

Abstract
The cytoskeleton, composed of actin microfilaments, microtubules, and intermediate filaments (IF), plays a key role in the control of cell shape, polarity, and motility. The organization of the actin and microtubule networks has been extensively studied but that of IFs is not yet fully characterized. IFs have an average diameter of 10 nm and form a network extending throughout the cell cytoplasm. They are physically associated with actin and microtubules through molecular motors and cytoskeletal linkers. This tight association is at the heart of the regulatory mechanisms that ensure the coordinated regulation of the three cytoskeletal networks required for most cell functions. It is therefore crucial to visualize IFs alone and also together with each of the other cytoskeletal networks. However, IF networks are extremely dense in most cell types, especially in glial cells, which makes its resolution very difficult to achieve with standard fluorescence microscopy (lateral resolution of ~250 nm). Direct STochastic Optical Reconstruction Microscopy (dSTORM) is a technique allowing a gain in lateral resolution of one order of magnitude. Here, we show that lateral dSTORM resolution is sufficient to resolve the dense organization of the IF networks and, in particular, of IF bundles surrounding microtubules. Such tight association is likely to participate in the coordinated regulation of these two networks and may, explain how vimentin IFs template and stabilize microtubule organization as well as could influence microtubule dependent vesicular trafficking. More generally, we show how the observation of two cytoskeletal components with dual-color dSTORM technique brings new insight into their mutual interaction.
Keywords: Biology, Issue 133, Intermediate filaments, vimentin, microtubule, STORM, super-resolution microscopy, cytoskeleton
Introduction
Cytoplasmic intermediate filaments (IFs) are 10 nm-diameter homo- or heteropolymers of a cell type specific subset of IF proteins. IFs participate in a large range of cellular functions such as cell motility, proliferation and stress responses. Their key role is highlighted by the fact that more than 90 human diseases are directly caused by mutations in IF proteins; for instance, changes in IF composition accompanies tumour growth and spreading1,2,3. There is growing evidence that the three cytoskeleton systems work in collaboration to control cellular functions such as cell polarization, division and migration2,3. Since there is a tight coupling between spatial architecture and functions, it is crucial to get insights into the structural organization of the IF with the other filaments and better understand the cytoskeletal cross-talk. this article provides a protocol to perform the super-resolution technique dual-color dSTORM (direct STochastic Optical Reconstruction Microscopy)4 and how it is used to investigate the interaction between IF and microtubules in fixed, cultured cells in 2 dimensions (2D).
Several super-resolution techniques have been developed over the past decade, and there discoveries were at the origin of the Nobel Prize in Chemistry in 20145. Among all these techniques lies the single molecule localization-based methods like PALM6 (Photo-Activated Localization Microscopy) which uses photoswitchable fluorescent proteins, STORM7 with pairs of fluorophores or dSTORM4 with conventional fluorophores. All these methods are based on the same principle which consists of (i) the switch of most of the fluorescent reporters into an "off" state" (non-fluorescent), (ii) the stochastic activation of a subset of them into a fluorescent state in order to localize their spatial position with nanometer accuracy, and (iii) the repetition of this process in order to activate as many subsets of fluorophores as possible. A final image is reconstructed using all the localizations of the activated molecules, providing a lateral resolution down to ~20-40 nm. Several commercialized optical systems allowing PALM/STORM are now available for biologists for routine experiments. Here, one such system was used to study the structural association of microtubules and IFs. Among all the single-molecule localization-based methods, the dual-color dSTORM technique was selected, because it is well suited to observe very thin lamellar regions (<1 µm) of adherent cells and can provide significant improvement of image resolution with minimal time and money investment. Indeed, dSTORM is a very convenient and versatile technique that is compatible with the standard organic dyes routinely used for cellular staining and immunofluorescence.
While one color dSTORM images are relatively simple to obtain using the fluorophore Alexa6478, dual color dSTORM requires to optimize the experimental conditions so that two dyes can blink properly in the chosen buffer, especially when there is limited laser power. Excellent papers are already available on the methods to set up multi-color imaging with dSTORM9,10,11,12,13, and these papers explain in detail the possible sources of artifacts and degraded image resolution as well as how to overcome them. In this article, the optimal experimental conditions in terms of cells fixation, immunostaining, sample preparation, imaging acquisition and image reconstruction are described in order to acquire images of dense cytoplasmic IF networks and microtubules in glial cells with dual-color dSTORM. Briefly, the extraction/fixation method described in Chazeau et al12 was adapted to glial cells and used with a post-fixation step, optimized antibodies concentration, a STORM buffer with 10 mM MEA described in Dempsey9 et al. which was found to be optimal for the experimental set up and sample type.
Glial cells mainly express three types of IF proteins: vimentin, nestin and GFAP (glial acidic fibrillary protein). These three proteins were shown to co-polymerize in astrocytes14. We previously showed using super-resolution structured illumination microscopy (SR SIM) that these three IF proteins can be found in the same single IF filament and that they display similar distribution and dynamics in glial cells 15. Due to the similarities between the three IF proteins, vimentin staining was used as a reporter for the whole IF network. Using dSTORM, we managed to resolve how IF form bundles along microtubules, which was not possible with diffraction limited microscopy techniques15. These observations may help to understand how vimentin IFs can template microtubules and regulate their growth trajectory, promoting the maintenance of a polarity axis during cell migration16. Super-resolution images provided key information on the mutual interaction of the two cytoskeletal subsystems, and brought insight into the link between spatial association and local function which could be cell-type specific17. In general, dual-color dSTORM can be used to study the crosstalk between cytoskeletal elements or other types of organelles, provided that good immunostaining conditions are reached in terms of density and specificity. This technique will be useful to better characterize the cytoskeleton changes observed during astrogliosis and in glioblastoma, the most common and most malignant tumor of the central nervous system, where the expression of IF proteins is altered 18,19,20,21.
Protocol
1. Coverslips preparation (Day 1, 30 min)
Place the 18-mm nº1.5H (170 µm +/- 5µm) glass coverslips on a plastic rack.
Put the rack in a 100-mL beaker filled with acetone and soak for 5 min.
Transfer the rack to a 100-mL beaker filled with absolute ethanol and soak for 5 min.
Transfer the rack to a new 100 mL beaker filled with absolute ethanol and place the beaker in an ultrasonic cleaner device (see Table of materials) at room temperature. Press "on" and wait for 10 min.
Put the rack under a laminar flow hood and let the coverslips dry or dry the coverslips with filtered air flow.
Put the rack with coverslips in a plasma cleaner device. Switch on the vacuum pump, close the door, wait for 3 min to make the vacuum and switch on the plasma for 1 min. Use the coverslips shortly after cleaning. Do not store them.
2. Cell plating (Day 1, 20 min)
Grow the glial cell line U373 in Minimum Essential Medium (MEM) with 10% Fetal Bovine Serum, 1% Penicillin-Streptomycin, and 1% non-essential amino acid in 10 cm diameter plastic petri dish.
Place clean glass coverslips in 12-well plates under the laminar flow hood and add 1 mL of preheated cell medium per well.
Take a dish containing cells from the cell incubator (37 °C, 5% CO2).
Remove the medium and add 10 mL PBS.
Remove the PBS and add 1 mL 0.05% Trypsin-EDTA.
Place the dish back in the incubator for 2-3 min.
Add 10 mL of warm medium preheated at 37 °C on the dish and resuspend the cells.
Seed about 100 000 cells per well (~150 µL of cells) in the 12-well plates that contain the coverslips.
Wait for at least 6 h (or overnight) to allow cell spreading.
3. Cell fixation (Day 2, 2h)
Prepare the cytoskeleton buffer with 80 mM PIPES, 7 mM MgCl2, 1mM EGTA, 150 mM NaCl, and adjust the pH to 6.9.
Wash the cells once with PBS.
Remove the PBS and gently add 1 mL per well of extraction/fixation solution (0.25% glutaraldehyde, 0.3% Triton X-100 in the cytoskeleton buffer) preheated at 37 °C.
Incubate for 60 s at room temperature.
Remove the solution and gently add 1 mL per well of preheated and freshly prepared 4% PFA (paraformaldehyde) solution diluted in the cytoskeleton buffer. Caution: Use gloves and work under the chemical hood.
Place the 12-well plate back in the incubator at 37°C for 10 min.
Remove the PFA and add 3 mL of PBS per well. Note: Work under the chemical hood with gloves and put the recycled PFA in a specific bin.
Wash twice with PBS.
Remove the PBS and add freshly prepared 10 mM NaBH4 solution diluted in PBS in order to quench the autofluorescence coming from glutaraldehyde.
Incubate for 7 min at room temperature.
Put the recycled NaBH4 in a specific bin.
Wash thrice 5 min with PBS.
Remove the PBS and add the blocking solution (5% BSA solution in PBS).
Incubate for 1 h at room temperature (or overnight at 4 °C). Note: The cytoskeleton buffer can be stored at room temperature for a few months. PFA, NaBH4 and glutaraldehyde should be handled with special care (hood, gloves, and specific recycling bins). Solutions should be added and removed gently in order to preserve the integrity of the cells. It is important to not let the cells dry.
4. Immunostaining (Day 2, 5h)
Prepare the solution of primary antibodies using anti-vimentin and anti-tubulin with a 1:500 dilution in the blocking solution.
Put 100 µL drops of diluted primary antibodies on a paraffin film layer and place the coverslips on top of them with cells facing downward.
Incubate cells with primary antibodies for 2 h. Put the coverslips back in a 12-well plate filled with PBS. Rinse thrice for 5 min each time in PBS at room temperature on an orbital shaker.
In the meantime, prepare the secondary antibodies solution using anti-mouse Alexa647 and anti-rat Alexa555 at a dilution of a 1:1, 000 in the blocking solution. Incubate cells with secondary antibodies for 1 h at room temperature. Proceed like 4.2. protecting the cells from light.
Put the coverslip back in a 12-well plate filled with PBS. Rinse thrice for 5 min each time in PBS at room temperature on an orbital shaker. Remove the PBS and add 0.5 mL of PBS mixed with 1 µL of 0.1 µm tetraspeck beads.
Put the wells on an orbital shaker for 30 min. Rinse with PBS. Remove the PBS and incubate the cells for 5 min with a 4% PFA solution in PBS. Rinse thrice in PBS. Note: Fixed cells can be stored for a couple of weeks in PBS at 4°C. Immunostaining should be done at the last minute.
5. Sample preparation (Day 3, 5 min)
Put aliquots of MEA (cysteamine, 1 M, pH 8.3), glucose oxidase (100x, 50 mg/mL), catalase (500x, 20 mg/mL) in an ice basket.
Prepare 50 mL of Tris-NaCl buffer (50 mM Tris, 10 mM NaCl, pH = 8) supplemented with 10% of glucose (100 mg/mL).
Prepare 1.2 mL of imaging buffer just before imaging with 10 mM MEA, 0.5 mg/mL (75 U/mL) glucose oxidase, 40 µg/mL catalase (80 - 200 U/mL) in the Tris-NaCl buffer with 10% glucose.
Mount the coverslip containing the labeled cells on the magnetic sample holder (e.g. chamlide chamber) and add the imaging buffer to fill entirely the chamber. Put the lid on and make sure that there is no air bubble between the imaging buffer and the lid.
Clean the bottom of the sample with absolute ethanol and verify that there is not leak. Use grease to seal the sample holder properly if necessary. Note: Prepare 1 M stock of MEA solution (pH 8.3 adjusted using HCl), stock of glucose oxidase at 50 mg/mL and stock of catalase at 20 mg/mL in water, make aliquots and store them at -20°C for up to one month for MEA and a year for glucose oxidase and catalase. Do not refreeze aliquots. Prepare the Tris-NaCl buffer in advance and store it at room temperature for several months. Add glucose on the day of the experiment (step 5.3).
6. Image acquisition (Day 3, ~1h per cell)
Switch on the microscope. Switch on the acquisition software and press Start system.
Select the Acquisition Tab. Expand the Imaging Setup tool in the Setup Manager tool group. Select the laser WF (wide field) mode of acquisition. Select the 100X NA 1.46 objective. Select the TIRF u-HP mode which uses a collimator to reduce the field of view being illuminated and concentrate the laser energy into a smaller area. Use the optovar lens 1.6x that gives a pixel size of 100nm.
Select the Time series tool and set up the time lapse with 50 000 frames and 0.0 ms between frames. Expand the Channels tool in the Acquisition parameter tool group and select "show all".
Set the Alexa647 track: check the 642 and 405 laser lines for excitation and accept to turn them on. Select the "655 LP" emission filter in the Imaging Setup. Rename "647".
Click on '+' in the Channels tool to add another light pass and select the mode "switch track every: frame" in the Imaging Setup tool. Check 561, 488 and 405 laser lines and accept to turn them on. Select the "BP 570-650+LP750" emission filter and use optovar lens 1.6x. Check that the TIRF-uHP mode is selected. Rename the track "555".
Select the Acquisition mode tool and set the frame size to 200 x 200 pixels. Select the Focus devices and strategy tool and set the definite focus to 1 000 frames (if there is a focus-lock system).
Put oil on the objective and put the sample on. Select the Locate tab, and open the shutter of the fluorescence lamp. Find the focus and look for the cell to image using the eyepiece. Put it in the center of the field of view using the joystick.
Select the Acquisition tab to go back to the acquisition mode. Select the "647" track, set the 642-laser power to 0.2%, set the 405-laser power to 0, set the exposure time to 50 ms and the gain of 50. Choose the Continuous acquisition mode to observe the cell on the screen. Adjust the focus. Make sure there is at least one tetraspeck bead in the field of view, increase the contrast to see them if necessary. Use an auto-scale mode for the display. Take a wide-field epifluorescence image of the vimentin network by clicking on snap and save it. To check the TIRF illumination, click on TIRF, adjust the angle of illumination and/or collimator if necessary to have a homogeneous field of illumination and save it. NOTE: It is important that the TIRF and epifluorescent images are of high quality before starting the raw data acquisition. Discontinuous, non-uniformly labeled filaments will give rise to poor quality dSTORM images.
Uncheck the "647" track and select and check the "555" track. Take an epifluorescence image of the microtubule network and save it. Take a TIRF image and save it too.
Uncheck the "555" track, select and check the "647" track, then press Continuous. Put the gain to 0 and exposure time to 10 ms. Set the 642-nm laser power to 100% in order to pump most of the Alexa647 dyes to the dark state (~2 kW/cm2). Once the fluorophores start blinking, put the exposure to 15 ms and gain to 300. Select the TIRF mode of illumination. Use the Range indicator mode without auto-scale to verify that single molecule signals do not saturate the camera (indicated by the red color). In that case, decrease the gain until the saturation disappears. Tetraspeck beads will remain saturated at the beginning of the acquisition. Press Stop to stop the continuous observation of the cell.
Expand the Online processing tool and check Online processing PALM to visualize the STORM image during acquisition. Use the following parameters: 9 for the Peak mask size (9 pixels diameter), and 6 for the Peak intensity to noise. Theses parameters will define the molecules that are taken into account in the processing (bright enough and not overlapping). In the PAL-Statistics tool, select Plot type: histogram, and Histogram Source: Precision. Press Start acquisition.
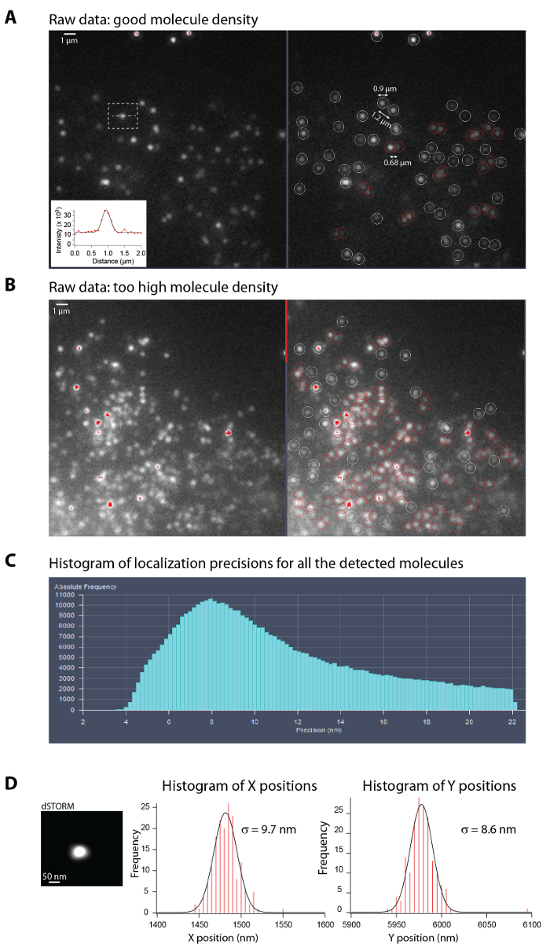
During acquisition, refocus if necessary (if there is no focus-lock device). Adjust the parameters (exposure, laser powers) and eventually switch on the 405-nm laser line (starting at 0.001%) to keep a medium density of fluorophores with a minimum distance of 1 µm between single molecules (> 9 pixels of 100 nm, the Peak mask size) (Figure 1A-B). If the molecule density is a little bit too high, use the HILO (Highly Inclined and Laminated Optical sheet) mode of illumination instead of TIRF. This will increase the laser power by 20%. Make sure that the peak of localization precision on the histogram below the reconstructed image is centered around or below 10 nm. Note: Usually the number of molecules decreases with time, and the 405-nm laser power is increased slowly accordingly. The goal is to decrease the localization precision (histogram displayed below the super-resolution image) as much as possible with a peak below 10 nm (Figure 1C). Increase the contrast of the PALM image if necessary, if too many bright beads are present in the field.
Press Stop to stop the movie when more than 106 localizations have been reached (typically after 40 000 frames). Put the lasers back to 0.2% (642 nm) and 0 (405 nm). Save the raw data of the STORM acquisition with the .czi format. Uncheck track "647" in the Channels tool, and select and check track "555".
Set the exposure time to 25 ms and gain to 0. Press the Continuous button and set both 488 and 561 lasers at 100% to pump Alexa555 dyes to the dark state (~2 kW/cm2 each). Once the fluorophores start blinking, put the gain to 250, use the HILO illumination mode and check that single molecules do not saturate the camera with the range indicator mode. If that is the case, decrease the gain. Press Stop. Decrease the 488-laser power to 50%.
Press Start acquisition. During acquisition, increase progressively the gain to always be at the limit of saturation. Increase step by step the 405-nm laser power to keep a medium density of single molecule in the field of view (See step 6.18). Decrease the 488 laser power to 20-30% when the 405 laser is higher than 15%. Then decrease gradually the 488 while increasing the 405 laser line in order to keep the blinking of the molecules as fast as possible. The 488 laser line coupled with the 561 excitation laser at maximal intensity increases both the on and off rates of the fluorophores, whereas the 405 laser line only increases the on rate.
Stop the acquisition when more than 500 000 localizations have been reached (after about 20 000 frames) or more when no more molecules are blinking. Save the raw data of the STORM acquisition with the .czi format. Note: Always start with the higher wavelength to avoid bleaching (or activation) of the other colors. Since the chamlide chamber is not sealed, it is possible to change the imaging buffer regularly, e.g. to test different buffer conditions. Using the 488 laser line to deplete the Alexa 555 dyes is necessary only when the 561 laser power is limited (with 100 mW lasers).
7. Image reconstruction (5 min)
In the PAL-Drift tool, use the Model-based correction type with automatic segments with a maximum size of 8. Press Apply multiple times in a row until the drift is corrected. This model-based correction applies an algorithm based on cross-correlation analysis which corrects the drift.
Using the PAL-Filter tools, improve the appearance of the STORM image by selecting only the molecules which were best localized. For example, limit the Localization precision to 30 nm and the PSF to 120-180 nm.
Use a pixel resolution of 5 or 10 nm in the PAL-render tool, as well as a Gaussian display mode, with an expansion factor of 1 PSF. Select Render auto dynamic range HR with a 95% value and Render auto dynamic range SWF with a 90% value. Select Render best quality.
Select PALM convert, in the PALM menu of the Processing tab (post-processing mode). Press Select and then Apply. Save the created image with a format. LSM (and not .czi, very important). Note: The image reconstruction can be performed immediately after the acquisition or later using the image processing mode of the acquisition software. In the off line post-processing mode, it is possible to reanalyze the stacks with different values of parameters (peak mask size, peak intensity to noise). Always choose to "discard overlapping molecules" and not "average before localization".
8. Estimation of the localization precision of a STORM image (10 min)
Open your STORM raw data and use the Image processing tab. Select the PALM method and then PALM again. Press Select.
Press Apply to start the reconstruction using a peak mask size of 9 and a peak intensity to noise of 6 (or the parameters you choose for the image).
Select the rectangle Graphics tool and draw a rectangle on the raw image around a single molecule present on the glass surface. There are usually a few single molecules there.
Press Apply again, and only the molecules present inside the rectangle region will be analyzed.
In the PAL-Statistics tool, select Plot type: histogram, and Histogram Source: X position or Y position, to visualize the position histograms.
Save the table with the list of localizations on the left below the images.
Using a software of data analysis, create histograms of X and Y positions (Figure 1D).
Fit the distributions by a Gaussian and save the σX and σY values. The localization precision σSMLM is estimated by (σX*σY)^0.5.
Repeat step 8.3-8.8 on as many molecules as possible and average the σSMLM
9. Channel registration (5 min)
Open the STORM image of each color using the Fiji (Image J) software.
Select the wand tool to measure the positions of each tetraspeck beads.
Calculate the X and Y shifts for each bead and average them.
Use the "Translate" command to translate the second image with the calculated X and Y shifts.
Merge the two images using the Merge Channels command.
Representative Results
A microscope equipped with 50 mW 405 nm and 100 mW 488, 561 and 642 nm solid-state lasers, an EMCCD 512x512 camera, an alpha Plan Apo 100X/1.46 objective and Band Pass 570-650 / Long Pass 655 emission filters was used for the representative results presented below.
Figure 1A gives an example of molecule density and signal to noise ratio that should be used during raw image acquisition. Good quality images are obtained with a minimum of ~1 µm between neighboring molecules. In good blinking conditions, fluorophores should be present in only one frame. Good blinking requires to adjust the imaging buffer conditions (pH, reductive agent, oxygen scavenging system) and laser powers, which influence the on- and off-rates of the fluorophores, and therefore the duty cycle (the fraction of time a fluorophore spends in the on state)9,11. Figure 1B, on the contrary, gives an example of raw image where the molecule density is too high and single molecule cannot be resolved.
Figure 1C shows a typical histogram of localization precisions for all the molecules detected and analyzed by the manufacturer software from a stack of 20 000 frames. This histogram corresponds to the dSTORM image of the microtubule network shown in Figure 3B. Values of localization precisions provided by the manufacturer software are in good agreement with values estimated following step 8, using the pointing precision of a single molecule localized at different time points22 (Figure 1D).
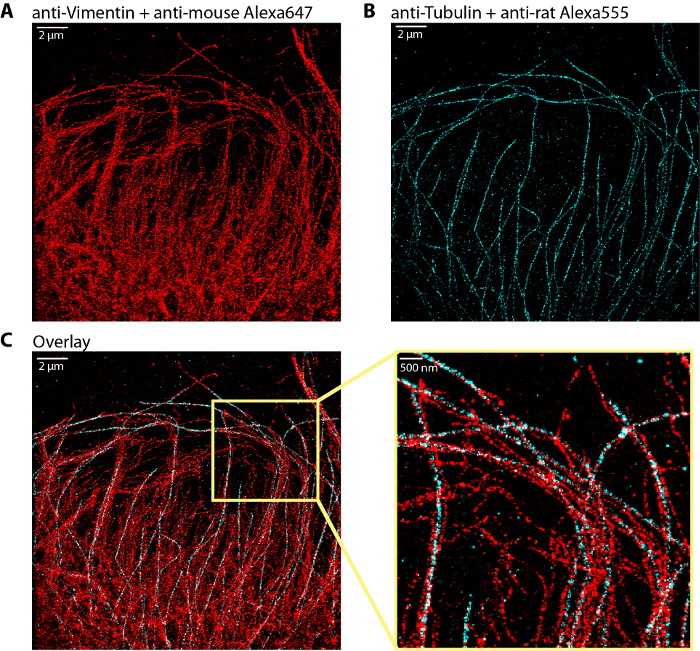
Figure 2A shows a typical example of dSTORM image of vimentin filaments immunolabeled with Alexa647. The increase in resolution can clearly be observed on comparison with the image acquired with a standard wide-field microscope (Figure 2B-C). The fluorescence intensity profiles represented in Figure 2D show that super-resolution microscopy imaging allows resolving vimentin bundles. The dSTORM resolution is sufficient to count the number of filaments present in the bundles.
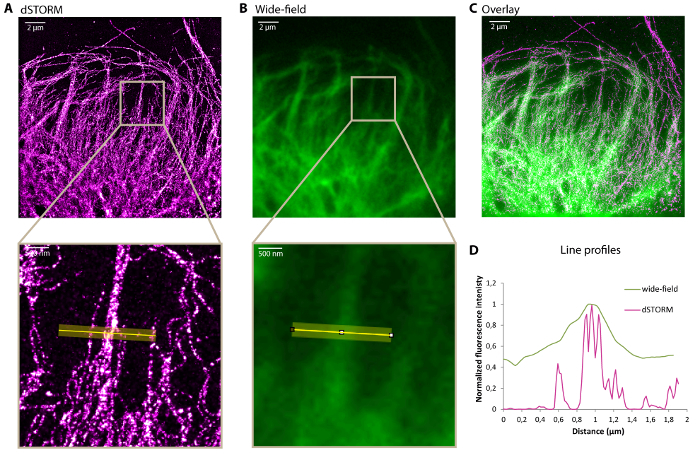
Figure 3 shows a typical example of dual-color dSTORM image of the vimentin and tubulin networks immunolabeled with Alexa647 and Alexa555 respectively. The overlay of the two networks shows that the vimentin bundles are often localized along microtubules, providing key structural information on the coupling between the two cytoskeleton subsystems.
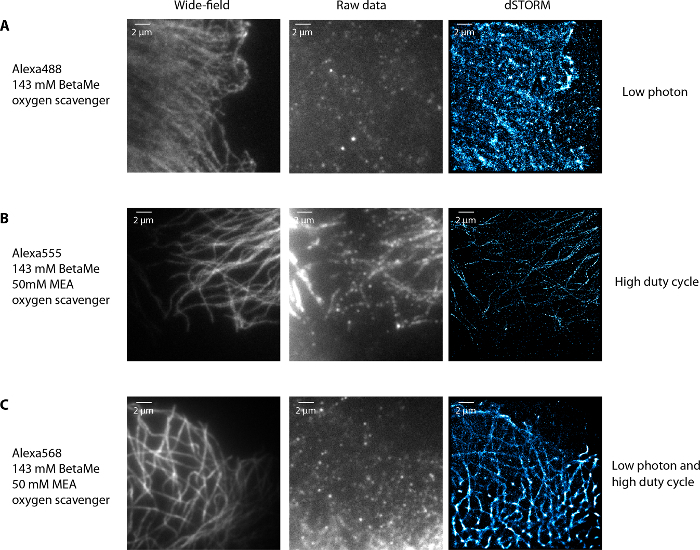
Figure 4 illustrates various examples of dSTORM images of microtubules obtained in non-optimal conditions. First, using Alexa488 and a buffer with 143 mM beta-mercaptoethanol (another reductive agent used instead of MEA9,10,11) and an oxygen scavenging system (composed of 0.5 mg/mL glucose oxidase and 40 µg/mL catalase), the fluorophores were blinking but were not bright enough to be precisely localized (too low photon number) (Figure 4A). Second, using Alexa555 with 143 mM beta-mercaptoethanol, 50 mM MEA and an oxygen scavenging system, the fluorophores could not be pumped to their dark state and therefore not be well spatially separated (too high duty cycle, i.e. the fraction of time in the "on" state is too long) (Figure 4B). Finally, using Alexa568, with 143 mM beta-mercaptoethanol, 50 mM MEA and an oxygen scavenging system, the fluorophores were not bright and were blinking slowly with very long "on" and "off" states (low photon number and too high duty cycle) (Figure 4C). In all these non-optimal conditions, the microtubule network was poorly rendered in the super-resolution images. In conclusion, optimal conditions require fluorophores with high photon number and low duty cycle9,10 in the corresponding imaging buffer. Note that optimizing labeling density and imaging buffer conditions to obtain good blinking conditions is a standard procedure for dual-color dSTORM and is not specific to IF-microtubule imaging.
According to the Nyquist-Shannon theorem, it is necessary to have fluorophores at least every 10 nm to have a resolution of 20 nm. Extended to 2D structures, Dempsey et al estimated that a labeling of ~104 fluorophores per µm2 is necessary. As the IF network is denser and its filaments are thinner (10 nm vs 25 nm diameter without the primary and secondary antibodies) compare to the microtubule network, we observed that twice more localizations are necessary to describe the IF network. We obtained good images with 5 000 localizations/µm2 for IF and 2500 localizations/µm² for microtubules, obtained with 40 000 frames and 20 000 frames respectively. The image with the highest resolution was obtained with a localization precision of σSMLM ~ 8-12 nm estimated following step 8 of the protocol (pointing precision of a single molecule localized at different time points22). This estimation of localization precision was in good agreement with the values provided by the manufacturer software shown in a histogram of localization precisions extracted from all the detected molecules in the raw data (example shown in Figure 1C). Such localization precision could provide an image with a resolution of ~30 nm (2.35*precision) if the labeling density is high enough. We routinely observed vimentin filament with a Full Width at Half Maximum (FWMH) of ~40 nm and microtubules with a width FWMH of ~55 nm. Since the image resolution depends on both the localization precision and the localization density, we provide experimental conditions which allow the best results taking into account both criteria.
Figure 1: Optimal density of single molecules during acquisition and localization precision estimation. (A-B) Left: Representative raw images from a single frame with an optimal density of single molecules in the bright state (A) and with a too high density (B) during STORM acquisition (step 6.12.) of fixed U373 cells stained with anti-vimentin and anti-mouse Alexa647. Right: Raw images where the single molecules with fluorescence level above the Peak intensity to noise threshold (set to 6) are surrounded by circles (white or red). The circle diameter is set by the Peak mask size (set to 9 pixel) and the color determines whether the molecules overlap (red) or not (white). Note that high density of single molecules (in (B)) lead to a high amount of overlapping molecules which are not taken into account for the STORM image reconstruction. A Inset: Graph showing a typical fluorescence intensity profile of a single molecule in red and its Gaussian fit in black. In this example, the localization precision is σx = 6.7 nm. Scale bar, 1 µm. (C) Typical histogram of localization precisions for all the molecules detected and analyzed by the manufacturer software. It can be visualized and is updated regularly during the acquisition of the raw data thanks to the online processing. This histogram corresponds to the dSTORM image of the microtubule network shown in Figure 3B. (D) Left: STORM image of a single molecule presents on the glass surface after reconstruction. Right: Histograms of the X and Y positions obtained after Gaussian fitting. Gaussian fitting the X and Y distributions give an estimate of the localization precision. Please click here to view a larger version of this figure.
Figure 2: Representative dSTORM image of a vimentin network. dSTORM image (A), corresponding standard wide-field image (B) and overlay (C) showing the leading edge of a U373 glial cell fixed as described in the protocol and stained for vimentin with Alexa647. Bottom: zoom of the highlighted region. (D) Transverse intensity profiles along the yellow line on the zoomed images in A and B. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
Figure 3: Representative dual color dSTORM image of vimentin and microtubule networks.(A) Vimentin network stained with Alexa647 (same as in Figure 2A). (B) Microtubule network labeled with Alexa555. (C) Overlay of the vimentin (red) and microtubule (cyan) networks with a zoom region on the right. The dSTORM image of vimentin was obtained with ~1.5 million of localizations out of 40 000 frames. The dSTORM image of microtubules was obtained with ~850 000 localizations out of 20 000 frames. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
Figure 4: Examples of non-optimal imaging conditions of microtubules. When the fluorophores blink but are not bright enough with an Alexa488 labeling of tubulin in U373 glial cells and 143 mM BetaMercaptoethanol and oxygen scavenger in the imaging buffer (A), when the fluorophores are bright enough but cannot be properly depleted in the dark state (at the maximum laser power available) and blink slowly with Alexa555 labeled tubulin in 143 mM BetaMercaptoethanol, 50 mM MEA and oxygen scavenger buffer (B) and when fluorophores blink slowly and are not bright using Alexa568 labeled tubulin in a storm buffer that contains 143 mM BetaMercaptoethanol, 50 mM MEA and oxygen scavenger (C). In all these cases, the dSTORM images have degraded resolution. Scale bar, 2 µm. Please click here to view a larger version of this figure.
Figure 5: Representative dual-color dSTORM image of vimentin and microtubule networks of a U373 cell fixed with cold methanol for 5 min. (A) Vimentin and (B) microtubules networks labeled with Alexa647 and Alexa555 respectively with highlighted zooms. Note the presence of single molecules at the front of the cells localized in the cytoplasm decreasing the global resolution of the filament networks. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
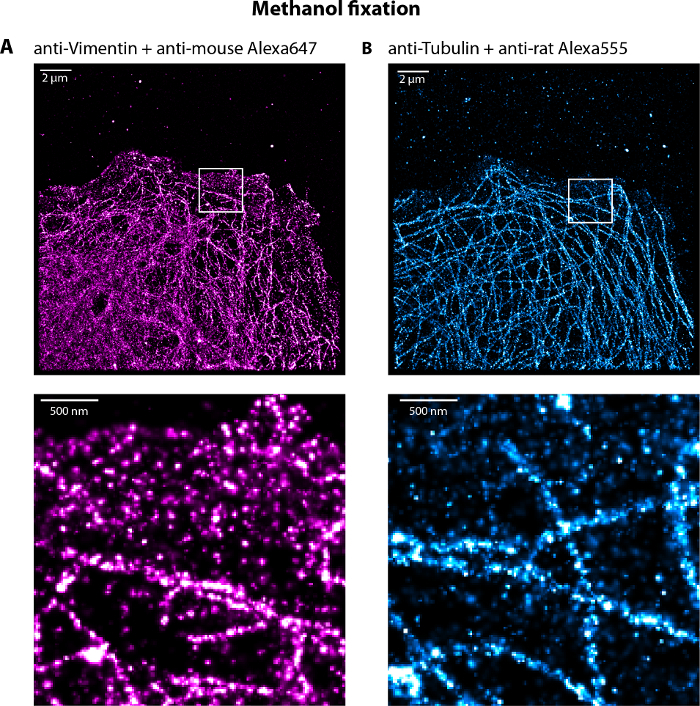
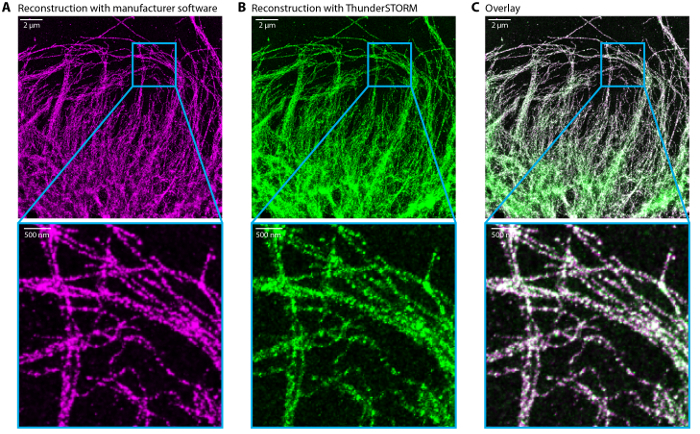
Figure 6:Comparison of dSTORM reconstructed images Reconstructed dSTORM image by the manufacturer software (A) and ThunderSTORM (FiJi plugin) (B). (C) Overlay of (A) in magenta and (B) in green. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
Discussion
Critical steps in the protocol
We present here a protocol which minimizes artifacts in the dSTORM images of microtubules and IFs in glial cells. Artifacts can be created at every step of the sample preparation and imaging: fixation, blocking, immunolabeling, drift during acquisition, non-optimal blinking conditions23. We list below the most critical steps.
Cleaning the coverslips is an important step to limit the non-specific binding of fluorophores on the surface and therefore to limit the background noise in the dSTORM image.
We suggest fixing the sample with a step of extraction and fixation solutions using a mix of glutaraldehyde and triton, and then fixation with PFA (paraformaldehyde) (step n°3). The fixation protocol was adapted from Chazeau et al12: we decreased the extraction/fixation step incubation time to 60 s (step 3.4), remove the glucose from the cytoskeleton buffer, and skipped the extra-step of permeabilization. The advantage of this method is that the freely moving cytoskeletal subunits are drained away, allowing super-resolution imaging with less background12. More generally the fixation step is critical because bad fixation (using old PFA, cold solutions, too long/short incubation steps) lead to partial destruction of the filament networks (broken microtubules, microtubules which do not reach the cell front and/or in a low density). Note that it is also possible to use methanol fixation, especially if only the vimentin network is imaged as described in Leduc et al15. However, background noise can be observed inside the cell (Figure 5). Although dSTORM images obtained with methanol fixed cells are of poorer quality, they still provide information on what both IF and microtubule networks should look like e.g. in term of filament density. Fixation with only glutaraldehyde gave very poor results for IFs, although it is the best protocol for microtubule fixation23.
In the immunostaining part, it is important to use anti-mouse antibodies which minimize cross-reaction with antibodies developed in rat. We could not use Fab fragments (anti-mouse) for this reason, because they cross-react non-specifically with the rat anti-tubulin in addition to the mouse anti-vimentin.
Due to the limited laser power available, it is necessary to use both 488 and 561 nm laser lines to pump the Alexa555 dyes to the dark state. High 488 laser power (~50%) is also necessary during acquisition to observe good blinking and low duty cycle (the fraction of time a fluorophore spends in the on state), although it increases the background noise as well. The TIRF illumination mode limits the excitation of fluorophores in a thin layer (~200 nm above the glass coverslip). It increases the signal-to-noise ratio by decreasing the background light, but also decreases the excitation power. The HILO mode allows axial sectioning and can be efficient to optimize the signal-to-noise ratio. It is therefore a good intermediate between epi and TIRF. Using the HILO mode is essential to excite efficiently the Alexa555 fluorophores, which require high laser power to blink properly.
Another critical step is the acquisition of raw data (step 6). It is necessary to adjust the acquisition parameters (exposure, gain, TIRF and even focus when no focus block device is available) while the movie is acquired. Keeping an optimal density of bright fluorophores is important (Figure 1A): it must be low enough to spatially separate each dye, but high enough to visualize the entire structure after 40 000 frames. If the number of activated molecules is too low, longer stacks should be acquired. We obtained good images with ~ 5000 localizations per µm² for IFs, and 2 500 localizations per cm² for microtubules.
Modifications and troubleshooting of the method
It is important to work with fresh samples and optimized imaging buffers. pH should be precisely adjusted, especially for the MEA.
Vimentin and microtubules should look uniformly labeled when imaged with standard wide-field microscopy technique (WF). If filaments look punctuated/dashed with classical microscopy, this will be amplified when using super resolution. In this case, it is better to prepare new samples. Non-optimized fixation conditions (old PFA etc.) may result in microtubules looking fragmented. High background on the glass surface may arise from an inefficient blocking. In that case, BSA (bovine serum albumin) can be replaced by FBS (fetal bovine serum), and used in each incubation steps.
If the labeling density is too high (Figure 1B) and cannot be sufficiently pushed in the dark off-state, even with the highest laser power, the amount of secondary antibodies should be lowered (better than decreasing the amount of primary antibodies). However, this requires the preparation of new samples. In general, it is necessary to evaluate empirically the quantity of secondary antibodies to use.
If the fluorophores do not blink normally (they should be in the bright state during only one frame), check that the pH of the imaging buffer is correct (pH 8.3). Prepare new solutions if the problem persists (especially MEA). Always check that the TIRF collimator is on (TIRF u_HP). With limited laser power, obtaining proper blinking may be challenging (the on and off rates as well as dye brightness depend on laser power, as described in van de Linde et al11). Other dyes like Alexa488 or Atto488 can be explored if more power of the excitation laser is available14.
If the fluorophores stop blinking before the end of the 40 000 frames, change the imaging buffer which may have been oxidized. Verify that there is no air bubbles between the buffer and the lid after the buffer change. Sealed samples could be imaged for a few hours in a raw.
If STORM movies are acquired on a regular TIRF microscope equipped with an EMCCD, it is possible to use ThunderSTORM24 to reconstruct the images. Similar options are available for the drift correction, image filtering and image rendering. ThunderSTORM is a plugin used with the Fiji/imageJ software. Similar results were obtained with the manufacturer software and ThunderSTORM (Figure 6).
COT (cyclooctatetraene) can be added to the imaging buffer to improve the localization precision of single molecules. It increases the number of photons emitted per cycle for Alexa64725. A better localization precision was indeed observed (down to 6 nm) when adding 2 mM COT to the STORM buffer described in step 5 or with 100 mM MEA as described in reference25. However, in these conditions, the number of bright molecules during acquisition decreases much faster than without COT, limiting the total number of molecule localizations at the end of the acquisition and potentially leading to under-sampling of the IF network. Other imaging buffers were reported to work well with Alexa dyes, for example using lactase and oxyrase as oxygen scavenging system26. In this protocol, we report the buffer composition which gave the best results in term of resolution for our type of samples and imaging set up, but each sample type requires buffer optimization, in addition to labeling and fixation conditions optimization.
Limitations of the method and possible improvements
The method presented here is limited to fixed cells and 2D images. Using a standard imaging set up with a TIRF microscope and an EMCCD, the most limiting factor was the laser powers (100 mW for the 561 nm laser was not sufficient to make the Alexa555 dyes blink). A 532-nm laser line may be more efficient to excite these dyes. A possible improvement of the method would be to perform 3D dSTORM using optical astigmatism27. This would require minimal modifications of the protocol, except for a lower labeling density and an increased frame number. 3D dual color STORM image would provide a better view of IFs wrapped around microtubules, especially in bundles. Note that actin filaments could also be observed using the same fixation method and an optimized amount of fluorescent phalloidin8. 3 color STORM could be used to observe the organization of the three cytoskeletal subsystems.
Using primary and secondary antibodies increases the size of the object of interest since the secondary antibody is localized up to 20 nm from the target. Typically, a 25-nm diameter microtubule will have a width of ~ 50 nm after labeling. One possibility to improve the distance between the target and the dye is to directly label the primary antibodies with the appropriate dyes. Ideally nanobodies labeled with organic dyes should be used28.
We provide here just a simple method to estimate the average localization precision of a single molecule following Endesfelder et al22. The overall resolution of the images that takes into account both localization precision and labeling density, can also be measured using the Fourier Ring Correlation approach29.
Regarding the channel registration, it is difficult to overlay perfectly all the beads present in the field of view. A more complex correction can be found in Chazeau et al 12.
Limitations arising from the blinking of fluorophores and bleaching of the probes can be overcome by using DNA-PAINT (DNA Points Accumulation for Imaging in Nanoscale Topography )30. In this method, the blinking necessary for single molecule localization is created by transient binding of short-dye-labeled oligonucleotides to their complementary targets.
Conclusion
This article presents a robust protocol to perform dual-color dSTORM imaging in 2 dimensions of cytoskeletal filaments in fixed cells.The experimental conditions that were found optimal in terms of fixation, immunolabeling and imaging buffer for our sample type are described in details. Then, the process of raw data acquisition and the type of blinking images that have to be recorded (molecule density, signal to noise ratio, fluorophore duty cycle etc) is presented as well as how to reconstruct dSTORM images, correct the drift and filter data. These steps are generic for dual-color dSTORM imaging and not sample type specific. Finally, it is shown how this technique helps to resolve the dense organization of the two coupled cytoskeletal networks.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
We thank Mickael Lelek, Orestis Faklaris and Nicolas Bourg for fruitful discussion, Andrey Aristov and Elena Rensen for help with the super-resolution technique and Shailaja Seetharaman for careful reading of the manuscript. We gratefully acknowledge the UtechS Photonic BioImaging (Imagopole) Citech of Institut Pasteur (Paris, France) as well as the France-BioImaging infrastructure network supported by the French National Research Agency (ANR-10-INSB-04; Investments for the Future), and the Région Ile-de-France (program Domaine d'Intérêt Majeur-Malinf) for the use of the Elyra microscope. This work was supported by the the Ligue Contre le Cancer and the French National Research Agency (ANR-16-CE13-0019).
References
- Herrmann H, Aebi U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb Perspect Biol. 2016;8(11) doi: 10.1101/cshperspect.a018242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber F, Boire A, Lopez MP, Koenderink GH. Cytoskeletal crosstalk: when three different personalities team up. Curr Opin Cell Biol. 2015;32:39–47. doi: 10.1016/j.ceb.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Leduc C, Etienne-Manneville S. Intermediate filaments in cell migration and invasion: the unusual suspects. Curr Opin Cell Biol. 2015;32:102–112. doi: 10.1016/j.ceb.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Heilemann M, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47(33):6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Gevaux D. Nobel Prize in Chemistry: seeing the nanoscale. Nat Nanotechnol. 2014;9(11):878. doi: 10.1038/nnano.2014.267. [DOI] [PubMed] [Google Scholar]
- Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3(10):793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf DJ, Edwards R, Kumarswami N, Knight AE. Test samples for optimizing STORM super-resolution microscopy. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8(12):1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey GT. A user's guide to localization-based super-resolution fluorescence imaging. Methods Cell Biol. 2013;114:561–592. doi: 10.1016/B978-0-12-407761-4.00024-5. [DOI] [PubMed] [Google Scholar]
- van de Linde S, et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc. 2011;6(7):991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- Chazeau A, Katrukha EA, Hoogenraad CC, Kapitein LC. Studying neuronal microtubule organization and microtubule-associated proteins using single molecule localization microscopy. Methods Cell Biol. 2016;131:127–149. doi: 10.1016/bs.mcb.2015.06.017. [DOI] [PubMed] [Google Scholar]
- Olivier N, Keller D, Rajan VS, Gonczy P, Manley S. Simple buffers for 3D STORM microscopy. Biomed Opt Express. 2013;4(6):885–899. doi: 10.1364/BOE.4.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson C, et al. Intermediate filament protein partnership in astrocytes. J Biol Chem. 1999;274(34):23996–24006. doi: 10.1074/jbc.274.34.23996. [DOI] [PubMed] [Google Scholar]
- Leduc C, Etienne-Manneville S. Regulation of microtubule-associated motors drives intermediate filament network polarization. J Cell Biol. 2017;216(6):1689–1703. doi: 10.1083/jcb.201607045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z, et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016;3(5):500–501. doi: 10.1016/j.cels.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuizen RP, et al. Co-Orientation: Quantifying Simultaneous Co-Localization and Orientational Alignment of Filaments in Light Microscopy. PLoS One. 2015;10(7):e0131756. doi: 10.1371/journal.pone.0131756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wang KK. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015;38(6):364–374. doi: 10.1016/j.tins.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslehaty H, Cordovi S, Hefti M. Symptomatic spinal metastases of intracranial glioblastoma: clinical characteristics and pathomechanism relating to GFAP expression. J Neurooncol. 2011;101(2):329–333. doi: 10.1007/s11060-010-0257-y. [DOI] [PubMed] [Google Scholar]
- Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121–130. doi: 10.1016/j.ceb.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Skalli O, et al. Astrocytoma grade IV (glioblastoma multiforme) displays 3 subtypes with unique expression profiles of intermediate filament proteins. Hum Pathol. 2013;44(10):2081–2088. doi: 10.1016/j.humpath.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Endesfelder U, Malkusch S, Fricke F, Heilemann M. A simple method to estimate the average localization precision of a single-molecule localization microscopy experiment. Histochem Cell Biol. 2014;141(6):629–638. doi: 10.1007/s00418-014-1192-3. [DOI] [PubMed] [Google Scholar]
- Whelan DR, Bell TD. Image artifacts in single molecule localization microscopy: why optimization of sample preparation protocols matters. Sci Rep. 2015;5:7924. doi: 10.1038/srep07924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesny M, Krizek P, Borkovec J, Svindrych Z, Hagen GM. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics. 2014;30(16):2389–2390. doi: 10.1093/bioinformatics/btu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier N, Keller D, Gonczy P, Manley S. Resolution doubling in 3D-STORM imaging through improved buffers. PLoS One. 2013;8(7):e69004. doi: 10.1371/journal.pone.0069004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahidiazar L, Agronskaia AV, Broertjes J, van den Broek B, Jalink K. Optimizing Imaging Conditions for Demanding Multi-Color Super Resolution Localization Microscopy. PLoS One. 2016;11(7):e0158884. doi: 10.1371/journal.pone.0158884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319(5864):810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova M, et al. Resolving bundled microtubules using anti-tubulin nanobodies. Nat Commun. 2015;6:7933. doi: 10.1038/ncomms8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuizen RP, et al. Measuring image resolution in optical nanoscopy. Nat Methods. 2013;10(6):557–562. doi: 10.1038/nmeth.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F, Jungmann R. Super-resolution microscopy with DNA-PAINT. Nat Protoc. 2017;12(6):1198–1228. doi: 10.1038/nprot.2017.024. [DOI] [PubMed] [Google Scholar]