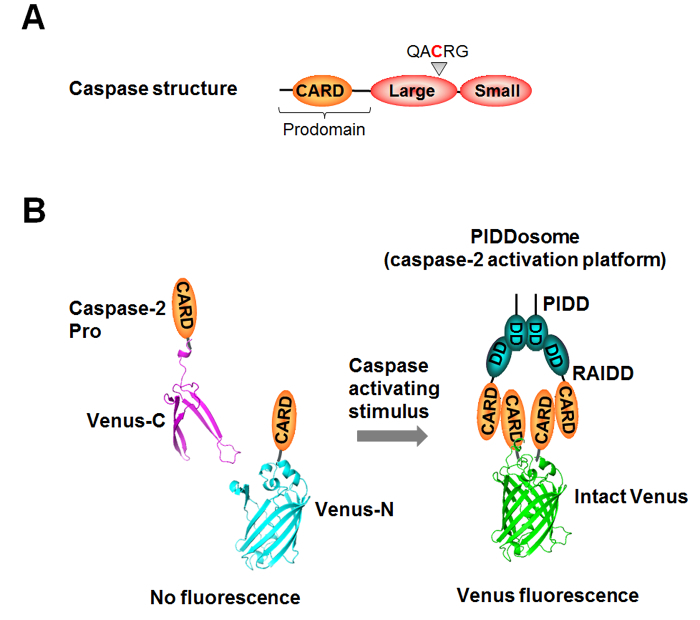
Caspase Bimolecular Fluorescence Complementation (BiFC) is an imaging-based assay that was developed to measure this first step in the activation of initiator caspases, allowing direct visualization of caspase induced proximity following activation platform assembly. This method takes advantage of the properties of the split fluorescent protein Venus. Venus is a brighter and more photostable version of yellow fluorescent protein (YFP) that can be separated into two non-fluorescent and slightly overlapping fragments: the N-terminus of Venus (Venus N or VN) and the C-terminus of Venus (Venus C or VC). These fragments retain the ability to refold and become fluorescent when in close proximity 13. Each Venus fragment is fused to the prodomain of the caspase, which is the minimal portion of the caspase that binds to the activation platform. This ensures that the caspases do not retain enzymatic activity and therefore concurrent analysis of downstream events associated with endogenous events is possible. The Venus fragments refold when the caspase prodomains are recruited to the activation platform and undergo induced proximity. (Figure 1B). The resulting Venus fluorescence can be used to accurately and specifically monitor the subcellular localization, the kinetics, and the efficiency of assembly of initiator caspase activation platforms in single cells. Imaging data can be acquired by confocal microscopy or by standard fluorescence microscopy and can be adapted to a number of different microscopy approaches including: time-lapse imaging to track caspase activation in real time; high resolution imaging for precise determinations of subcellular localization; and end point quantification of efficiency of activation.
This technique was first developed to investigate the activation of caspase-214. While the activation platform for caspase-2 is thought to be the PIDDosome, consisting of the receptor PIDD (p53-induced protein with a death domain) and the adaptor RAIDD (RIP-associated ICH-1/CAD-3 homologous protein with a death domain), PIDDosome-independent caspase-2 activation has been reported. This suggests that additional activation platforms for caspase-2 exist 15,16. Despite not knowing the full components of the caspase-2 activation platform, the caspase BiFC technique has allowed for successful interrogation of the caspase-2 signaling pathways at the molecular level 14,17. We have also successfully adapted this protocol for the inflammatory caspases (caspase-1, -4, -5 and -12) 18 and, in principle, the same approach should be sufficient to similarly analyze each of the remaining initiator caspases. This protocol could similarly be adapted to investigate other pathways were dimerization is a major activating signal. For example, STAT proteins are activated by dimerization following phosphorylation by Janus kinase (JAK) 19. Thus, the BiFC system could be used to visualize for STAT activation as well as many other pathways regulated by dynamic protein interactions. The following protocol provides step-by-step instructions for the introduction of the reporters into cells as well as methodologies for image acquisition and analysis.
Protocol
1. Preparation of cells and culture dishes
NOTE: Perform Steps 1-3 in a tissue culture laminar flow hood. Wear gloves.
- If using glass bottom dishes, coat the glass with fibronectin. Skip to Step 1.2 if using plastic dishes.
- Make a 0.1 mg/mL solution of fibronectin: dilute 1 mL of 1 mg/mL fibronectin solution in 9 mL of 1 X PBS.
- Cover the glass bottom of each well with 0.5-1 mL of fibronectin and incubate for 1-5 min at room temperature.
- Using a pipette, collect the fibronectin solution and store at 4 °C. NOTE: The fibronectin solution can be reused multiple times.
- Wash the glass once with 1-2 mL 1 X PBS, and aspirate off PBS.
Plate 1 x 105 cells per well of a 6-well plate in 2 mL of the appropriate cell culture media (see Table 1 for cell numbers to use for different sized wells). If using a cell line that stably expresses the caspase BiFC components (see Discussion), plate 2 x 105 cells and proceed to the treatment step (Step 3).
Allow cells to adhere to the dish overnight at 37 °C in a tissue culture incubator.
2. Transfection of cells to introduce the caspase BiFC components
- Transfect cells with the appropriate transfection reagent. NOTE: This protocol outlines the instructions for Lipofectamine 2000, which has been optimized for minimal toxicity. This protocol has been successfully used in Hela cells, MCF7 cells and LN18 cells. If using additional transfection reagents follow the manufacturer's instructions and optimize as needed.
- Add 12 µL of the transfection reagent to 750 µL of reduced serum media in a sterile tube.
- Incubate at room temperature for 5 min.
- Add 10 ng of a reporter plasmid (a plasmid encoding a fluorescent protein, e.g. red fluorescent protein [RFP], that will label the transfected cells) to a sterile 1.5 mL tube for each well to be transfected.
- Add 20 ng of each caspase BiFC plasmid (e.g., 20 ng of caspase-2 prodomain [C2 Pro]-VC and 20 ng of C2 Pro-VN) to each tube (Table 2).
- Bring each tube up to a total volume of 100 µL by adding reduced serum media.
- Using a p200 pipette, add 100 µL of transfection reagent solution from Step 2.1.2 to the plasmid mixture in a dropwise manner.
- Incubate the plasmid-transfection reagent mix for 20 min at room temperature.
- Aspirate the media from the cells using a pipette or with suction and then, using a p1000 pipette, pipette 800 µL of serum free media gently down the side of the well.
- Using a p200 pipette, add 200 µL of the DNA-lipid complex dropwise to each well.
- Incubate the cells in a tissue culture incubator at 37 °C for 3 h.
- Taking care not to disrupt the monolayer, remove the serum free media containing the DNA-lipid complexes by aspiration using suction or with a pipette.
- Pipette 2 mL of pre-warmed (37 °C) complete growth media gently down the side of each well.
Incubate the cells at 37 °C in a tissue culture incubator for 24-48 h for optimal protein expression.
3. Induction of caspase activation
- Treat with a drug or stimulus that induces caspase activation approximately 24 h post-transfection.
- Add the desired concentration of the drug to pre-warmed (37 °C) imaging media (complete growth media supplemented with Hepes [20 mM, pH 7.2-7.5] and 2-mercaptoethanol [55 µM]) and mix gently (Table 3).
- Aspirate the media from the cells using a pipette or with suction and then, using a pipette, pipette 2 mL of the solution from 3.1.1 gently down the side of the well.
- Add imaging media without the drug to one well as an untreated control.
Incubate the cells in a tissue culture incubator at 37 °C or proceed to Step 4.3 for a time-lapse experiment.
4. Caspase BiFC data acquisition
- Caspase BiFC for single time point acquisition
- Turn on the microscope and fluorescent light source following the manufacturer's instructions.
- Find the cells under the RFP filter and focus on the reporter gene fluorescence.
- Using a hand-tally counter, count all of the RFP-positive cells in the visual field. Record the number.
- While maintaining the same visual field, switch to the GFP filter (or YFP filter if available) and, using a hand-tally counter, count the number of red cells that are also green (Venus-positive). Switch back and forth between the two filters to ensure that only the red cells are evaluated for Venus-positivity. Record the number (Figure 3).
- Count at least 100 RFP-positive cells from each of three individual areas of the plate.
- In each area, calculate the percentage of Venus-positive transfected cells (i.e. red cells that are also green). Average the resulting percentages to get the standard deviation.
- Three-dimensional imaging of caspase BiFC
- Turn on the microscope following the manufacturer's instructions and launch the acquisition software.
- Select the 60x or 63x oil objective and place a drop of oil on the objective
- Place the culture dish on the microscope stage using the correct plate holder.
- Using either an epifluorescence light source and the microscope eye piece or the confocal lasers and the computer screen, navigate to a field of interest.
- Focus on the reporter fluorescence using the RFP laser or filter.
- If epifluorescence has been used up to this point, switch the light source to the confocal lasers and visualize the live image of the cells as acquired by the camera and displayed by the acquisition software on the computer screen.
- Using the joystick control and the focus wheel, fine tune the focus and position of the cells so that the cells are in the center of the view finder. Choose cells that are separated from each other and are not overlapping.
- Input the percentage laser power to 50% and the exposure time at 100 ms for both Venus and RFP as a starting point to determine the optimal settings to use for the experiment.
- Turn on the live capture and inspect the resulting image and the accompanying display histograms for both channels.
- If the signal is low and the image is hard to make out, increase the percentage laser power and/or exposure time in increments until the signal in the image looks good.
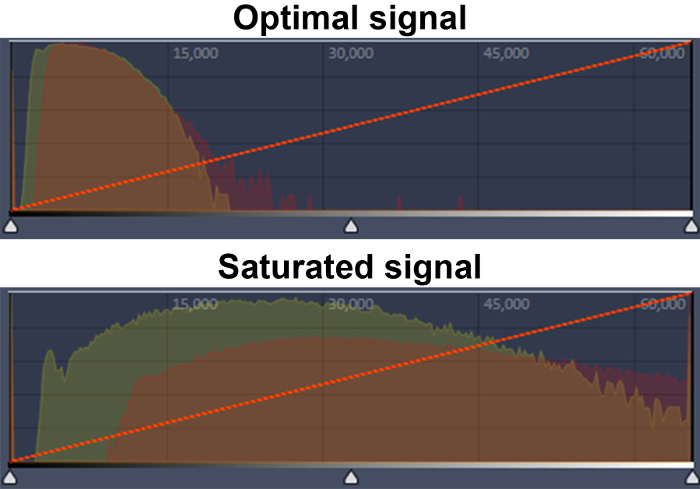
- Ensure that the signal does not reach saturation. Inspect the display histogram to make sure a distinct peak is visible for each fluor. If the peak encompasses the entire histogram display, input lower laser power and exposure settings (Figure 2).
- Visualize control (untreated or non-transfected) cells under the same conditions to ensure that the signal is specific. NOTE: Single color transfectants can also be used to control for crossover, but if the signals are spatially distinct (e.g. mitochondrial versus cytosolic) this is not necessary.
- Select or open the Z stack module in the microscope software.
- Using the focus knob, approximate the focal position corresponding to the center of the cell.
- Adjust the focus in one direction until the cell is no longer visible, select this as the top position.
- Adjust the focus in the opposite direction until the cell is again no longer visible and select this as the bottom position.
- Choose the optimal step size (usually approximately 20 µm) and start the acquisition to instruct the camera to automatically take a single image in each channel at each of the 20 µm steps between the top and bottom positions.
- Use 3D rendering software to reconstruct the 3D image (Figure 4, Movie 1).
- Time-lapse imaging of caspase BiFC
- At least 1 h prior to the experiment, turn on the microscope and set the temperature to 37 °C.
- Select the 40x, 60x, or 63x oil objective and place a drop of oil on the objective.
- Place the culture dish on the microscope stage using the correct plate holder.
- If a CO2 source is available, place the CO2 delivery device (usually a plexiglass lid attached to the tube that delivers the CO2) on top of the plate holder. Set the CO2 level to 5% and turn on the CO2 controller from within the software. If a CO2 source is not available, include 20 mM Hepes to buffer the media.
- Navigate to a field of transfected cells and visualize the live image of the cells as acquired by the camera and displayed by the acquisition software on the computer screen using the RFP laser.
- Following Steps 4.2.8-4.2.12, input the settings for percentage laser power and exposure time for the RFP laser. Keep these values as low as possible while still being able to detect the fluorescent signal.
- Using a positive control sample (see Table 3 for examples), follow Steps 4.2.8-4.2.12 to input the settings for percentage laser power and exposure time for the YFP laser light. Keep these values as low as possible while still being able to detect Venus signal.
- For each well of the plate, choose a number of different positions that contain one or more cells expressing the reporter.
- Input the time interval between each frame of the time-lapse and total number of frames to be taken. Ensure there is time to acquire all the selected positions before the next frame is to be taken.
- Re-visit each position and correct and update the focus as needed.
- To correct for focal drift that can result from changes in temperature or external vibrations, turn on the focus drift correction system (if available).
- Begin the time-lapse and save the data.
- Analyze the data using available imaging software (Figure 5, Movie 2).
Representative Results
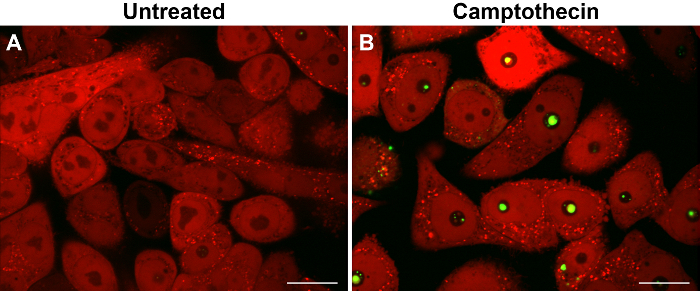
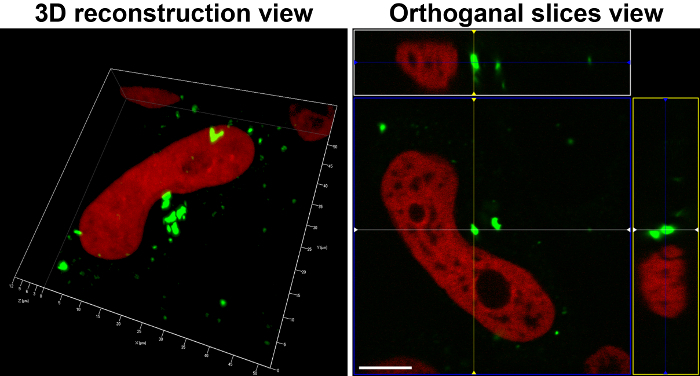
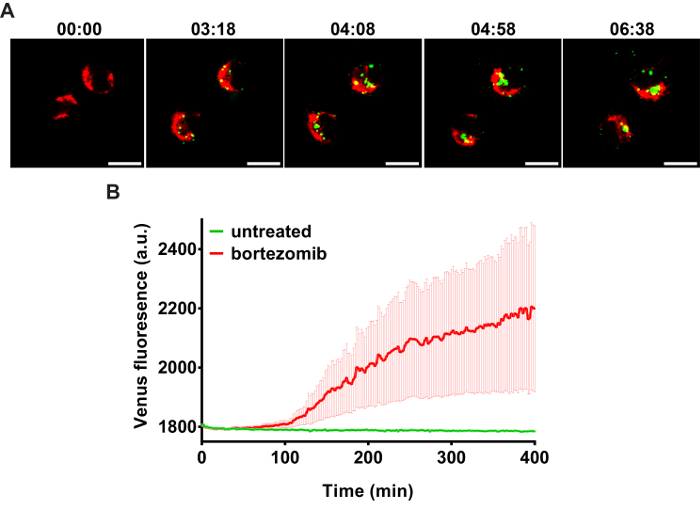
An example of caspase-2 BiFC induced by DNA damage is shown in Figure 3. Camptothecin, a topoisomerase I inhibitor, was used to induce DNA damage and caspase-2 activation. The red fluorescent protein mCherry was used as a reporter to show that the cells express the BiFC probe and to help visualize the total number of cells. Venus fluorescence is shown in green and the large puncta represent caspase-2 induced proximity. These cells can be counted to determine the percentage of Venus-positive cells. Conclusions about subcellular localization can also be made from such images. In the example shown, caspase-2 induced proximity was detected in the nucleolus as well as in the cytoplasm 17. To provide high resolution visualization of the subcellular localization of the caspase activation complex, single cells can be imaged in three dimensions. Figure 4 shows two ways to represent such 3D images. The first is to generate a 3D reconstruction of the cell. The example shows camptothecin-induced caspase-2 BiFC in green and nucleus in red. The 3D reconstruction shows the relative localization of both fluorophores throughout the cells. This type of results display is especially effective when presented as a movie, rotating the cell around a single axis (Movie 1). The second panel is the orthogonal view of the same cell. This provides the xy, xz, and yz visualization of the cell at a particular point in the cell. Depiction of results in this manner can be more suited to a paper presentation such as in a journal article as it is easier to interpret the 3D data presented in a 2D format. Figure 5 shows an example of time-lapse imaging of caspase-2 BiFC in a glioblastoma cell line treated with the proteasome inhibitor bortezomib (see also Movie 2). The graph shows the increase in the average Venus intensity per cell averaged across a number of cells (adapted from 20). This type of data can also be displayed as a number of single cell traces on one graph. In the example shown, the onset of caspase induced proximity is about 2 h following treatment.
Figure 1: Schematic of caspase structure and bimolecular fluorescence complementation (BiFC) methodology.(A) General structure of an initiator caspase. The prodomain, large and small subunits, and the conserved QACRG motif that contains the active site cysteine are depicted. (B) BiFC uses split fluorescent proteins that alone are not fluorescent but can associate to reform the fluorescent molecule when fused to interacting proteins. For caspase BiFC, the prodomain (Pro) or full-length caspase is fused to the N-terminal half (Venus-N) and to the C-terminal half (Venus-C) of Venus. In their monomeric state the caspase fusion proteins are not fluorescent. Upon recruitment to an activation platform, such as the PIDDosome, as shown here, association of the two halves of Venus is enforced representing caspase induced proximity that is measured as an increase in Venus fluorescence. Please click here to view a larger version of this figure.
Figure 2: Representative image for recognizing saturation using the display histogram. Examples of optimal (upper panel) and saturated signals (lower panel) are shown. The Venus (yellow peak) and RFP (red peak) signals are shown. The x-axis indicates intensity and the y-axis is number of pixels. Please click here to view a larger version of this figure.
Figure 3: Caspase-2 bimolecular fluorescence complementation (BiFC) following DNA damage. Hela cells expressing the caspase-2 BiFC components were left untreated (A) or treated with camptothecin (100 µM) (B) in the presence of qVD-OPH (20 µM). mCherry was co-expressed as a fluorescent reporter. Representative images show cells (red) with caspase-2 BiFC (green) in camptothecin-treated cells 16 h following treatment. Scale bars represent 20 µm. Please click here to view a larger version of this figure.
Figure 4: Localization of caspase-2 bimolecular fluorescence complementation (BiFC). Hela cells expressing the caspase-2 BiFC components and a fluorescent nuclear reporter were treated with camptothecin (20 µM) in the presence of qVD-OPH (20 µM). 3D reconstructions composed from 0.1 μm serial confocal images through the z-plane of the cell were made after 24 h and show the nucleus in red and caspase-2 BiFC in green. The left panel shows a 3D maximum intensity projection rendering reconstruction (see also Movie 1). The right panel shows the orthogonal slices view of the same cell. The middle box (blue) is the xy plane, the right box (yellow) is the yz plane, and top box (white) is the xz plane. The yz and xz planes intersect according to the cross hairs. Scale bars represent 10 µm. Please click here to view a larger version of this figure.
Figure 5: Time-lapse of caspase bimolecular fluorescence complementation (BiFC)(A) LN18 glioblastoma cells transfected with the caspase-2 BiFC components and a red fluorescent mitochondrial marker (DsRed-mito, red) were treated with bortezomib (15 nM) in the presence of qVD-OPH (20 µM). The cells were placed on a confocal microscope at 37 °C and images were taken every 2 min for 8 h. Images of representative cells from the time-lapse show that the induced proximity of the two caspase-2 BiFC components resulted in an increase in Venus fluorescence (green) over time. (B) The average intensity of Venus in each cell at each time point was measured. Venus fluorescence increased over time after bortezomib treatment. Division in the untreated control indicated low or negligible toxicity conditions from laser light (Figure adapted from 20). Scale bars represent 20 µm. Error bars represent standard error of the mean (SEM) of 8 (control) and 14 (bortezomib) individual cells (see also Movie 2). Please click here to view a larger version of this figure.
| Size of plate | Number of cells | Volume of media |
| 6 well plate | 1 x 105 cells/ well | 2 mL |
| 12 well plate | 5x 104 cells/ well | 1 mL |
| 24 well plate | 2.5 x 104 cells/ well | 0.5 mL |
| 4 chamber dish | 2.5 x 104 cells/ chamber | 1 mL |
| 8 chamber dish | 1.25 x 104 cells/ chamber | 0.5 mL |
Table 1: Guidelines for numbers of cells to plate. If using cells stably expressing the caspase bimolecular fluorescence complementation (BiFC) components, plate double the amounts shown here.
| Size of plate | Transfection reagent | Reduced serum media | Reporter plasmid | Caspase-pro VC plasmid | Caspase-pro VN plasmid | Reduced serum media added to plasmid mix | Transfection reagent solution added to plasmid mix | Serum free media added to cells |
| 6 well plate | 12 µL/plate | 750 µL | 10 ng | 20 ng | 20 ng | to total 100 µL | 100 µL | 800 µL |
| 12 well plate | 12 µL/plate | 750 µL | 5 ng | 10 ng | 10 ng | to total 50 µL | 50 µL | 400 µL |
| 24 well plate | 12 µL/plate | 750 µL | 2.5 ng | 5 ng | 5 ng | to total 25 µL | 25 µL | 200 µL |
| 4 chamber dish | 4 µL/plate | 250 µL | 2.5 ng | 5 ng | 5 ng | to total 25 µL | 25 µL | 200 µL |
| 8 chamber dish | 4 µL/plate | 250 µL | 1.25 ng | 2.5 ng | 2.5 ng | to total 12.5 µL | 12.5 µL | 100 µL |
Table 2: Recommended volumes of reagents used for transient transfection. Note: the amount of plasmid suggested is only a starting point. If no signal is detected, it is recommended to titrate the plasmid from 20 - 200 ng per well of a 6-well plate.
| Caspase | Treatment | Concentration | Time | Expected results |
| Caspase-1 | Over-expressed ASC | 250 ng/well of a 6 well plate | 48 h | 50% BiFC positive |
| Caspase-2 | Over-expressed RAIDD | 500 ng/well of a 6 well plate | 24 h | 90% BiFC positive |
| Camptothecin | 100 µM | 16 h | 30-60% BiFC positive | |
| Heat shock | 1 h at 45 °C | 16 h | 80% BiFC positive | |
| Caspase-4 | Overexpressed NALP1 | 250 ng/well of a 6 well plate | 48 h | 30% BiFC positive |
| Caspase-5 | Overexpressed IPAF | 250 ng/well of a 6 well plate | 48 h | 15% BiFC positive |
Table 3: Examples of positive controls for caspase bimolecular fluorescence complementation (BiFC). The conditions and expected results are based on published data using the Pro constructs 14,17,18. Co-expressed plasmids should be transfected alongside the BiFC plasmids (Step 2.1.4)
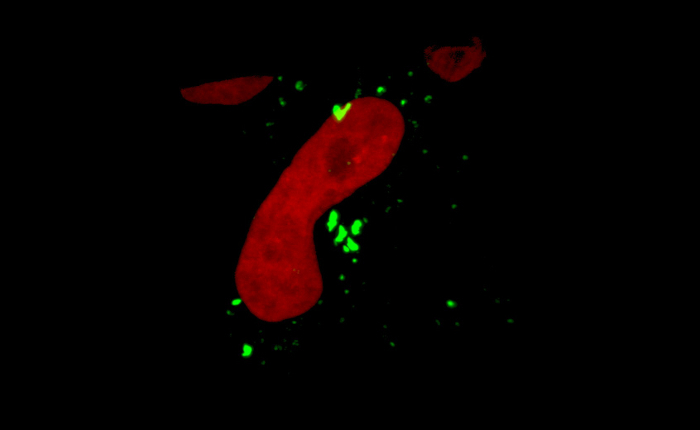 Movie 1: 3D localization of caspase-2 bimolecular fluorescence complementation (BiFC). 3D reconstruction, rotated around the y-axis of confocal images through the z-plane of a Hela cells expressing the caspase-2-BiFC components and a fluorescent nuclear reporter were treated with camptothecin (20 µM) for 16 h. The movie shows caspase-2 BiFC (green), and the nucleus (red). Please click here to view this video. (Right-click to download.)
Movie 1: 3D localization of caspase-2 bimolecular fluorescence complementation (BiFC). 3D reconstruction, rotated around the y-axis of confocal images through the z-plane of a Hela cells expressing the caspase-2-BiFC components and a fluorescent nuclear reporter were treated with camptothecin (20 µM) for 16 h. The movie shows caspase-2 BiFC (green), and the nucleus (red). Please click here to view this video. (Right-click to download.)
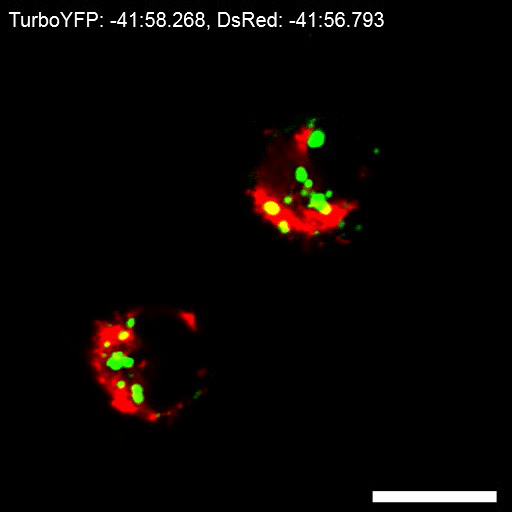 Movie 2: Time-lapse of caspase-2 bimolecular fluorescence complementation (BiFC). LN18 glioblastoma cells transfected with the caspase-2 BiFC components and treated with bortezomib (15 nm) were imaged every 2 min for 8 h. The movie shows caspase-2 BiFC (green), and the mitochondria (red). Please click here to view this video. (Right-click to download.)
Movie 2: Time-lapse of caspase-2 bimolecular fluorescence complementation (BiFC). LN18 glioblastoma cells transfected with the caspase-2 BiFC components and treated with bortezomib (15 nm) were imaged every 2 min for 8 h. The movie shows caspase-2 BiFC (green), and the mitochondria (red). Please click here to view this video. (Right-click to download.)
Discussion
This protocol discusses the use of split fluorescent proteins to measure caspase induced proximity. Split Venus was chosen for this technique because it is very bright, highly photostable and the refolding is fast 13. Thus, the analysis of Venus refolding upon caspase induced proximity can provide close to real-time estimations of caspase protein interaction dynamics. Venus is split into two slightly overlapping fragments, the N terminus of Venus (Venus-N or VN) comprising amino acids 1-173 and the C terminal fragment (Venus-C or VC) comprising residues 155-239. Other split fluorescent proteins exist but some, such as split mCherry, require incubation at 4 °C for up to 2 h for refolding 21. Additional split fluorescent proteins including split Cerulean (a version of cyan fluorescent protein) and the far red split mLumin have yet to be tested in this context 13,22. Ultimately, if these fluorescent proteins prove suitable for measuring caspase induced proximity, this could allow for the simultaneous evaluation of multiple caspases.
Generally, the prodomain (Pro) region of the caspase is fused to the split fluorescent fragments. This region contains the minimal portion of the caspase required to bind to the activation platform and encompasses the CARD or DED interaction motif. The advantage of using the prodomain is that it does not introduce any enzymatic activity to the cells. This allows for the simultaneous monitoring of downstream events due to the endogenous caspase. Research to date has shown little difference in the kinetics and localization of caspase BiFC between the prodomain and the full length caspase 14. However, the full-length constructs tend to express with lower efficiency, necessitating the transfection of increased amounts of plasmid DNA. In addition, it is recommended to mutate the catalytic cysteine to a serine to prevent caspase-induced apoptosis upon expression. Despite these drawbacks, certain investigations may require the concurrent analysis of the full length BiFC pairs to determine if there are context-dependent differences in caspase induced proximity in the presence or absence of the catalytic domains.
This protocol is specifically adapted for HeLa cells. The same protocol works well for additional adherent cell lines including the mammary carcinoma cell line, MCF-7, the glioblastoma cell line, LN18, and mouse embryonic fibroblasts (MEF). It is recommended that the user optimizes both plating and transfection conditions when adapting this protocol to a new cell type. The protocol describes two main microscopy approaches that can be used to evaluate the cells: epifluorescence and confocal microscopy. When using confocal microscopy, cells must be plated on glass coverslips because the majority of high numerical aperture objectives that are used in conjunction with confocal microscopy will not penetrate the thickness of plastic dishes or glass slides. Thus, it is recommended to use dishes with embedded no. 1.5 glass coverslips that have the optimal coverslip thickness of 0.17 mm. A number of different types of these dishes are available that range from individual 3 cm dishes, to chamber slides, to standard size multi-well plates. Tables 1 and 2 outline how to adapt the plating and transfection instructions for the different well sizes. For the epifluorescence imaging that is described here (4.1), standard plastic tissue culture plates are sufficient as long as the microscope is equipped with long pass objectives.
Because this approach is effectively a "lights on" method to measuring caspase activation, the inclusion of a fluorescent reporter is essential both to allow visualization of the cells prior to or in the absence of activation of the caspase reporter and to identify the transfected cells, when transient transfection is used. A number of different reporters can be used and the only criteria when choosing one are: 1) the fluorophore is sufficiently bright so that it can be easily detected by the imaging protocol; and 2) its fluorescent spectrum does not overlap with that of YFP. For example, green fluorescent protein (GFP) should not be used as a reporter because it will be detected by the YFP filter or laser used to detect Venus. This is due to the spectral overlap of the two fluorophores. Choosing a fluorophore that localizes to a specific organelle can provide additional qualitative information. For example, use of a protein that targets the mitochondria, such as the red fluorescent protein DsRed-mito, can provide additional information on the overall viability of the cell. Mitochondria tend to undergo fragmentation (fission) during the early stages of cell death 23 and this can be visualized with a reporter like DsRed-mito. The use of an organelle marker as a reporter can also allow conclusions about subcellular localization of caspase BiFC to be made.
There are a number of ways that caspase BiFC data can be acquired and analyzed. Deciding which method to use will be determined by the parameters to be explored (endpoint population analysis versus single cell positional or kinetic information) and the availability of instrumentation and imaging software. This protocol outlines a few microscope-based approaches to collecting the results of a caspase BiFC experiment for acquiring both single time points and time-lapse data. However, it should be noted that a number of variations and combinations of these approaches to be used to creatively interrogate caspase activation pathways.
Single time point data acquisition (4.1) is used to determine the percentage of cells with caspase induced proximity at a single time point. For this protocol, one simply counts the number of transfected cells that are Venus-positive. Thus, highly specialized equipment is not required and only the most basic epifluorescence microscope with a GFP (or YFP) and RFP filter and a hand tally counter is needed. Some imaging systems can be automated to automatically take a set number of images per well of a plate. If such instrumentation is available, the cells in the acquired images can be similarly counted by visual inspection, or quantitated using imaging software. Three-dimensional imaging (4.2) provides high resolution images of caspase BiFC throughout the focal planes of the cell. A confocal microscope with lasers that excite Venus and RFP (or the chosen transfection reporter) and Z-stack capabilities is required. A number of 3D rendering software modules are available that provide different ways to visualize the data. For both these types of analysis, it is possible to fix the cells and image or assess them at a later point. However, the process of fixation does diminish the Venus signal, which could add artifacts to the analysis and therefore this approach is not recommended. Time-lapse imaging (4.3) can be used to provide simultaneous kinetic and positional analysis of caspase BiFC. A microscope equipped with a motorized stage can be set up to take images of multiple positions and even compare different treatment groups in the same experiment.
For time-lapse, a number of additional considerations should be taken into account. The laser light can cause artifacts due to phototoxicity or photobleaching and both need to be controlled for. In general, both can be avoided by using the lowest possible amount of light (input low percentage laser power and short exposure times in Steps 4.3.6-4.3.7). Phototoxicity results when the laser light induces death in the cells and can be controlled for by imaging untreated cells under the same conditions and confirming that cell division proceeds as normal. Photobleaching occurs when multiple images taken of the same cell decreases the fluorescence. The Venus fluorescent protein is quite photostable, so this is rarely a problem in practice. The effects of photobleaching can be determined by taking numerous sequential images of a Venus-positive cell and ensuring that the fluorescence remains stable. This only needs to be done once, as long as the same laser power and exposure settings are used for every experiment. Effects due to phototoxicity or photobleaching can also be mitigated by reducing the total number of images captured. This can be achieved by lengthening or shortening the time interval between captures (Step 4.3.9). Using 5-10 min intervals over a 16 h time-lapse is a good place to start. If no photoxicity is observed or experiments of shorter duration are required, the interval between frames can be reduced. If caspase activation is rapid, shorter experiments with shorter time intervals can be used. For example, for a 16 h experiment with 10 min intervals, a total of 192 images will be taken (96 for each fluorophore). A 2 h experiment with 1.5 m intervals will yield 160 images. Therefore, the long and short experiments would each expose the cells to similar total amounts of laser light. For short duration experiments the caspase activating stimulus can be added directly to the media immediately prior to initiation of the time-lapse capture (after Step 4.3.11 instead of at Step 3). To do this, remove the lid from the plate and gently drop in a solution containing the stimulus using a pipette. Take care not to jostle the plate and quickly check that the cells are still in focus before proceeding.
When carrying out time-lapse experiments, it is also critical to make sure the immediate environment of the cells closely resembles that of a tissue culture incubator. A number of environmental controls are available on different microscope set ups. These include smaller stage top incubators as well as environmental chambers that encapsulate the entire microscope. Both of these devices usually deliver heat, CO2, and humidity to the chamber in a manner that can be precisely controlled. The precise control of CO2 and temperature is essential to the success of the experiment because unexpected temperature fluctuations can lead to focal drift and absence of CO2 causes the pH of the media to rise that can be toxic to the cells. If a CO2 source is not available, the media can be buffered by the addition of Hepes (see Step 3.1.1). Even if a CO2 source is available, it is recommended to include Hepes in the imaging medium as an extra measure to keep the cells healthy. However, even in the presence of Hepes, the absence of CO2 for prolonged periods of time can cause cell death. Therefore, it is important to inspect untreated cells, imaged under the same conditions, and the color of the media at the conclusion of the experiment to ensure that the pH has remained stable. Note that phenol red does not need to be omitted from any of the media used in this protocol since the contribution of autofluorescence to the Venus signal is minimal.
There are a number of available methods to analyze the imaging data resulting from a time lapse experiment. Measuring the mean intensity of Venus pixels per transfected cell is the most straightforward way to analyze the timing of caspase induced proximity. As shown in Figure 3, Figure 4, Figure 5, the caspase-2 BiFC often appears as one or more puncta located in different subcellular compartments. Imaging analysis approaches that measure numbers of foci, object size, or additional similar factors can yield informative quantitative data about these structures. Not all caspase BiFC appears as puncta. For example, the inflammatory caspases have shown different BiFC patterning including diffuse fluorescence, filamentous structures, and single or multiple puncta per cell 18. These patterns were dependent on the caspase activated as well as the upstream activating signals. Therefore, the type of analysis that is most suitable for the time-lapse results will largely be dictated by the type of fluorescent patterning that is observed.
Many of the treatment stimuli that are assessed when using this protocol will induce apoptosis either directly through the caspase analyzed or indirectly by engaging a different cell death pathway. This causes the cells to shrink and detach from the plate, which makes it very difficult to image the cells and almost impossible to determine any specific localization of the caspase-BiFC. Inclusion of a pan-caspase inhibitor will prevent this cell death. The recruitment of caspases to their activation platforms and the associated caspase BiFC is not dependent on the catalytic activity of the caspase. Therefore, caspase inhibition will have no effect on this step, but will inhibit the downstream physical characteristics of apoptosis including blebbing, detachment, and eventual loss in plasma membrane integrity. Thus, if carrying out endpoint analysis (Step 4.1) or Z stacks (Step 4.2), the inclusion of a caspase inhibitor is essential. For time-lapse experiments in contrast, if apoptosis-related events such as MOMP, annexin V binding (to detect phosphatidyl serine exposure) or propidium iodide uptake (to detect plasma membrane permeabilization) are to be analyzed alongside caspase BiFC, the caspase inhibitor should be omitted to prevent inhibition of these events. A caspase inhibitor such as qVD-OPh (10-20 µM) should be added to in the media that contains the apoptosis-inducing stimulus (Step 3.1.1). If co-expressing a pro-apoptotic protein with the caspase BiFC components, the caspase inhibitor should be added at Step 2.1.12.
While caspase BiFC is one of the few available methods that can be used to visualize events in caspase activation pathways in live cells, a number of considerations must be taken into account when designing these experiments. Because this approach is overexpression-based, controls for specificity are important. This can be accomplished in one of two ways. First, introduction of single point mutations into the CARD or DED of the caspase prodomain such that they disrupt the binding between the caspase and its activation platform should decrease both the intensity of caspase BiFC and the percentage of Venus-positive cells. For example, mutation of the residue D59 to E in the prodomain of caspase-1 disrupts binding to the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) and equally disrupts ASC-induced caspase-1 BiFC 18,24. Second, use of cells that are deficient in the adaptor proteins that recruit the caspase to the activation platform will decrease specific BiFC signals. This can be achieved using knockout cells, if available, or using siRNA or CRISPR/Cas9-based approaches to deplete the cells of the adaptor. Accordingly, depletion of the caspase-2 adaptor protein RAIDD completely blocked caspase-2 BiFC induced by heat shock or DNA damage 14,17. For these experiments, the knockout or knockdown cells should be compared with matched control cells (i.e. litter-matched wild type MEF or control knockdown cells) and, if a new cell line is used, optimal transfection conditions should be assessed to ensure equal expression of the BiFC reporter across cell lines.
A second limitation of this approach is that, when using transient transfection, it is difficult to ensure that each cell is expressing equal amounts of each protein. If expression is unequal, one Venus fragment could be recruited to the platform at higher frequencies. This could mask the full fluorescent signal and lead to false negative results. To overcome this problem, a new version of the caspase BiFC reporter for stable cell line generation was recently reported 17. This new version consisted of a construct that expressed the C2 Pro-BiFC components on a single open reading frame separated by the viral 2A self-cleaving peptide 25. Therefore, the BiFC components are transcribed as a single mRNA from the same promoter but translated to produce stoichiometrically equal amounts of the VC and VN fusion proteins. The cell lines generated to stably express this construct demonstrated similar trends of activation to the transiently expressed reporters but were both more sensitive and displayed lower background fluorescence. Thus, the above protocol can be greatly simplified if used in conjunction with reporter cell lines that stably express the caspase BiFC components.
Data to date suggest minimal interference between the caspase BiFC components and the endogenous proteins. For example, caspase-2 is known to be activated upstream of MOMP but caspase-2 BiFC did not block the progression of MOMP 14. Thus, these reporters do not appear to act in a dominant negative fashion. However, if simultaneously evaluating downstream events, this should be investigated within each set of experiments. Including a control non-transfected well in the experiment is the best way to approach this. If the kinetics of cell death are unchanged with or without expression of the caspase sensor, this will indicate that the caspase-BiFC components are not interfering with endogenous function. Conversely, the endogenous caspase could possibly affect the BiFC readout, either in efficiency or in the size or shape of the structures. Expressing the reporter in cells deficient for the caspase will control for this.
Despite these limitations, the caspase BiFC technique is a useful method to interrogate caspase activation pathways that can complement and even improve on existing approaches to measure initiator caspase activation. By measuring induced proximity, the first step in activation, this technique overcomes problems associated with measuring later steps such as caspase auto-processing associated with activation. For example, while using western blot to monitor the cleavage of the executioner caspases, caspase-3 and caspase-7, provides an accurate measure of activation 2, using the same approach for initiator caspases can be flawed. This is because cleavage of initiator caspases, is not sufficient for activation 4,26,27. In fact, many initiator caspases are cleaved during apoptosis by caspase-3 without producing active enzymes 28. Similarly, techniques based on using peptide substrates designed as targets for specific caspases need to take into account the overlapping specificities of these sequences for the caspases 29. This does not mean that such approaches cannot be used to measure caspase activation, but the drawbacks should be acknowledged when interpreting the results. Caspase BiFC provides an alternative approach that can help confirm results using more conventional methods. In addition, the ability to track the assembly of the caspase activation complexes in real time and to distinctly determine the size, shape and localization of the complexes is a significant advantage of this technique that is not possible to assess by other means.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We would like to acknowledge all the previous members of the Bouchier-Hayes lab who contributed to the development of this technique. This work was funded in part by a Texas Children's Hospital Pediatric Pilot award to LBH. We thank Joya Chandra (MD Anderson, Houston, Texas) for permission to include data published in collaboration with her team. The development of the reagents described was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (NIAID P30AI036211, NCI P30CA125123, and NCRR S10RR024574) and the assistance of Joel M. Sederstrom
References
- Salvesen GS, Riedl SJ. Caspase mechanisms. Adv Exp Med Biol. 2008;615:13–23. doi: 10.1007/978-1-4020-6554-5_2. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15(6):725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007;14(4):641–650. doi: 10.1038/sj.cdd.4402103. [DOI] [PubMed] [Google Scholar]
- Baliga BC, Read SH, Kumar S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 2004;11(11):1234–1241. doi: 10.1038/sj.cdd.4401492. [DOI] [PubMed] [Google Scholar]
- Boatright KM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11(2):529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Aravind L, Dixit VM, Koonin EV. The domains of death: evolution of the apoptosis machinery. Trends Biochem Sci. 1999;24(2):47–53. doi: 10.1016/s0968-0004(98)01341-3. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A. 1999;96(20):10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, et al. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285(22):16632–16642. doi: 10.1074/jbc.M109.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8(5):405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Kischkel FC, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14(22):5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304(5672):843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- Shyu YJ, Liu H, Deng X, Hu CD. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 2006;40(1):61–66. doi: 10.2144/000112036. [DOI] [PubMed] [Google Scholar]
- Bouchier-Hayes L, et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell. 2009;35(6):830–840. doi: 10.1016/j.molcel.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, et al. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol. 2009;185(2):291–303. doi: 10.1083/jcb.200811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ. 2012;19(10):1722–1732. doi: 10.1038/cdd.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, et al. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J Cell Biol. 2017. [DOI] [PMC free article] [PubMed]
- Sanders MG, et al. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis. 2015;6:e1813. doi: 10.1038/cddis.2015.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296(5573):1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Manton CA, et al. Induction of cell death by the novel proteasome inhibitor marizomib in glioblastoma in vitro and in vivo. Sci Rep. 2016;6:18953. doi: 10.1038/srep18953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JY, et al. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem Biophys Res Commun. 2008;367(1):47–53. doi: 10.1016/j.bbrc.2007.12.101. [DOI] [PubMed] [Google Scholar]
- Chu J, et al. A novel far-red bimolecular fluorescence complementation system that allows for efficient visualization of protein interactions under physiological conditions. Biosens Bioelectron. 2009;25(1):234–239. doi: 10.1016/j.bios.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10(8):870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J. 2013;449(3):613–621. doi: 10.1042/BJ20121198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Vignali DA. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin Biol Ther. 2005;5(5):627–638. doi: 10.1517/14712598.5.5.627. [DOI] [PubMed] [Google Scholar]
- Chang DW, Xing Z, Capacio VL, Peter ME, Yang X. Interdimer processing mechanism of procaspase-8 activation. EMBO J. 2003;22(16):4132–4142. doi: 10.1093/emboj/cdg414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke HR, et al. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;274(13):8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- Slee EA, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144(2):281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15(2):322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]