

Abstract
Antibody-conjugates (ACs) modified with virus-derived peptides are a potentially powerful class of tumor cell delivery agents for molecular payloads used in cancer treatment and imaging due to increased cellular accumulation over current ACs. During early AC in vitro development, fluorescence techniques and radioimmunoassays are sufficient for determining intracellular localization, accumulation efficiency, and target cell specificity. Currently, there is no consensus on standardized methods for preparing cells for evaluating AC intracellular accumulation and localization. The initial testing of ACs modified with virus-derived peptides is critical especially if several candidates have been constructed. Determining intracellular accumulation by fluorescence can be affected by background signal from ACs at the cell surface and complicate the interpretation of accumulation. For radioimmunoassays, typically treated cells are fractionated and the radioactivity in different cell compartments measured. However, cell lysis varies from cell to cell and often nuclear and cytoplasmic compartments are not adequately isolated. This can produce misleading data on payload delivery properties. The intravenous injection of radiolabeled virus-derived peptide-modified ACs in tumor bearing mice followed by radionuclide imaging is a powerful method for determining tumor targeting and payload delivery properties at the in vivo phase of development. However, this is a relatively recent advancement and few groups have evaluated virus-derived peptide-modified ACs in this manner. We describe the processing of treated cells to more accurately evaluate virus-derived peptide-modified AC accumulation when using confocal microscopy and radioimmunoassays. Specifically, a method for trypsinizing cells to remove cell surface bound ACs. We also provide a method for improving cellular fractionation. Lastly, this protocol provides an in vivo method using positron emission tomography (PET) for evaluating initial tumor targeting properties in tumor-bearing mice. We use the radioisotope 64Cu (t1/2 = 12.7 h) as an example payload in this protocol.
Keywords: Bioengineering, Issue 133, Viral-derived peptides, Antibody-conjugates, SDS-PAGE, Confocal microscopy, Cellular fractionation, Copper-64, PET imaging
Introduction
Antibody-conjugates (ACs) are biopharmaceuticals that are maturing into a transformative class of effective drugs for improving cancer treatments and for detecting tumors. Composed of a monoclonal antibody (mAb) conjugated to molecular payloads such as radioisotopes, small molecules, and biological toxins, ACs are able to deliver these payloads to cancer cells with exquisite target antigen affinity and specificity. Thus ACs have the potential to significantly reduce nonspecific toxicity and increase payload activity at the tumor site. Therapeutically, ACs transporting cytotoxic small molecules (commonly referred to as antibody-drug conjugates) have been approved for treating patients with breast cancer and Hodgkin's lymphoma who have failed conventional treatments 1,2. In addition, ACs transporting radioisotopes (commonly referred to as radioimmunoconjugates) are also in development. An AC transporting a radioisotope for imaging is approved for identifying prostate cancer metastasis 3. With many more therapeutic ACs submitted for approval 4, optimism is high for the future of ACs to improve cancer care 5.
Nonetheless, when delivering chemotherapeutics or radioisotopes, ACs have difficulty effectively accumulating these payloads inside target cells. This aspect significantly contributes in many cases in the inability of ACs to provide long-lasting disease-free survival or high contrast tumor imaging 6,7. In general, once ACs bind their target antigen they are internalized through a process known as receptor-mediated endocytosis. The ACs are then entrapped inside endosomes and trafficked to lysosomes for degradation and payload release 8. The intracellular trafficking process poses challenges for ACs to achieve a high payload specificity and efficacy against target cancer cells. For example, many antigens such as Her2 (target for therapeutic AC Trastuzumab-emtansine) can recycle up to 85% of bound antibodies in the first 30 min 9. Furthermore, once degradation occurs, released chemotherapeutics and radioisotopes can be actively exported by increased expression and/or activity of membrane associated transport proteins 10,11. Lysosome degradation also impedes the delivery of novel biological payloads such as therapeutic enzymes and oligonucleotides that can be deactivated 12,13. In essence, the cancer cell is highly effective at abrogating the necessary intracellular accumulation of payloads delivered by ACs.
This protocol describes how to implement the concept of ACs-coupled to virus-derived peptides, specifically for escaping endosome entrapment and localizing to the cell nucleus. With such sophistication to manipulate host cell systems, it is not surprising that the development of virus-derived proteins and peptides as potential biopharmaceuticals has long been ingrained in therapeutic research 14. For millions of years viruses have evolved to acquire an exceptional collection of proteins able to exploit normal physiological mammalian cell systems in order to effectively enter host cells. For viruses that are internalized via receptor-mediated endocytosis, they are also challenged with escaping trafficking to the lysosome where the onslaught of a localized concentration of proteases can be problematic for survival. A well characterized viral-derived peptide utilized in drug delivery for escaping endosome entrapment is the human immunodeficiency virus transactivator of transcription (Tat) protein 15. Tat is able to escape endosome entrapment by sensing low-pH at which point protein conformational changes occur enabling Tat to insert itself into and disrupt the endosomal membrane 16. This results in Tat-payload conjugates able to access the cytoplasm. The second viral manipulation element related to this protocol is the approach used to deliver therapeutic genes and drugs to the nucleus 17. Viruses have evolved to successfully manipulate host cell machinery for progressing past the nuclear membrane by passing through the nuclear pore complex (NPC). Cellular macromolecules contain (or bind to proteins that contain) nuclear localization signals (NLSs) necessary for binding to nuclear transport proteins (e.g. karyopherins α and β), which provide the required movements through the NPC. Viruses have developed proteins to contain NLS sequences that provide them with the ability to utilize host cell transport proteins for shuttling into the nucleus 18.
Numerous ACs have previously been functionalized with Tat- and NLS-derived peptides and tested for their ability to accumulate inside cancer cells and for targeting tumors 19,20,21,22,23,24,25,26,27,28,29,30 (Table 1). Studies delivering cytotoxic payloads have demonstrated that ACs modified with virus-derived peptides are able to significantly increase cellular accumulation, cytotoxicity, and tumor killing over unmodified ACs 22,26. A common feature for this novel class of AC is their construction. Typically, peptides contain a terminal cysteine providing a free sulfhydryl group. MAbs are first reacted with a noncleavable bifunctional crosslinker containing N-hydroxysuccinimide (NHS) and maleimide groups at opposite ends. The NHS esters react with primary amines on the mAb to form amide bonds. The reacted mAb with free maleimide groups is then reacted with the sulfhydryl groups on the peptides to form a thioester bond and thus linking the peptide and mAb. Although homobifunctional crosslinkers have been used 28, heterobifunctional crosslinker are more commonly used in the construction of virus-derived peptide-ACs 22,23,26,31,32. This protocol specifically uses the crosslinker sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) for its ease of use and because it is used in the approved antibody-drug conjugate Trastuzumab-emtansine and in many virus-derived peptide-ACs 8,22,23,26,31,32. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) is the primary method for initially determining conjugation efficiency and for semi-quantifying the number of peptides per mAb. Confocal microscopy using a fluorescently-labeled secondary antibody specific to the mAb is typically the method for initially evaluating intracellular distribution properties of virus-derived peptide-modified ACs. Thus far, radioisotopes are the primary payloads delivered by virus-derived peptide-modified ACs. Radioisotopes are advantageous because radioactivity in cells is easily quantified by gamma counting. In addition, ACs that are translated into mouse models of human cancers provide researchers with the ability to evaluate tumor targeting using molecular imaging modalities such as single photon emission computed tomography and positron emission tomography (PET) 23,32,33. In general, the construction and validation testing methods primarily used by researchers provide a very good assessment of ACs modified with virus-derived peptides during the initial development stage to effectively enter and deliver the payload inside target cells and to target tumors.
Tat- and NLS-modified ACs have illuminated key areas for further improving payload delivery inside cancer cells and to tumors. With respect to NLS-modified ACs, the efficiency in intracellular accumulation can be modest 23,31,34. Inefficient intracellular accumulation is caused by continued endosomal entrapment. In vivo tumor targeting can also be diminished with both Tat- and NLS-modified ACs. The active sequences of Tat and NLS contain several positive charged residues. When attached to mAbs, the overall cationic charge can be significantly increased 35. As a consequence, the Tat- and NLS-modified ACs have increased uptake in healthy tissues and increased rapid blood clearance.
Our group developed a composite compound consisting of cholic acid linked to NLS (ChAcNLS; Figure 1). ChAcNLS-modified ACs are able to increase intracellular accumulation of delivered radioisotopes and improve tumor targeting compared to NLS-modified and traditional ACs 33,34. The mechanism behind cholic acid is inspired by the ability of select nonenveloped viruses that cannot rely on membrane fusion to utilize cholic acid to trigger endosome escape through the formation of ceramide. For example, porcine enteric virus recruits cholic acid that activates sphingomyelinase, which catalyzes the hydrolysis of sphingomyelin into ceramide 36,37,38. This destabilizes endosomal membrane and allows for virus escape. Thus, cholic acid is another virus-derived component that complements NLS.
As this field moves forward and future advancements occur in payload delivery by ACs modified with virus-derived peptides, it is an opportune time to provide visual demonstrations of their biochemical and functional characteristics during initial development. Here, we describe our protocol for the initial evaluation of virus-derived peptide-modified ACs for the efficient yet simple determination of intracellular accumulation and tumor targeting during early stage development. We use the commercially available mAbs 7G3 and A14 as example model systems. Procedure 1 describes the use of SDS-PAGE as a method that allows for ‘go/no go’ decisions for constructed ACs. Procedure 2 describes a method using trypsinization allowing for improved visualization of AC intracellular distribution and accumulation. Procedure 3 describes a method for improved intracellular fractionation to accurately determine nuclear localization. In this procedure we utilize the payload 64Cu (t1/2 = 12.7 h) because it is vulnerable to cellular efflux and is a positron emitter 10. Thus, Procedure 4 describes in vivo tumor targeting characterization by PET imaging to visualize tumor uptake relative to background (i.e. nontarget healthy tissues) and determine whether the example AC can specifically and effectively target tumors. These methods are sufficient for investigators developing ACs modified with virus-derived peptides to identify candidates for further advancement.
Protocol
The in vivo animal experiments described were performed according to an approved protocol and under the ethical guidelines of the Centre Hospitalier Universitaire de Sherbrooke Ethics Committee for Animal Experiments.
1. Antibody Peptide Conjugation
NOTE: ChAcNLS can be synthesized at any commercial peptide manufacturer or university-affiliated peptide synthesis service platform. The synthesis of ChAcNLS can be found in reference 34. For Procedures 1 and 2 use the mAb 7G3, which is specific for the leukemia antigen IL-3Rα 39.
In a 1.7 mL microcentrifuge tube, prepare a 10 mg/mL (~1 mg total antibody) solution of 7G3 in phosphate buffered saline (PBS), pH 7.6.
Dissolve sulfo-SMCC in dimethyl sulfoxide (DMSO) at a concentration of 5-10 mM. Vortexing the solution thoroughly works best in order to achieve complete dissolution.
Add to the tube containing 7G3 the dissolved sulfo-SMCC solution (typically between 5-20 μL depending on the molar ratio of sulfo-SMCC-to-7G3 desired). Testing sulfo-SMCC-to-7G3 ratios of 10-, 25-, and 50-to-1 is recommended. Incubate at room temperature for 1 h.
- Purify and concentrate the maleimide-derivatized 7G3 by using an ultrafiltration device and buffer exchange in PBS, pH 7.0. Centrifuge at 8,000 x g for approximately 5 min. Discard flow through and fill with PBS, pH 7.0 and centrifuge again.
- Perform this 7-8 times to completely exchange the buffer. Recover the concentrated 7G3 protein by placing the filter device upside down in a clean microcentrifuge tube. Centrifuge for 2 min at 1,000 x g. Recovery volume should be approximately 0.3 mL
In the tube containing the maleimide-activated 7G3, add 2-fold molar excess of ChAcNLS relative to the sulfo-SMCC-to-7G3 ratio used in the previous step and incubate overnight at 4 °C. For example, if a 10-to-1 sulfo-SMCC-to-7G3 ratio was used, then use a 20-to-1 ChAcNLS-to-7G3 ratio. Total volume should not be significantly changed. NOTE: Although ChAcNLS does not cause precipitation during the conjugation process this is a possibility that could occur with other peptides. Observe the tube during the reaction for signs of precipitation, which is most likely caused when peptides are hydrophobic. In these cases it may be worth revisiting the peptide of interest in order to design changes to provide increased hydrophilicity.
Concentrate the 7G3-ChAcNLS using an ultrafiltration device to desired concentration and buffer exchange in PBS pH 7.4 (see step 1.4). The recovery yield of total protein should be ≥ 75% of the original starting material.
Perform SDS-PAGE to evaluate the constructed 7G3-ChAcNLS candidates using a 12% polyacrylamide gel (provides sufficient separation of the ChAcNLS conjugated heavy and light chains from nonconjugated chains). Mix 10 μg of 7G3-ChAcNLS in PAGE loading buffer containing 2 μL β-mercaptoethanol and load into well.
Set the appropriate voltage (120 V for 1 h for a 12% gel) and run the electrophoresis. Stop the electrophoresis when the Coomassie dye front reaches the bottom of the gel. NOTE: Do not let the Coomassie dye front run off the gel.
Remove gel from electrophoresis apparatus and rinse with destaining solution containing 10% v/v of glacial acetic acid and 20% v/v methanol.
Take a photo of the gel and with a ruler and measure the distance (cm) migrated for the standards and 7G3 conjugates. Calculate the retention factor (Rf) value, which is the distance migrated divided by the gel length (from where protein is loaded to the Coomassie migration front). Plot the molecular weight (MW) in log against the Rf values from each protein standard and extrapolate the size of the 7G3 conjugates.
Calculate the number of peptides per antibody by dividing the difference in MW between 7G3 conjugates and unmodified 7G3 by 1768.5 g/mol (MW of ChAcNLS).
Take digital images of the Coomassie-stained gel and perform densitometry analysis. At this point, selection of a lead candidate can be based on the AC with the maximum number of ChAcNLS molecules that does not contain significant aggregate species.
2. Confocal Microscopy for Intracellular Accumulation Evaluation
NOTE: It is important to test the cell selectivity of intracellular accumulation of the peptide-modified mAbs prior to developing formulations with a payload of interest. Because Tat has also been shown to have a propensity for nonspecific cell penetration, it is worth first analyzing intracellular accumulation and cell selectivity prior to undertaking costly development steps with expensive payloads. For this reason, Procedure 1 should also modify isotype specific irrelevant control mAbs. For the rest of the protocol we will work with ACs modified with 10 ChAcNLS molecules per antibody. A schematic including key steps for Procedures 2 and 3 is described in Figure 2.
CAUTION: This step of the protocol involves the handling and manipulation of paraformaldehyde. Please follow manufacturer instructions when handling.
Treat target TF-1a cells with 200 nM of 7G3-ChAcNLS including modified control mAbs. Incubate 5 x 106 antigen-positive cells with the conjugates for 1 h at 37 °C.
After 1 h, remove supernatant and wash with 1 mL 3x in ice-cold PBS. Add fresh media and incubate for an additional hour at 37 °C.
- After the additional hour, remove supernatant and wash 3x in 1 mL ice-cold PBS. Add 0.5 mL of PBS containing 0.25% trypsin and ethylenediaminetetraacetic acid (EDTA) at 37 °C for 3 up to 30 min. NOTE: During initial pilot testing it is recommended to not trypsinize cells in order to determine the differences in AC intracellular accumulation. This protocol promotes the use of trypsin and we demonstrate how this improves assessment of intracellular accumulation efficiency of different AC candidates.
- Test the different times for incubation by visually checking for all cells being detached. In addition, incubate cell aliquots with viability staining solutions and perform flow cytometric analysis as previously described 40.
- Neutralize trypsin with 1.5 mL of cell culture RPMI 1640 media/10% fetal bovine serum. Centrifuge cells at 500 x g for 5 min, remove supernatant, and wash 3x in 0.5 mL ice-cold PBS.
Fix cells in 0.5 mL PBS containing 1% paraformaldehyde and 1% sucrose on ice for 30 min. Wash cells 3x in 0.5 mL ice-cold PBS and centrifuge at 250 x g for 5 min. Permeabilize cells in 0.5 mL PBS containing 0.15% Triton X-100 for 5 min on ice. Wash cells in ice-cold PBS and repeat centrifugation.
- Suspend cells in 0.1 mL PBS containing 2 μg/mL (or the manufacturer recommended concentration) of an anti-murine (isotype specific) Fc secondary polyclonal antibody conjugated to AlexaFluor 647 for 1 h at room temperature in the dark.
- Centrifuge at 250 x g for 5 min and wash cells 3x in 0.5 mL ice-cold PBS. Suspend cells in 0.5 mL PBS. PAUSE: Cells can be stored in refrigerator. NOTE: It is essential to select a secondary antibody that recognizes the correct isotype of the mAb of interest. AF647 is selected because of its far-infrared emission, which does not interfere with propidium iodide (PI) staining of the nucleus.
Add PI to a concentration of 10 μg/mL to the container with cells. Next, mount 1 x 105 cells onto glass slides using mounting media and cover with a glass coverslip.
Examine cells with a Plan Apo 60X oil immersion objective NA 1.42 on an inverted laser scanning confocal microscope. Detect PI fluorescence using the 488 nm argon laser and the spectral scanning prism set for 600 - 650 nm. For AF647 fluorescence use the 633 nm helium-neon laser and the spectral scanning prism set for 650 - 700 nm. Collect fluorescence emissions from PI and AF647 sequentially. NOTE: Use the same setting throughout the evaluation and comparison of the cells. However, you will have to adjust settings for trypsinized cells compared to non-trypsinized cells.
Use microscope software to acquire images. Collect images using serial horizontal optical sections of 1,024 x 1,024 pixels with 2X line averaging taken at 0.5 μm intervals through the entire cell thickness. Present the images as stacked z-projections from four consecutive slices at the maximum fluorescence intensity.
Analyze cells with microscope software. Record the cellular distribution pattern of the conjugates. Specifically, evaluate whether intracellular fluorescence in the cytoplasm is grouped and near the cell surface or diffuse and homogeneous. Also evaluate the relative fluorescence intensity per cell.
3. Radiolabeled AC Construction and Cellular Fractionation for the Evaluation of 64 Cu Intracellular Delivery Efficiency
CAUTION: Procedures 3 and 4 involve the handling and manipulation of radioactivity. Before performing these steps, researchers should have approved safety training and protocols approved from their home institution's radiation safety authority.
NOTE: For Procedures 3 and 4 we use the mAb A14, which is specific for the invasive bladder cancer antigen IL-5Rα41. Also, all experiments are time sensitive due to the short half-life of 64Cu. In general, it is best to not wait past 1 week to perform in vitro experiments and no longer than 72 h for in vivo studies.
In a 1.7 mL tube suspend 2,2'-(7-(2-((2,5-dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NOTA-NHS) in DMSO to a concentration of 10 mM.
React 50-fold molar excess of NOTA-NHS with A14 (2 mg starting material) in 0.1 M sodium bicarbonate at pH 8.6 for 1 h at room temperature in a volume ≤ 0.5 mL (the final NOTA-NHS volume should not exceed 2% v/v of total volume).
Purify and buffer-exchange NOTA-A14 using an ultrafiltration device in PBS, pH 7.6 (see step 1.4).
For subsequent attachment of ChAcNLS, follow steps 1.2-1.6.
React NOTA-A14-ChAcNLS (250 μg batches) with 100 megabecquerel (MBq) of 64CuCl2 in 0.1 M sodium bicarbonate, pH 5.5 for 1 h at 37 °C ≤ 0.1 mL. 64Cu is produced on the TR-PET cyclotron at the CIMS 42.
Concentrate 64Cu-A14-ChAcNLS in PBS pH 7.4 using an ultrafiltration device to the desired concentration (see step 1.4).
- Determine the radiolabeling efficiency of 64Cu-A14-ChAcNLS by instant thin-layer chromatography (ITLC) using chromatography strips and 0.1 M, pH 5.5 sodium citrate (ITLC eluent) as solvent. Apply 0.5 μL aliquot of the reaction solution to the ITLC strip. Perform an autoradiograph of the strip and obtain a digital image.
- Perform densitometry to obtain the proportion of bound and free 64Cu. If free 64Cu content is > 5%, return to previous step and perform additional purification.
- In addition, perform SDS-PAGE with Coomassie staining to evaluate aggregations. Perform a second SDS-PAGE followed by an autoradiograph of the gel to determine if there are radiolabeled aggregate species.
- Affinity binding quality check point: Incubate 64Cu-A14 conjugates at increasing concentrations (0-100 nM) in duplicate with 1 x 106 target cells per mL. To estimate nonspecific binding, mix duplicate samples of 64Cu-A14 conjugates with cells in the presence of 100-fold molar excess of unlabeled A14.
- After 1 h incubation on ice, wash cells and count total bound antibody samples and total nonspecific bound antibody samples in a gamma counter. Graph specific binding (total bound antibody - nonspecific bound antibody) against reactive free 64Cu-A14 (nM) used in the assay. Determine the dissociation constant (Kd) by nonlinear regression using graphing software.
For 64Cu cellular accumulation studies: Treat cells with 100 nM of the 64Cu-labeled A14 conjugates for 1 h, 6 h, and 24 h at 37 °C as previously described33. Include additional parameters such as treatment in the presence of unmodified A14 to block IL-5Rα sites and at 4 °C to block receptor-mediated internalization.
At the defined time points, as previously described in step 2.3 - 2.3.2, add 0.5 mL of PBS containing 0.25% trypsin and EDTA at 37 °C followed by neutralization and washing.
Incubate cells in plasma membrane lysis buffer containing 1% NP-40, 10 mM Tris pH 7.5, 10 mM NaCl, 3 mM MgCl2 buffer on ice for 10 min. Centrifuge the plasma membrane-lysed cells at 90 x g for 5 min.
Remove supernatant and place in fresh tube. This represents cytoplasmic fraction. Wash nuclei 3x in ice-cold PBS and add washes to the cytoplasmic fraction.
Ensure vials containing the nuclear and cytoplasmic fractions are sealed. Then place them in a gamma counter calibrated for 64Cu in order to convert raw counts to MBq.
- Determine the quality of nuclei isolation by performing Western blot analysis for Lamin A/C, a restricted nuclear protein, and Rab7, an abundant cytoplasmic protein on whole cell lysate, nuclear and cytoplasmic fractions.
- Treat 5 x 106 cells with 500 μL of radio-immunoprecipitation assay (RIPA) buffer and the plasma membrane lysis buffer in step 3.12 containing 1%, 2%, and 4% NP-40. Furthermore, treat the isolated nuclei in 500 μL of RIPA buffer to obtain isolated nuclear proteins.
- Precipitate isolated nuclear, cytoplasmic and whole cell proteins using 4x the sample volume of cold acetone for 60 min at -20 °C. Centrifuge at 13,000 x g for 10 min and decant supernatant being careful not to dislodge the protein pellet. Add 100 μL of PBS to dissolve proteins.
- Load 10 μg of protein into each well of a 10% SDS-PAGE gel. Perform electrophoresis as described in 1.7-1.9. Perform Western Blot transfer as previously described in refeference 43.
- Identify the ladder markers that best correspond to a MW of ~40 kDa. Cut the membrane in half at this marker. Probe the blot containing the higher MW proteins with Lamin A/C-specific antibodies. Probe the membrane with the lower MW proteins with the Rab 7-specific antibodies. Western blot is a well known technique and not described in this protocol.
4. PET Imaging Evaluation of Tumor Targeting
NOTE: The techniques of implanting tumor cells in mice to generate heterotopic xenografts are well known and most laboratories have in-house protocols tailored for their tumor system. Thus, this is not covered in the protocol. The xenograft model will be nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice bearing IL-5Rα-positive invasive bladder tumors HT-1376 and HT-B9. IL-5Rα-positive invasive bladder tumor cells comprise > 66% of total cells in the xenograft. In contrast, only ~11% of IL-5Rα-positive HT-B9 cells were contained in developed xenografts41. Thus this model provides an excellent example for evaluating AC tumor targeting in two tumors that represent foreseeable patient tumor heterogeneity.
In groups containing ≥ 4 (NOD/SCID) mice, inject 20-30 μg (6-9 MBq) of 64Cu-A14-ChAcNLS, 64Cu-A14-NLS, and 64Cu-A14 into the tail vein.
On the same day place a cylindrical phantom (24.8 mL) containing 5 MBq of 64Cu to convert the radioactive counts per second into injected dose per gram of tissue (%ID/g).
Wait 48 h and then anesthetize mice by placing them in an induction chamber with 1.5 L/min of oxygen flow with 2% isoflurane. Apply sterile ophthalmic ointment to mouse eyes after induction and before placement into PET scanner.
Rapidly transfer the mouse to a PET scanner table in the headfirst prone position with a nose cone for isoflurane. Position the respiration and rectal probes and monitor temperature and respiration rate to ensure that physiological conditions are maintained during the experiment as previously described 44.
Start the PET data acquisition using a regular sampling mode setting and an energy window of 250 - 650 keV. Typically, a 45-minute static scan per mouse is sufficient to provide sufficient coincident events for reconstruction and production of high quality PET images.
After the 64Cu-AC scan, remove mouse from the scanner and place it back in the cage. Allow for the mouse to sufficiently recover before returning to the animal facility.
Obtain reconstructed images using 20 iterations of a three-dimensional Maximum Likelihood Expectation Maximization algorithm.
- Perform region of interest (ROI) analysis of the tumor and visually assessable organs using the AMIDE software.
- Draw ROIs to obtain quantitative %ID/g data by manually using the 3D Freehand tool setting and carefully delineate the tumors or the adjacent thigh muscle. The counts per pixel in the ROI will be converted to %ID/g from previous step 4.2.
Compare 64Cu-A14, 64Cu-A14-ChAcNLS, 64Cu-A14-NLS images for tumor contrast, and quantitative ROI %ID/g, and tumor-to-background ratios.
Representative Results
For Procedure 1, the construction of 7G3 modified with ChAcNLS using sulfo-SMCC as a crosslinker is very reliable. Typically, when loaded onto a 12% gel and analyzed by SDS-PAGE, this results in distinguishable stepwise increases in MW proportional to increasing sulfo-SMCC-to-7G3 ratios used and allows for the heavy and light chains to be individually assessed for ChAcNLS conjugation (Figure 3). 7G3 reacted at 10-, 20-, 25-, and 50-to-1 sulfo-SMCC-to-7G3 ratios followed by Rf measurement and MW extrapolation results in 3, 7, 10, and 20 ChAcNLS molecules per 7G3. By densitometry, the percent monomer species is > 90% for 7G3 modified with 3-10 ChAcNLS. For 7G3-ChAcNLS20 the aggregate species is 45%. This demonstrates SDS-PAGE is simple yet effective for selecting a 7G3-ChAcNLS conjugate for further development. Although, slight smearing with antibody chains can occur due antibody glycosylation this does not interfere with the identification of aggregate species.
For Procedure 2, the representative data demonstrates differences between using trypsin and no trypsin when evaluating virus-derived peptide-modified ACs for intracellular efficiency accumulation. In this example, the ability of 7G3-ChAcNLS to increase its intracellular accumulation can be evaluated in cells treated with and without trypsin (Figure 4). However, the importance of trypsinization is evident when interpreting accumulation of conjugates used for comparison to determine AC efficiency and selectivity. Trypsinization allows for the baseline intracellular fluorescence level to be established as observed with unmodified 7G3. One can detail that 7G3 is limited to the cytoplasm in small foci. In contrast, cells that are not trypsinized, the 7G3-specific fluorescence is limited to the cell surface due to the overexpression of IL-3Rα on the cell surface. This makes it difficult to investigate intracellular accumulation and distribution because the fluorescence at the cell surface dominates and thus blocks the intracellular fluorescence from being visualized. Trypsinizing cells is also important when evaluating specificity as evidence with IgG-ChAcNLS. We can see that there is some non-specific fluorescence from the cell surface but would not be able to determine the level of intracellular accumulation caused by the peptide if the cells were not trypsinized. Lastly, without trypsinization one could falsely interpret that a newly developed AC does not accumulate inside target cells. As seen with 7G3-NLS, cells that are not trypsinized the majority of fluorescence is again from the cell surface. After trypsinization, one can observe that 7G3-NLS does accumulate, albeit limited, inside the target cells.
For Procedure 3, the representative data demonstrates quality control for construction, affinity, and in vitro payload delivery efficiency and specificity for the radioisotope 64Cu with A14-ChAcNLS. Densitometry performed on radiographic images taken from a 12% polyacrylamide gel containing 64Cu-A14, 64Cu-A14-NLS, or 64Cu-A14-ChAcNLS show that the radiolabeled conjugates are 100% monomer species (Figure 5A). 64Cu-A14-ChAcNLS shows nanomolar affinity for IL-5Rα. A14-ChAcNLS as a function of increasing concentrations of 64Cu-A14-ChAcNLS revealed specific binding approached saturation at concentrations of 3-5 nM in both HT-1376 and HT-B9 cells (Figure 5B). The Kd for 64Cu-A14-ChAcNLS on HT-1376 and HT-B9 cells was 6.4 ± 1.7 nM and 3.1 ± 0.8 nM, respectively. ChAcNLS conjugation reduced the affinity of A14 approximately 2.5-fold for IL-5Rα on HT-1376 and HT-B9 cells. Gamma counting reveals that the increase of radioactivity in the nucleus and in the cytoplasm delivered by 64Cu-A14-ChAcNLS is specific and increases over time (Figure 6). Gamma counting typically demonstrates approximately >6-fold increase in nuclear and intracellular accumulation in cells treated with 64Cu-A14-ChAcNLS compared to treatment with 64Cu-A14-ChAcNLS in the presence of excess unmodified A14 or when treatment studies are performed at 4 °C (Figure 6A). There is also a ≥ 3-fold increase in nuclear and intracellular accumulation of 64Cu relative to cells treated with 64Cu-A14 and 64Cu-IgG-ChAcNLS at all time points tested (Figure 6B).
Figure 7 demonstrates the importance of the cell fractionation protocol. HT-1376 and HT-B9 cells were incubated with plasma membrane lysis buffer containing 1%, 2%, or 4% NP-40. Isolated nuclei should exclusively contain Lamin A/C. Accordingly cytoplasmic fractions should be exclusive for Rab7. As a control, whole cells lysed with RIPA buffer should be positive for Lamin A/C in both fractions. This protocol shows that when evaluating the nuclear fraction of lysed HT-1376 cells, there is no Rab7 present. In addition, there is no Lamin A/C present in the cytoplasmic fraction. However, there is a reduced amount of Rab7 in the cytoplasm taken from cells lysed with 4% NP-40 (Figure 7A). This most likely suggests that 4% NP-40 lysis buffer is disrupting the nuclear membrane in addition to the plasma membrane. This results in the mixing of nuclear proteins with cytoplasmic proteins and thus dilutes the concentration of Rab 7. This explains the reduced amount of Rab 7 present. HT-B9 cells are even more sensitive than HT-1376 cells. When treated with plasma membrane lysis buffer containing 4% NP-40, Lamin A/C is clearly seen in the cytoplasmic fraction (Figure 7B). There is a corresponding reduction in the amount of Lamin A/C in the nuclear fraction. HT-B9 cells are also sensitive at 2% NP-40 lysis buffer as the amount of Rab7 in the cytoplasmic fraction is visibly reduced relative to 1% NP-40. Thus, one should optimize cellular fractionation with the particular cells of interest so that quantitative results of payload accumulation in the nucleus and intracellular space are accurate. This is important for evaluating the efficiency of nuclear localization of a novel AC.
For Procedure 4, PET imaging at 48 h post-injection of either 64Cu-A14, 64Cu-A14-NLS, or 64Cu-A14-ChAcNLS in mice bearing HT-1376 and HT-B9 heterotopic xenografts on opposite hind legs allows for the visualization of tumor targeting properties (Figure 8). As in our example, visual inspection of the PET images reveals the HT-1376 and HT-B9 tumor targeting capabilities of 64Cu-A14-ChAcNLS, 64Cu-A14, and 64Cu-A14-NLS (Figure 8A). However, in cases where visual inspection is difficult as with the HT-B9 tumors, ROI analysis can be used to determine tumor-to-muscle ratios (Figure 8B).
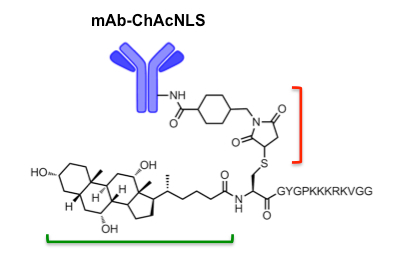 Figure 1: ChAcNLS-modified mAbs. NLS residues from SV-40 Large T-antigen (KKKRKV) are sandwiched between spacer residues capped by an N-terminal cysteine. Cholic acid (green bracket) is coupled to cysteine as previously described 34. After construction and purification of ChAcNLS, the cysteine sulfhydryl group is used for conjugation via sulfo-SMCC (red bracket; shown already attached to the mAb). NOTE: mAb (150 kDa) and ChAcNLS (1.8 kDa) are not to scale. Adapted and reprinted with permission from Paquette, M. et al. NLS-cholic acid conjugation to IL-5Rαspecific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. doi:10.102/acs.bioconjchem.8b00077. (2018). -
Figure 1: ChAcNLS-modified mAbs. NLS residues from SV-40 Large T-antigen (KKKRKV) are sandwiched between spacer residues capped by an N-terminal cysteine. Cholic acid (green bracket) is coupled to cysteine as previously described 34. After construction and purification of ChAcNLS, the cysteine sulfhydryl group is used for conjugation via sulfo-SMCC (red bracket; shown already attached to the mAb). NOTE: mAb (150 kDa) and ChAcNLS (1.8 kDa) are not to scale. Adapted and reprinted with permission from Paquette, M. et al. NLS-cholic acid conjugation to IL-5Rαspecific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. doi:10.102/acs.bioconjchem.8b00077. (2018). -
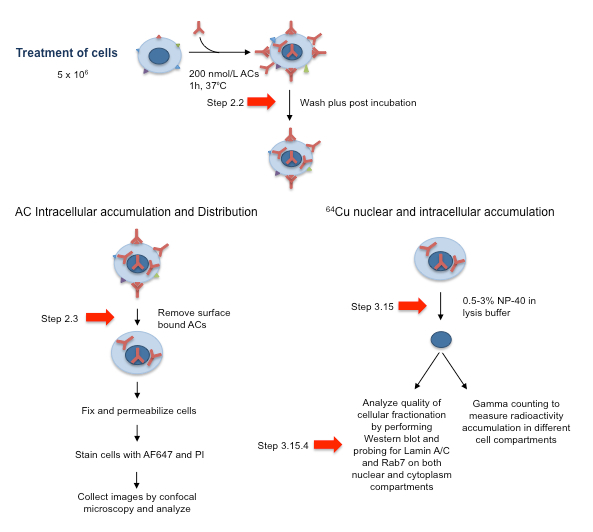 Figure 2: Schematic of AC Cell Treatment Approach and Subsequent Processing for Analysis by Confocal Microscopy and for Analysis of the Cell Fractionation Quality. Red arrows correspond to essential steps 2.2, 2.3, 3.15, and 3.15.4. Adapted and reprinted with permission from Beaudoin, S. et al. ChAcNLS, a Novel Modification to Antibody-Conjugates Permitting Target Cell-Specific Endosomal Escape, Localization to the Nucleus, and Enhanced Total Intracellular Accumulation. Molecular Pharmaceutics.13 (6), 1915-1926, doi:10.1021/acs.molpharmaceut.6b00075 (2016).
Figure 2: Schematic of AC Cell Treatment Approach and Subsequent Processing for Analysis by Confocal Microscopy and for Analysis of the Cell Fractionation Quality. Red arrows correspond to essential steps 2.2, 2.3, 3.15, and 3.15.4. Adapted and reprinted with permission from Beaudoin, S. et al. ChAcNLS, a Novel Modification to Antibody-Conjugates Permitting Target Cell-Specific Endosomal Escape, Localization to the Nucleus, and Enhanced Total Intracellular Accumulation. Molecular Pharmaceutics.13 (6), 1915-1926, doi:10.1021/acs.molpharmaceut.6b00075 (2016).
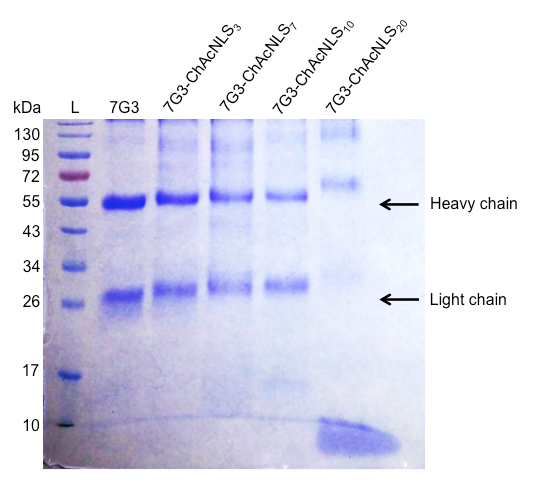 Figure 3: Analysis of ChAcNLS-modified mAbs. A reducing SDS-PAGE showing the running ladder (L) with corresponding molecular weights in kilodalton (kDa) and unmodified 7G3 heavy and light chains as references. The following lanes are the migration bands of the heavy and light chains from 7G3 modified with 3, 7, 10, and 20 ChAcNLS.
Figure 3: Analysis of ChAcNLS-modified mAbs. A reducing SDS-PAGE showing the running ladder (L) with corresponding molecular weights in kilodalton (kDa) and unmodified 7G3 heavy and light chains as references. The following lanes are the migration bands of the heavy and light chains from 7G3 modified with 3, 7, 10, and 20 ChAcNLS.
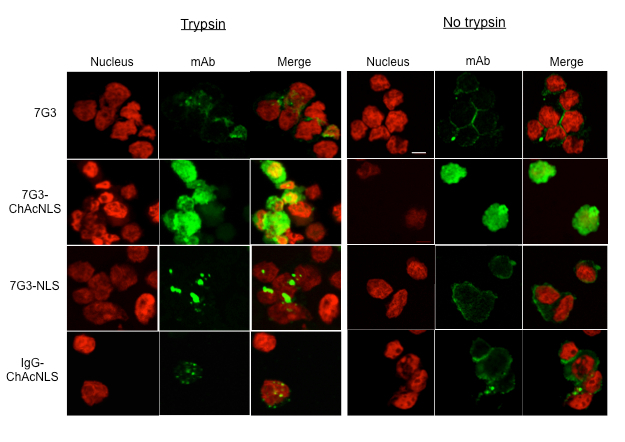 Figure 4: Analysis of mAb Intracellular Distribution and Accumulation. Confocal microscopy images illustrating the intracellular 7G3 distribution and relative fluorescence accumulation in IL-3Rα-positive leukemia cells treated with 7G3, 7G3-ChAcNLS, IgG, and IgG-ChAcNLS. Cells were trypsinized or not trypsinized prior to fixation. Nuclei are stained with propidium iodide (PI; red), anti-murine-Fc-AF647 dye (mAb; green), and PI/mAb merged. Scale is 50 μm. Within the trypsin and no trypsin groups, images were acquired with identical instrument settings between the different ACs shown. The trypsin portion of Figure 4 is adapted from refefence 34 with permission. Adapted and reprinted with permission from Beaudoin, S. et al. ChAcNLS, a Novel Modification to Antibody-Conjugates Permitting Target Cell-Specific Endosomal Escape, Localization to the Nucleus, and Enhanced Total Intracellular Accumulation. Molecular Pharmaceutics. 13 (6), 1915-1926, doi:10.1021/acs.molpharmaceut.6b00075 (2016).
Figure 4: Analysis of mAb Intracellular Distribution and Accumulation. Confocal microscopy images illustrating the intracellular 7G3 distribution and relative fluorescence accumulation in IL-3Rα-positive leukemia cells treated with 7G3, 7G3-ChAcNLS, IgG, and IgG-ChAcNLS. Cells were trypsinized or not trypsinized prior to fixation. Nuclei are stained with propidium iodide (PI; red), anti-murine-Fc-AF647 dye (mAb; green), and PI/mAb merged. Scale is 50 μm. Within the trypsin and no trypsin groups, images were acquired with identical instrument settings between the different ACs shown. The trypsin portion of Figure 4 is adapted from refefence 34 with permission. Adapted and reprinted with permission from Beaudoin, S. et al. ChAcNLS, a Novel Modification to Antibody-Conjugates Permitting Target Cell-Specific Endosomal Escape, Localization to the Nucleus, and Enhanced Total Intracellular Accumulation. Molecular Pharmaceutics. 13 (6), 1915-1926, doi:10.1021/acs.molpharmaceut.6b00075 (2016).
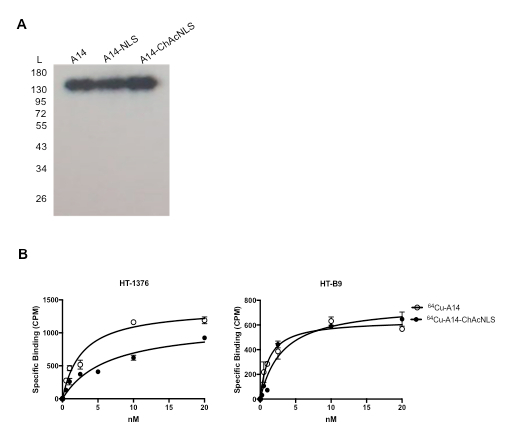 Figure 5: 64Cu-A14-ChAcNLS Purity and Affinity. (A) The radiochemical purity of 64Cu-labeled A14, A14-NLS, A14-ChAcNLS were checked by SDS-PAGE followed by autoradiography. (B) Specific binding curves for 64Cu-A14 and 64Cu-A14-ChAcNLS on HT-1376 and HT-B9 cells. Error bars indicate standard deviation of experiment performed in replicate. Adapted and reprinted with permission from Paquette, M. et al. NLS-cholic acid conjugation to IL-5Rα-specific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. doi:10.102/acs.bioconjchem.8b00077. (2018).
Figure 5: 64Cu-A14-ChAcNLS Purity and Affinity. (A) The radiochemical purity of 64Cu-labeled A14, A14-NLS, A14-ChAcNLS were checked by SDS-PAGE followed by autoradiography. (B) Specific binding curves for 64Cu-A14 and 64Cu-A14-ChAcNLS on HT-1376 and HT-B9 cells. Error bars indicate standard deviation of experiment performed in replicate. Adapted and reprinted with permission from Paquette, M. et al. NLS-cholic acid conjugation to IL-5Rα-specific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. doi:10.102/acs.bioconjchem.8b00077. (2018).
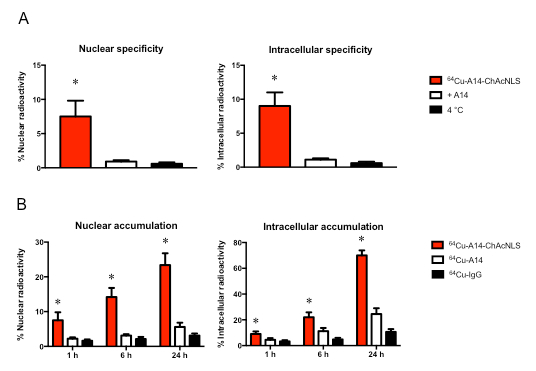 Figure 6: 64Cu Nuclear and Intracellular Accumulation. Representative example of cell treatment performed in triplicate (error bars denote standard deviation) to demonstrate (A) specificity and (B) accumulation enhancement of 64Cu-A14-ChAcNLS. The % accumulation is presented in the nucleus (left panels) and total intracellular space (right panels) relative to the total amount of radioactivity used during treatment of IL-5Rα-positive invasive bladder cancer cells. Error bars indicate standard deviation of experiment performed in replicate. * indicates p ≤0.05.
Figure 6: 64Cu Nuclear and Intracellular Accumulation. Representative example of cell treatment performed in triplicate (error bars denote standard deviation) to demonstrate (A) specificity and (B) accumulation enhancement of 64Cu-A14-ChAcNLS. The % accumulation is presented in the nucleus (left panels) and total intracellular space (right panels) relative to the total amount of radioactivity used during treatment of IL-5Rα-positive invasive bladder cancer cells. Error bars indicate standard deviation of experiment performed in replicate. * indicates p ≤0.05.
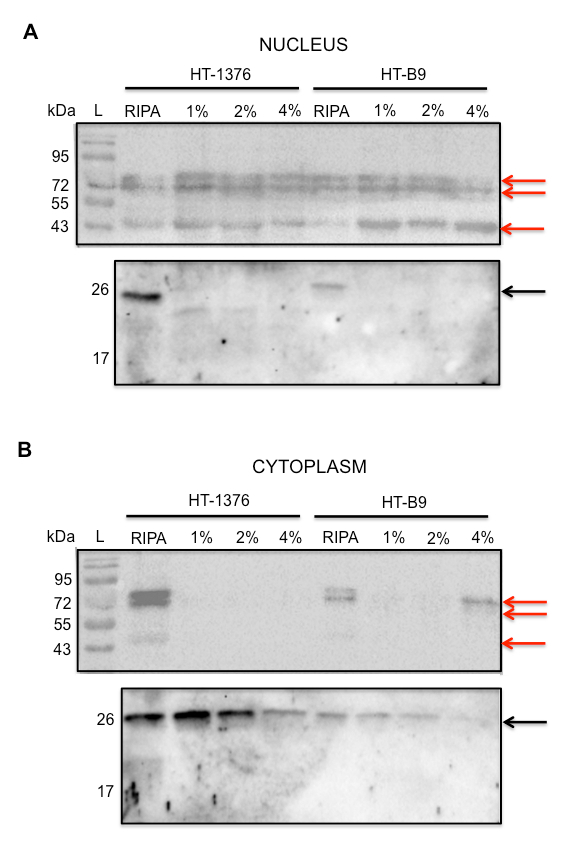 Figure 7: Analysis of Cell Fractionation Procedure. Western blot of (A) Nuclear and (B) Cytoplasmic fractions for Lamin A/C (top blots; red arrows) and Rab 7 (bottom blots; black arrows) obtained from treatment of HT-1376 and HT-B9 cells in plasma membrane lysis buffer containing either 1%, 2%, or 4% NP-40. Whole cells lysed with RIPA buffer was used as positive control for Lamin A/C and Rab7 in both fractions. Running standards (L) are only depicted in top blots. Three arrows are shown for Lamin A/C due to its multiple isoforms. Adapted and reprinted with permission from Paquette, M. et al. NLS-cholic acid conjugation to IL-5Rα-specific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. doi:10.102/acs.bioconjchem.8b00077. (2018).
Figure 7: Analysis of Cell Fractionation Procedure. Western blot of (A) Nuclear and (B) Cytoplasmic fractions for Lamin A/C (top blots; red arrows) and Rab 7 (bottom blots; black arrows) obtained from treatment of HT-1376 and HT-B9 cells in plasma membrane lysis buffer containing either 1%, 2%, or 4% NP-40. Whole cells lysed with RIPA buffer was used as positive control for Lamin A/C and Rab7 in both fractions. Running standards (L) are only depicted in top blots. Three arrows are shown for Lamin A/C due to its multiple isoforms. Adapted and reprinted with permission from Paquette, M. et al. NLS-cholic acid conjugation to IL-5Rα-specific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. doi:10.102/acs.bioconjchem.8b00077. (2018).
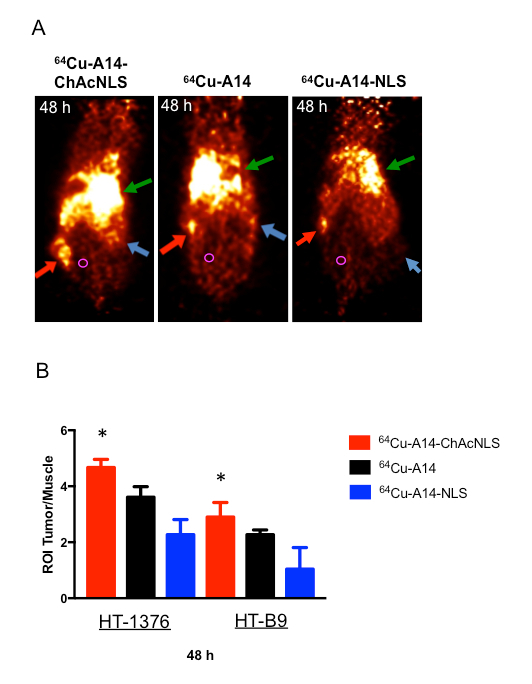 Figure 8: In Vivo Analysis of Tumor Targeting by PET Imaging and ROI Analysis. (A) PET images at 48 h post-injection of NOD/SCID mice bearing HT-1376 (red arrows) and HT-B9 (blue arrows) tumors intravenously injected with 64Cu-A14, 64Cu-A14-NLS, and 64Cu-A14-ChAcNLS. Green arrow is liver uptake. Muscle ROI drawing shown with magenta circle. (B) ROI HT-1376 and HT-B9 tumor-to-muscle ratios of the ACs at 48 h post-injection. Error bars indicate standard deviation of experiment in n = 10 ROIs per mouse (n = 5). * indicates p 0.05.
Figure 8: In Vivo Analysis of Tumor Targeting by PET Imaging and ROI Analysis. (A) PET images at 48 h post-injection of NOD/SCID mice bearing HT-1376 (red arrows) and HT-B9 (blue arrows) tumors intravenously injected with 64Cu-A14, 64Cu-A14-NLS, and 64Cu-A14-ChAcNLS. Green arrow is liver uptake. Muscle ROI drawing shown with magenta circle. (B) ROI HT-1376 and HT-B9 tumor-to-muscle ratios of the ACs at 48 h post-injection. Error bars indicate standard deviation of experiment in n = 10 ROIs per mouse (n = 5). * indicates p 0.05.
| Tat |
| CGGQVCFITKALGISYGRKKRRQRRRPPQGS 20 |
| CFITKALGISYGRKKRRQRRRPPQGSQTHQVSLSKQ 30 |
| CGISYGRKKRRQRRR 29 |
| GRKKRRQRRRPPQGYC 22,25 |
| TRQARRNRRRRWRERQRGC 26 |
| NLS |
| CGYGPKKKRKVGG 21,23,24,27 |
| Cholic acid-CGYGPKKKRKVGG 34 |
Table 1: List of Tat and SV-40 Large T-antigen Derived Peptides used in AC Modifications.
Discussion
Major goals of systemic delivery of anti-cancer agents are to increase accumulation at the tumor site, and uptake within cancer cells, and decrease unwanted side effects in healthy tissues. AC targeted delivery of molecular payloads to tumor cells is a highly promising approach to treat and detect tumors. However, the lack of efficacy caused by endosome entrapment and down stream lysosomal degradation remains an important challenge. While this protocol utilizes the ChAcNLS peptide as an example for the construction of next generation ACs, the described procedures are adaptable to any peptide-modified AC being developed with the aim of increasing the amount of intracellular delivered payload to targeted cancer cells.
The most critical steps in this protocol are: 1) identifying and limiting high MW aggregate species; 2) the trypsinization of cells prior to analysis for determining intracellular accumulation; 3) performing quality assurance of cellular fractionation procedures; and 4) performing PET imaging to evaluate tumor targeting in vivo.
It is important the conjugation of virus-derived peptides to mAbs does not result in high MW aggregate species that is most likely caused by intramolecular crosslinking between mAbs. This is the initial concern and should be dealt with before progressing further. Moving forward with peptide-modified ACs that contain unwanted aggregates could give results that provide false evidence on aspects such as the intracellular and in vivo payload delivery profiles. Reactive maleimides on mAbs is what is most likely responsible for covalent aggregation. One solution is to reduce the sulfo-SMCC-to-mAb ratio. Another alternative is to use N-acetyl derivatives of amino acids such as Cys, Lys, His, Ser, and Thr during peptide conjugation to the maleimide-activated mAb 45. The nucleophilic side-chains are able to react with the maleimide moiety of SMCC and decrease intra-antibody crosslinking. Alternatively, separation by size exclusion chromatography after conjugation can be used to isolate monomer species 45. Site-specific conjugation of the peptide to the antibody can also be performed. For example, antibody carbohydrates can be modified for conjugation to peptides, which has shown to efficiently produce monomer species 24.
SDS-PAGE with 12% polyacrylamide gels is an inexpensive and effective method to gain a broad perspective of large size differences in AC species. If an increase in % polyacrylamide is used, separation of heavy chains and intramolecular species will be difficult to distinguish. Conversely, if a reduced % polyacrylamide is used, the light chains are likely to run off the gel. Gradient gels containing 4-20% polyacrylamide also provide good retention shifts for the heavy and light chains on a single gel. Among the various analytical techniques available for biomolecular analysis, high-performance liquid chromatography and capillary electrophoresis-SDS are superior to SDS-PAGE for the separation and characterization of ACs 46. Nonetheless, SDS-PAGE is an early and inexpensive method to analyze the initial construction of these novel agents and then decide on the following course of action.
The incubation of cells in solution containing 0.25% trypsin (step 2.3), which removes a maximum number of cell-surface proteins and bound ACs is important because it allows for improved visualization and relative increases in intracellular accumulation between reference antibodies. The camera parameters are set to capture images at maximum fluorescence intensity. This is because of variation from cell to cell in the amount of AC that is accumulated. Thus, capturing images at maximum intensity allows for cells with less AC accumulation to also be evaluated. However, cancer antigens are typically highly expressed on tumor cells. If the cells were not subjected to digestion by trypsin, then the fluorescence emanating from ACs at the cell-surface would dominate the image. In addition, without trypsinization development could be limited with ACs with subtle but significant intracellular accumulation properties.
Ensuring high quality cellular fractionation is important to accurately evaluate the accumulation of 64Cu in the nucleus versus the cytoplasm. The ability of ChAcNLS to increase AC and payload intracellular accumulation is due to the active localization to the nucleus 34. As future virus-peptides are developed, these peptides may also localize to the nucleus as part of their mechanism to increase intracellular payload efficiency. Thus, it is important that cellular fractionation be of high quality to accurately detail this process. This protocol emphasizes the importance of evaluating the nuclei for cytoplasmic markers and also the cytoplasmic fraction for nuclear markers. For example, Hu et al., developed an AC modified with a Tat peptide and explored nuclear accumulation of a radioisotope payload 24. Western blot analysis for calpain, an abundant cytoplasmic protein, was present in the nuclear fraction. This was most likely because commercial nuclear isolation kits were used. This protocol demonstrated that HT-1376 and HT-B9 cells required different concentrations of NP-40 in order to isolate enriched nuclei with intact membranes. When HT-B9 cells were lysed with buffer containing 4% NP-40, Lamin A/C was present in the cytoplasmic fraction indicating the treatment disrupted nuclear membranes and allowed for Lamin A/C to enter the cytoplasm. If the nuclear and cytoplasmic fractions are not isolated carefully, it could be difficult to identify the contribution of nuclear localization to total intracellular accumulation. There are reports of biological- and artificially-derived peptides that specifically target other intracellular compartments such as the endoplasmic reticulum, Golgi apparatus, and mitochondria 47,48,49. Thus, the isolation of intracellular compartments is becoming increasingly relevant.
This protocol finishes with its description of 64Cu-A14-ChAcNLS targeting of HT-1376 and HT-B9 tumors. From the visual inspection of the PET images in Figure 8, it is evident by the increased tumor PET signal relative to the adjacent reduced surrounding background 64Cu-A14-ChAcNLS has superior targeting of HT-1376 and HT-B9 tumors (Figure 8A). ROI analysis also demonstrates its ability to evaluate tumor targeting. In our example, we use ROI tumor-to-muscle ratios to quantify tumor targeting and demonstrate 64Cu-A14-ChAcNLS is the superior AC (Figure 8B). PET imaging also allows for the evaluation of AC sequestering in healthy organs. The liver is the primary organ that metabolizes antibodies. Figure 8A demonstrates uptake in the liver is comparable in mice injected with 64Cu-A14-ChAcNLS and 64Cu-A14. At this early development phase, this data suggests ChAcNLS does not cause unwanted sequestering. 64Cu has a half-life of 12.7 h, which is not ideal for ACs that have half-lives of several days. As previously described in the visual protocol by Zeglis and Lewis, the use of the long-lived radioisotope zirconium-89 (89Zr) is a payload whose physical half-life is ideal for mAbs and can be imaged by PET 50. Thus, for evaluating tumor delivery over several days, 89Zr is a preferred choice. This is particularly important if therapy is a goal where high tumor uptake is favorable and increases over time with antibodies 7. Biodistribution should also be performed to determine individual organ uptake to further explore the targeting or sequestering properties of newly developed ACs. In addition, mice can be predosed with unmodified specific antibody to block receptor sites on tumor cells to evaluate targeting specificity. Nonetheless, 64Cu with PET imaging and ROI analysis sufficiently provide an initial assessment of the tumor delivery properties of the developed ACs presented in this protocol.
Thus far, ACs modified with virus-derived peptides have been restricted to the delivery of radioisotopes. This is partly due to historical reasons and also because radioisotopes are easily quantified during in vitro and in vivo testing, and can be visualized using noninvasive whole-body imaging modalities. Naturally, as this field expands to the transportation and delivery of payloads other than radioisotopes, such as chemotherapeutics, enzymes, and oligonucleotides, modifications to the techniques described in this protocol will have to be developed. For example, novel methods for evaluating the intracellular accumulation of alternative payloads will have to be developed. In addition, modifications for in vivo evaluation will bee necessary as these payloads cannot be easily imaged and therefore it will be difficult to assess biodistribution, specificity, and tumor uptake efficiency. For these reasons, radionuclide payloads should remain as surrogate payloads for future development with alternative cargos.
Disclosures
The authors have nothing to disclose
Acknowledgments
This work was funded by the Cancer Research Society (Canada) and the CIMS. The authors thank Dr. Samia Ait-Mohand and Jean-Francois Beaudoin for assistance. Dr. Angel Lopez (University of South Australia) for mAb A14.
References
- Verma S, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. New England Journal of Medicine. 2012;367(19):1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. Journal of Clinical Oncology. 2012;30(18):2183–2189. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchelouche K, Choyke PL, Capala J. Prostate specific membrane antigen- a target for imaging and therapy with radionuclides. Discovery Medicine. 2010;9(44):55–61. [PMC free article] [PubMed] [Google Scholar]
- Hamilton GS. Antibody-drug conjugates for cancer therapy: The technological and regulatory challenges of developing drug-biologic hybrids. Biologicals. 2015;43(5):318–332. doi: 10.1016/j.biologicals.2015.05.006. [DOI] [PubMed] [Google Scholar]
- Deonarain MP, Yahioglu G, Stamati I, Marklew J. Emerging formats for next-generation antibody drug conjugates. Expert Opinion on Drug Discovery. 2015;10(5):463–481. doi: 10.1517/17460441.2015.1025049. [DOI] [PubMed] [Google Scholar]
- Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Research. 2014;16(2) doi: 10.1186/bcr3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nature Biotechnology. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Current Opinion in Chemical Biology. 2010;14(4):529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- Austin CD, et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Molecular Biology of the Cell. 2004;15(12):5268–5282. doi: 10.1091/mbc.E04-07-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan JN, et al. Comparative uptakes and biodistributions of internalizing vs. noninternalizing copper-64 radioimmunoconjugates in cell and animal models of colon cancer. Nuclear Medicine and Biology. 2005;32(8):851–858. doi: 10.1016/j.nucmedbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Chen R, et al. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Molecular Cancer Therapeutics. 2015;14(6):1376–1384. doi: 10.1158/1535-7163.MCT-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs H, Weng A, Gilabert-Oriol R. Augmenting the Efficacy of Immunotoxins and Other Targeted Protein Toxins by Endosomal Escape Enhancers. Toxins (Basel) 2016;8(7) doi: 10.3390/toxins8070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsnes S, Sandvig K, Petersen OW, van Deurs B. Immunotoxins--entry into cells and mechanisms of action. Immunology Today. 1989;10(9):291–295. [PubMed] [Google Scholar]
- Zheng D, et al. Virus-derived anti-inflammatory proteins: potential therapeutics for cancer. Trends in Molecular Medicine. 2012;18(6):304–310. doi: 10.1016/j.molmed.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Kristensen M, Birch D, Morck Nielsen , H Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. International Journal of Molecular Sciences. 2016;17(2) doi: 10.3390/ijms17020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yezid H, Konate K, Debaisieux S, Bonhoure A, Beaumelle B. Mechanism for HIV-1 Tat insertion into the endosome membrane. Journal of Biological Chemistry. 2009;284(34):22736–22746. doi: 10.1074/jbc.M109.023705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escriou V, Carriere M, Scherman D, Wils P. NLS bioconjugates for targeting therapeutic genes to the nucleus. Advanced Drug Delivery Reviews. 2003;55(2):295–306. doi: 10.1016/s0169-409x(02)00184-9. [DOI] [PubMed] [Google Scholar]
- Pouton CW, Wagstaff KM, Roth DM, Moseley GW, Jans DA. Targeted delivery to the nucleus. Advanced Drug Delivery Reviews. 2007;59(8):698–717. doi: 10.1016/j.addr.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Anderson DC, et al. Tumor cell retention of antibody Fab fragments is enhanced by an attached HIV TAT protein-derived peptide. Biochemical Biophysical Research Communications. 1993;194(2):876–884. doi: 10.1006/bbrc.1993.1903. [DOI] [PubMed] [Google Scholar]
- Chen P, et al. Nuclear localizing sequences promote nuclear translocation and enhance the radiotoxicity of the anti-CD33 monoclonal antibody HuM195 labeled with 111In in human myeloid leukemia cells. Journal of Nuclear Medicine. 2006;47(5):827–836. [PubMed] [Google Scholar]
- Cornelissen B, Hu M, McLarty K, Costantini D, Reilly RM. Cellular penetration and nuclear importation properties of 111In-labeled and 123I-labeled HIV-1 tat peptide immunoconjugates in BT-474 human breast cancer cells. Nuclear Medicine and Biology. 2007;34(1):37–46. doi: 10.1016/j.nucmedbio.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Costantini DL, Chan C, Cai Z, Vallis KA, Reilly RM. (111)In-labeled trastuzumab (Herceptin) modified with nuclear localization sequences (NLS): an Auger electron-emitting radiotherapeutic agent for HER2/neu-amplified breast cancer. Journal of Nuclear Medicine. 2007;48(8):1357–1368. doi: 10.2967/jnumed.106.037937. [DOI] [PubMed] [Google Scholar]
- Fasih A, et al. (1)(1)(1)In-Bn-DTPA-nimotuzumab with/without modification with nuclear translocation sequence (NLS) peptides: an Auger electron-emitting radioimmunotherapeutic agent for EGFR-positive and trastuzumab (Herceptin)-resistant breast cancer. Breast Cancer Research and Treatment. 2012;135(1):189–200. doi: 10.1007/s10549-012-2137-y. [DOI] [PubMed] [Google Scholar]
- Hu M, et al. Site-specific conjugation of HIV-1 tat peptides to IgG: a potential route to construct radioimmunoconjugates for targeting intracellular and nuclear epitopes in cancer. European Journal of Nuclear Medicine and Molecular Imaging. 2006;33(3):301–310. doi: 10.1007/s00259-005-1908-7. [DOI] [PubMed] [Google Scholar]
- Kameyama S, et al. Effects of cell-permeating peptide binding on the distribution of 125I-labeled Fab fragment in rats. Bioconjugate Chemistry. 2006;17(3):597–602. doi: 10.1021/bc050258k. [DOI] [PubMed] [Google Scholar]
- Kersemans V, Cornelissen B, Minden MD, Brandwein J, Reilly RM. Drug-resistant AML cells and primary AML specimens are killed by 111In-anti-CD33 monoclonal antibodies modified with nuclear localizing peptide sequences. Journal of Nuclear Medicine. 2008;49(9):1546–1554. doi: 10.2967/jnumed.107.047399. [DOI] [PubMed] [Google Scholar]
- Mie M, et al. Intracellular delivery of antibodies using TAT fusion protein. Biochemical Biophysical Research Communications. 2003;310(3):730–734. doi: 10.1016/j.bbrc.2003.09.071. [DOI] [PubMed] [Google Scholar]
- Niesner U, et al. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjugate Chemistry. 2002;13(4):729–736. doi: 10.1021/bc025517+. [DOI] [PubMed] [Google Scholar]
- Stein S, et al. A disulfide conjugate between anti-tetanus antibodies and HIV (37-72)Tat neutralizes tetanus toxin inside chromaffin cells. FEBS Letters. 1999;458(3):383–386. doi: 10.1016/s0014-5793(99)01186-2. [DOI] [PubMed] [Google Scholar]
- Tolstikov VV, Cole R, Fang H, Pincus SH. Influence of endosome-destabilizing peptides on efficacy of anti-HIV immunotoxins. Bioconjugate Chemistry. 1997;8(1):38–43. doi: 10.1021/bc9600729. [DOI] [PubMed] [Google Scholar]
- Gao C, Leyton JV, Schimmer AD, Minden M, Reilly RM. Auger electron-emitting 111In-DTPA-NLS-CSL360 radioimmunoconjugates are cytotoxic to human acute myeloid leukemia (AML) cells displaying the CD123/CD131 phenotype of leukemia stem cells. Applied Radiation and Isotopes. 2015;110:1–7. doi: 10.1016/j.apradiso.2015.12.043. [DOI] [PubMed] [Google Scholar]
- Leyton JV, et al. Auger electron radioimmunotherapeutic agent specific for the CD123+/CD131- phenotype of the leukemia stem cell population. Journal of Nuclear Medicine. 2011;52(9):1465–1473. doi: 10.2967/jnumed.111.087668. [DOI] [PubMed] [Google Scholar]
- Paquette M, et al. NLS-cholic acid conjugation to IL-5Rα-specific antibody improves cellular accumulation and in vivo tumor-targeting properties in a bladder cancer model. Bioconjugate Chemistry. 2018. [DOI] [PubMed]
- Beaudoin S, et al. ChAcNLS, a Novel Modification to Antibody-Conjugates Permitting Target Cell-Specific Endosomal Escape, Localization to the Nucleus, and Enhanced Total Intracellular Accumulation. Molecular Pharmaceutics. 2016;13(6):1915–1926. doi: 10.1021/acs.molpharmaceut.6b00075. [DOI] [PubMed] [Google Scholar]
- Boswell CA, et al. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjugate Chemistry. 2010;21(12):2153–2163. doi: 10.1021/bc100261d. [DOI] [PubMed] [Google Scholar]
- Shivanna V, Kim Y, Chang KO. The crucial role of bile acids in the entry of porcine enteric calicivirus. Virology. 2014;456-457:268–278. doi: 10.1016/j.virol.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivanna V, Kim Y, Chang KO. Ceramide formation mediated by acid sphingomyelinase facilitates endosomal escape of caliciviruses. Virology. 2015;483:218–228. doi: 10.1016/j.virol.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancevic B, Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Letters. 2010;584(9):1728–1740. doi: 10.1016/j.febslet.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomarker Research. 2014;2(1):4. doi: 10.1186/2050-7771-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MA. Detection of dead cells and measurement of cell killing by flow cytometry. Journal of Immunological Methods. 2000;243(1-2):155–166. doi: 10.1016/s0022-1759(00)00232-5. [DOI] [PubMed] [Google Scholar]
- Paquette M, et al. Targeting IL-5Rα with antibody-conjugates reveals a strategy for imaging and therapy for invasive bladder cancer. Oncoimmunology. 2017;6(10):e1331195. doi: 10.1080/2162402X.2017.1331195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisler S. Production of 64Cu on the Sherbrooke TR-PET cyclotron. Journal or Radioanalytical and Nuclear Chemistry. 2004;257(1) [Google Scholar]
- Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. North American Journal of Medical Sciences. 2012;4(9):429–434. doi: 10.4103/1947-2714.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, et al. A dual tracer PET-MRI protocol for the quantitative measure of regional brain energy substrates uptake in the rat. Journal of Visualized Experiments. 2013. p. e50761. [DOI] [PMC free article] [PubMed]
- Wakankar AA, et al. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: changes due to modification and conjugation processes. Bioconjugate Chemistry. 2010;21(9):1588–1595. doi: 10.1021/bc900434c. [DOI] [PubMed] [Google Scholar]
- Liu J, Abid S, Lee MS. Analysis of monoclonal antibody chimeric BR96-doxorubicin immunoconjugate by sodium dodecyl sulfate-capillary gel electrophoresis with ultraviolet and laser-induced fluorescence detection. Analytical Biochemistry. 1995;229(2):221–228. doi: 10.1006/abio.1995.1406. [DOI] [PubMed] [Google Scholar]
- Rege K, Patel SJ, Megeed Z, Yarmush ML. Amphipathic peptide-based fusion peptides and immunoconjugates for the targeted ablation of prostate cancer cells. Cancer Research. 2007;67(13):6368–6375. doi: 10.1158/0008-5472.CAN-06-3658. [DOI] [PubMed] [Google Scholar]
- Zhan J, Ge L, Shen J, Wang K, Zheng S. A trans-Golgi network retention signal YQRL fused to ricin A chain significantly enhances its cytotoxicity. Biochemical Biophysical Research Communications. 2004;313(4):1053–1057. doi: 10.1016/j.bbrc.2003.12.037. [DOI] [PubMed] [Google Scholar]
- Zhan J, Stayton P, Press OW. Modification of ricin A chain, by addition of endoplasmic reticulum (KDEL) or Golgi (YQRL) retention sequences, enhances its cytotoxicity and translocation. Cancer Immunology, Immunotherapy. 1998;46(1):55–60. doi: 10.1007/s002620050460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeglis BM, Lewis JS. The bioconjugation and radiosynthesis of 89Zr-DFO-labeled antibodies. Journal of Visualized Experiments. 2015. [DOI] [PMC free article] [PubMed]