

Abstract
Pharmacological targeting of G protein-coupled receptors (GPCRs) is of great importance to human health, as dysfunctional GPCR-mediated signaling contributes to the progression of many diseases. The ligand/receptor pair CXC chemokine ligand 12 (CXCL12)/CXC chemokine receptor 4 (CXCR4) has raised significant clinical interest, for instance as a potential target for the treatment of cancer and inflammatory diseases. Small molecules as well as therapeutic antibodies that specifically target CXCR4 and inhibit the receptor's function are therefore considered to be valuable pharmacological tools. Here, a flow cytometry-based cellular assay that allows identification of compounds (e.g., small molecules) that abrogate CXCL12 binding to CXCR4, is described. Essentially, the assay relies on the competition for receptor binding between a fixed amount of fluorescently labeled CXCL12, the natural chemokine agonist for CXCR4, and unlabeled compounds. Hence, the undesirable use of radioactively labeled probes is avoided in this assay. In addition, living cells are used as the source of receptor (CXCR4) instead of cell membrane preparations. This allows easy adaptation of the assay to a plate format, which increases the throughput. This assay has been shown to be a valuable generic drug discovery assay to identify CXCR4-targeting compounds. The protocol can likely be adapted to other GPCRs, at least if fluorescently labeled ligands are available or can be generated. Prior knowledge concerning the intracellular signaling pathways that are induced upon activation of these GPCRs, is not required.
Keywords: Molecular Biology, Issue 133, G protein-coupled receptor, CXC chemokine receptor 4, fluorescently labeled chemokine ligand 12, flow cytometry, cell-based assay, receptor binding, drug discovery, small molecules, Jurkat cells
Introduction
G protein-coupled receptors (GPCRs) are cell surface proteins that can be activated by extracellular ligands (e.g., peptides, protein hormones, amines), thereby regulating many physiological and developmental processes1. When an agonist occupies its GPCR binding pocket, the induced conformational change in the receptor protein promotes the binding of intracellular receptor-associated heterotrimeric G proteins, consisting of Gα-GDP and Gβγ subunits. The subsequent exchange of GTP for GDP on the Gα subunit results in the dissociation of the G protein subunits (Gα-GTP and Gβγ) that, in turn, will further initiate downstream signaling pathways2,3. When the Gα-GTP becomes hydrolyzed, re-association of the Gα-GDP and Gβγ subunits will convert the G protein back into its resting state3,4. Distinct types of G proteins exist (Gs, Gi/o, Gq, G12/13), which are categorized based on sequence similarity with the Gα subunit5. All of these G proteins induce defined intracellular signaling pathways that underlie the biological response to receptor activation. Subsequent to receptor activation, GPCR kinases (GRKs) phosphorylate the intracellular tail of GPCRs, thereby promoting interaction with β-arrestins. This process leads to the termination of G protein signaling, receptor desensitization and internalization6. β-arrestins are also part of multi-molecular complexes that trigger signaling cascades independent of G protein signaling7.
GPCRs are amongst the most validated molecular targets for therapeutic intervention, as deregulated GPCR-mediated signaling, for instance due to gain-of-function mutations in the receptor gene or receptor overexpression, contributes to the etiology of many human diseases8. Therefore, GPCRs represent one of the most important classes of drug targets investigated by the pharmaceutical industry8,9,10. A notable example of a clinically relevant GPCR is the CXC chemokine receptor 4 (CXCR4), which can be activated by a sole natural ligand, the CXC chemokine ligand 12 (CXCL12)11. Due to its established role as a major co-receptor for human immunodeficiency virus 1 (HIV-1) entry and infection in cluster of differentiation 4 (CD4) positive T-lymphocytes12, CXCR4 was first investigated as an antiviral drug target. CXCL12-CXCR4 interaction in the bone marrow further regulates the retention and homing of stem and progenitor cells13. Also, given its involvement in many aspects of cancer biology (e.g., tumor cell survival, metastasis, tumor-related angiogenesis)14 and several other human diseases (e.g., inflammatory diseases)15, CXCR4 raised significant interest as a promising target for drug discovery. AMD3100, a small molecule that specifically targets CXCR4, was initially discovered as an anti-HIV drug candidate16 and is still one of the most potent CXCR4 antagonists described to date17. Its development as an antiviral drug was, however, discontinued18. Currently this molecule is used as a stem cell mobilization agent during the treatment of multiple myeloma and lymphoma patients18. Several other chemically unrelated small molecules and biologics that inhibit CXCR4 function with varying potency have been described19.
Receptor binding methods are valuable tools in pharmacology that allow the identification of compounds (e.g., small molecules) that directly interact with the GPCR of interest. In order to perform binding studies, there is no need for prior knowledge concerning the intracellular signaling properties or functionality of a given GPCR. Although this can be considered to be an advantage, it implies that compounds for which receptor binding can be demonstrated need to be further characterized by evaluating their potential agonistic or antagonistic activity. This activity can be evaluated using pharmacological or biological assays related to the GPCR under study. Dependent on their activity profile, receptor binding molecules might then potentially evolve to become novel lead compounds for investigation in pre-clinical and clinical studies. Molecules that specifically bind to a receptor with high affinity can also serve as scaffolds to generate therapeutic or diagnostic tools, for instance by radiolabeling them for noninvasive in vivo imaging of tumor cells20, or as potential vehicles for targeted delivery of therapeutics21. In case of CXCR4, in vivo imaging of tumor cells has already been demonstrated using mouse models wherein labeled CXCR4-targeting molecules allowed the visualization of human cancer xenografts20,22,23.
In this report, we describe a detailed protocol for a competition binding assay that enables the identification of small molecules and biologics that directly interfere with agonist (CXCL12) binding to CXCR4. The basic principle of the assay is the competition between a fixed amount of fluorescently labeled ligand (CXCL12AF647, see Table of Materials and Reagents) and unlabeled compounds for binding to the receptor protein17,24. The specific fluorescent signal from labeled ligand bound to single cells expressing CXCR4 is then analyzed by flow cytometry. This fluorescent signal will be reduced when unlabeled small molecules disrupt the interaction between CXCL12AF647 and CXCR4. The assay uses non-manipulated living cells that endogenously express CXCR4 (i.e., Jurkat cells). Hence, no cell membrane preparation is required, which makes the assay convenient, fast and compatible with increased throughput. Since a fluorescently labeled ligand is used, radioactivity is avoided.
Because CXCL12 is the natural agonist for CXCR4, small molecule compounds that interfere with CXCL12AF647 binding in the assay are likely to interact with the orthosteric receptor binding site (i.e., the binding site occupied by the natural agonist). Molecules that would interact with receptor binding sites topographically distinct from the orthosteric binding site remain undetected, if they do not influence binding of CXCL12. For instance, positive and negative allosteric modulators, an important and emerging category of GPCR targeting molecules acting on allosteric binding sites25, will potentially not be picked up with this assay. In addition, whether the compounds identified with this binding assay function as receptor antagonists or as agonists cannot be derived. Investigation of the identified compounds in additional pharmacological or functional receptor-related assays will thus be required. These assays might include (a combination of) cellular fluorescence- or luminescence-based assays for the detection of second messengers (e.g., Ca2+, cyclic adenosine monophosphate (cAMP)), phenotypic or biological assays and β-arrestin recruitment assays, the choice of which depends on the specific signaling properties of the GPCR under study. Hence, the competitive binding assay described herein mainly serves as an initial screening assay that needs to be complemented with other cell-based assays to enable an in-depth characterization of compounds with receptor binding potency.
Protocol
1. Maintenance of Cell Culture
NOTE: All steps described under 1 and 2 are carried out under sterile conditions in a laminar flow cabinet.
Grow cells in T75 culture flasks at 37 °C and 5% CO2 in a humidified incubator. NOTE: In this assay, Jurkat cells (i.e., human leukemic T lymphocyte cells that endogenously express CXCR417) are used. Expression of CXCR4 at the cell surface should be evaluated throughout cell culturing by means of flow cytometry. A description of the flow cytometry procedure and reagents to determine receptor expression levels at the cell surface is, however, not within the scope of this protocol, but has been described previously17.
Let Jurkat cells grow in suspension until they reach 80 - 85% confluency. Before passaging the cells to a novel flask, allow all reagents to reach room temperature (RT).
Add 20 mL of fresh complete growth medium (RPMI-1640 medium, 10% fetal bovine serum (FBS), 2 mM glutamine) to a novel T75 culture flask.
Add 5 mL of Jurkat cell suspension from the original T75 flask (containing 25 mL of cell suspension) to the novel T75 culture flask. Incubate at 37 °C and 5% CO2 in a humidified incubator.
2. Preparation of CXCL12, Assay Buffer, and Jurkat Cells for the Competition Binding Assay.
Prepare a stock solution of CXCL12AF647 (20 µg/mL; see Table of Materials and Reagents) by dissolving the lyophilized reagent (stored at -80 °C, in the dark) in ultrapure water supplemented with 0.01% (volume/volume) of Polysorbate 20. Store single use aliquots from this stock solution at -80 °C, protected from light.
Prepare assay buffer by adding 40 mL HEPES (1 M, 20 mM final concentration) to 200 mL Hank's Balanced Salt Solution (HBSS, 10x, without phenol red and without sodium bicarbonate, 1x final concentration). Add ultrapure water to obtain a final volume of 2 L. Add 4 g (0.2% weight/volume) bovine serum albumin (BSA), and dissolve the BSA via magnetic stirring. Finally, adjust the pH to 7.4 (use NaOH for this) and filter the solution through 0.2 µm pores (see Table of Materials and Reagents) using a vacuum manifold. NOTE: This assay buffer will be used in all further steps of the protocol.
Count the number and the viability of the cells. For this, take a sample of the cell suspension and dilute it in phosphate buffered saline (PBS). NOTE: We routinely use an automated cell viability analyzer (see Table of Materials and Reagents) capable of counting cell suspensions at varying concentrations. For cell counting, dilute 0.5 mL of cell suspension in 1.5 mL PBS (other dilutions, e.g., 0.1 mL in 1.9 mL PBS, are also possible). The use of this method, which is based on the trypan blue dye exclusion method, has been described previously26. Several other devices are commercially available for counting cell number and viability that should work equally well.
Collect the desired number of cells (i.e., ~24 x 106 cells to run the assay with one complete 96-well plate) in a sterile 50 mL tube by centrifugation (see Table of Materials and Reagents for the type of centrifuge used) at 400 x g for 5 min at RT.
Gently pour off the supernatant without disturbing the cell pellet. Add fresh assay buffer (e.g., 20 mL) and resuspend the cells by gently pipetting up and down.
Centrifuge the cells again at 400 x g for 5 min at RT.
Pour off the supernatant again and resuspend the cell pellet in fresh assay buffer to obtain a density of 5 x 106 cells/mL.
3. Competition Binding Assay
NOTE: The actual competition binding assay is performed at RT and can be performed under non-sterile conditions.
Dilute the compounds under investigation in assay buffer (see 2.2) to obtain the desired concentration. Either prepare a fixed concentration of compound for initial screening (e.g., 10 µM final concentration) or, alternatively, a serial dilution series of concentrations for more detailed characterization of the compounds (e.g., a 1/3, 1/4 or 1/5 dilution series starting at 1 µM, final concentration). Keep in mind that the compound solution will ultimately become 2x diluted in the assay; therefore, prepare a 2x concentrated solution.
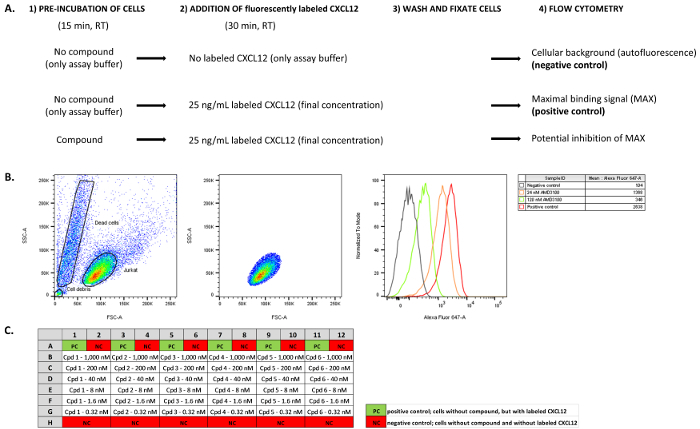
Dispense 100 µL of compound solution (2x concentrated) into a clear 96-well round bottom plate (see Table of Materials and Reagents) according to a pre-defined experimental lay out (e.g., Figure 1C). NOTE: At this stage, negative and positive control samples are included in the assay. In the negative control sample, 100 µL of assay buffer is added instead of compound to the wells of the 96-well plate. For the positive control sample, assay buffer is also added during this step. See also Figure 1C for a typical experimental layout in which a dilution series of several compounds is tested.
Add 50 µL of cell suspension (see 2.7; 0.25 x 106 cells) from a reagent reservoir into the 96-well plate using a multichannel pipette. Incubate the plate for 15 min at RT in the dark.
Add 50 µL of fluorescently labeled CXCL12 (i.e., 100 ng/mL of CXCL12AF647 in assay buffer, 4x concentrated, 25 ng/mL final concentration) from a similar reagent reservoir to the wells of the 96-well plate. Incubate for 30 min at RT in the dark. NOTE: For the negative control samples, add assay buffer instead. Hence, the fluorescent signal detected in the negative control samples will correspond to the autofluorescent background signal (Figure 1A). For the positive control samples, add 50 µL/well of CXCL12AF647. The positive control samples will yield the maximal fluorescence signal detected, since no potential inhibition by pre-incubation with compounds was included (Figure 1A).
Centrifuge the 96-well plate at 400 x g for 5 min at RT. Remove the supernatant from the pelleted cells by flipping over the plate. Dry the plate on a tissue.
Add 200 µL of fresh assay buffer from a reagent reservoir to the wells using a multichannel pipette. Immediately proceed.
Centrifuge the plate again for 5 min at 400 x g at RT. Remove the supernatant by flipping over the plate and again dry it on tissue.
Gently resuspend the cell pellet in 200 µL of 1% paraformaldehyde dissolved in PBS. This step will fix the cells.
Continue the protocol immediately with the quantification of the fluorescence by flow cytometry.
4. Analysis of the samples by flow cytometry
CXCL12AF647 stained and fixated cells are now ready to be analyzed using flow cytometry. Several types of flow cytometers can be used, but they need to be equipped with the correct laser (i.e., a red laser, excitation range ~630 nm) for excitation and suitable filters for fluorophore detection (emission filters ~660 nm). They need to be capable of handling samples in a 96-well plate format. Examples of suitable flow cytometry devices are given in the Table of Materials and Reagents.
Start up the device and open the corresponding software (see Table of Materials and Reagents).
Select the following cellular parameters to be visualized in a dot blot format: forward scatter (FSC), side scatter (SSC) and the fluorophore (CXCL12AF647) detection channel. NOTE: With the FSC parameter, cells are discriminated based on their size, since the detected light absorption is proportional to the cell's diameter. The SSC parameter, measuring light scattering at a 90° angle, provides information about the granularity of the cells.
- Choose one sample (e.g., a negative control sample) to perform gating of a defined homogenous cell population based on the FSC and SSC parameters.
- Select automatic injection of ~100 µL of fixated cells from this negative control sample into the flow cytometer.Select the option "mixing" before injection and use a sample flow rate of 1.5 µL/s.
- Run this sample by selecting "Acquire Data." The FSC and SSC parameters for this sample will now appear on the screen.
- Select the software's gating tool. Based on the FSC and SSC dot blot visualization, pre-define a homogenous and viable cell population by gating. To do so, create a polygon (using the software's gating tool) that includes the homogenously distributed single cells ("events") based on these two dimensions. NOTE: The gating procedure aims to define a homogenous and viable cell population that will be used for further analysis. Gating relies on the assumption that the majority of viable cells will form a homogenous cell population based on the FSC and SSC parameters. By performing this step, cellular debris, dead cells, and cell aggregates can largely be excluded from further analysis. An illustration of the gating process is given in Figure 1B.
Select to analyze 20,000 "events" (i.e., single cells) per sample. NOTE: This means that for each sample, 20,000 cells that fall within the pre-defined gate will eventually be analyzed. Data acquisition for each sample will continue until this number of events is analyzed.
Start the run (select "Record Data"). The flow cytometry device will now analyze all samples one-by-one by recording the mean fluorescence intensity (MFI) for each sample. This MFI corresponds to the mean fluorescent signal corresponding to the 20,000 cells that fall within the pre-defined gate.
5. Data analysis
Use the MFI obtained for each sample to perform all further calculations. Analysis of the flow cytometry data can be performed by several commercially available software packages (see Table of Materials and Reagents).
To determine the percentage inhibition of the fluorescent binding signal, as a result of compound pre-incubation, apply the following formula:
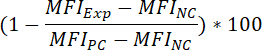 Where: MFIExp = the MFI of the experimental (= compound-treated) sample MFIPC = the MFI of the positive control MFINC = the MFI of the negative control
Where: MFIExp = the MFI of the experimental (= compound-treated) sample MFIPC = the MFI of the positive control MFINC = the MFI of the negative controlTo determine the IC50 value of a compound (i.e., the concentration of compound that can reduce the fluorescent binding signal by 50%), apply the following formula:
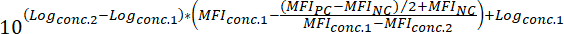 Where: Logconc.2 = the log of a concentration of compound that results in less than 50% inhibition of the difference between the MFI value of the PC and NC Logconc.1 = the log of a concentration of compound that results in more than 50% inhibition of the difference between the MFI value of the PC and NC NOTE: Alternatively, in case of highly active compounds, a dose-response curve covering several logs of magnitude can be generated based on the MFI corresponding to each tested concentration of compound. By applying non-linear regression curve fitting using appropriate software (see Table of Materials and Reagents), IC50 values can then be deduced from the generated curves. An example of this analysis approach is shown in Figure 3.
Where: Logconc.2 = the log of a concentration of compound that results in less than 50% inhibition of the difference between the MFI value of the PC and NC Logconc.1 = the log of a concentration of compound that results in more than 50% inhibition of the difference between the MFI value of the PC and NC NOTE: Alternatively, in case of highly active compounds, a dose-response curve covering several logs of magnitude can be generated based on the MFI corresponding to each tested concentration of compound. By applying non-linear regression curve fitting using appropriate software (see Table of Materials and Reagents), IC50 values can then be deduced from the generated curves. An example of this analysis approach is shown in Figure 3.
Representative Results
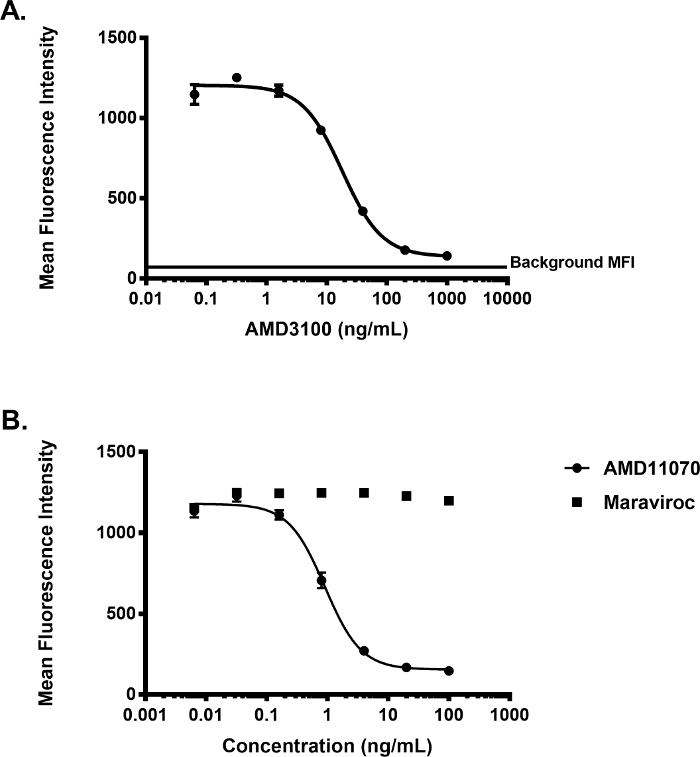
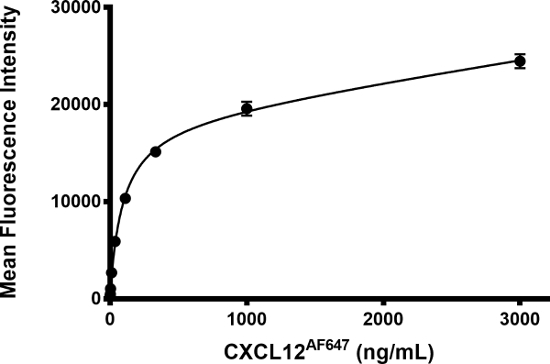
The general workflow of the binding assay is presented in Figure 1A. An illustration of the type of flow cytometry data obtained for different sample types in a standard experiment (i.e., negative control, positive control, and experimental sample) is depicted in Figure 1B, and a possible plate layout to perform the assay in a 96-well plate format is given in Figure 1C. Incubation of Jurkat cells, which endogenously express the chemokine receptor CXCR4 (i.e., the GPCR of interest in this assay), with increasing amounts of fluorescently labeled CXCL12 (i.e., CXCL12AF647) resulted into increased levels of fluorescence (Figure 2), reflecting increased levels of ligand binding to CXCR4. To demonstrate the receptor-specificity of CXCL12AF647 binding, a well-established and receptor-specific CXCR4 antagonist (the small molecule AMD3100) was used. Jurkat cells were first pre-incubated with a 1/5 dilution series of AMD3100 ranging from 0.064 ng/mL to 1,000 ng/mL, followed by an incubation with a final concentration of 25 ng/mL CXCL12AF647 (i.e., the fixed amount of labeled chemokine routinely used in the assay). Then, Jurkat cells were further processed as described in the protocol. The fluorescent signal (the mean fluorescent intensity, MFI) after addition of the fixed amount of CXCL12AF647 to the pre-incubated Jurkat cells, was obtained by flow cytometry analysis for all samples. This fluorescent signal could almost be entirely inhibited in case Jurkat cells were pre-treated with the highest concentration of AMD3100 (1,000 ng/mL). Under this condition, the inhibited level of fluorescence was only slightly above the background level corresponding to the level of autofluorescence for Jurkat cells. In conclusion, applying 25 ng/mL (final concentration) of CXCL12AF647 in the binding assay generated a large fluorescent signal window (when compared to the negative control) with a minimal residual level of non-specific receptor binding (Figure 3A).
This binding assay has primarily been used to screen for compounds disrupting ligand binding in panels of unlabeled compounds at a single fixed concentration, or to study the dose-dependent effect of active compounds. In the latter case, the goal is to obtain IC50 values (i.e., the concentration of compound that inhibits the binding signal by 50%), which reflect the binding potency of the compounds. Figure 3 illustrates how flow cytometry data obtained with this binding assay can be used to determine the inhibitory potency (in terms of IC50) of small molecules disrupting the binding of CXCL12 to CXCR4. Based on the dose-dependent inhibition of CXCL12AF647 binding by AMD3100, the IC50 value was determined here by applying non-linear regression analysis. In this example, the calculated IC50 value for AMD3100 corresponds to 18.12 ng/mL. Because in screening campaigns, the majority of randomly selected compounds are expected not to inhibit the binding of CXCL12AF647 to CXCR4+ Jurkat cells, an example of an inactive compound was also included in this study. Here, the small molecule maraviroc, which exhibits potent anti-HIV-1 activity by acting as a specific antagonist for the CC chemokine receptor 5 (CCR5)27, was tested in the binding assay. No inhibition of the maximal fluorescent binding signal was detected after pre-incubating Jurkat cells with maravoric, even at the highest concentration tested (100 ng/mL; Figure 3B). In the same experiment, a serial dilution series of AMD11070, another small molecule related to AMD3100, but with improved pharmacokinetic properties28, was included. Unlike maraviroc, a 1/5 dilution series from AMD11070 ranging from 0.0064 to 100 ng/mL clearly and dose-dependently inhibited CXCL12AF647 binding (Figure 3B).
Figure 1: Overview of the workflow and illustration of the type of data obtained. (A). The main steps of the protocol are schematized for the negative and positive control samples, and compound-treated samples. Samples without compound pre-incubation and without addition of fluorescently labeled chemokine (CXCL12AF647) are included to determine the background (auto)fluorescence (negative control samples). Samples without compound pre-incubation, but with addition of a fixed amount of CXCL12AF647 are used to determine the maximal binding signal in the assay (positive control samples). In compound-treated samples, cells are pre-incubated with compound before addition of a fixed amount of CXCL12AF647. (B). The mean fluorescent binding signal is determined by flow cytometry analysis of Jurkat cells. From all events (i.e., Jurkat cells) analyzed (left panel) only the fluorescent signal from a gated subpopulation of cells is used for further analysis (middle panel). Pre-incubation of cells with small molecules (e.g., AMD3100) inhibits the binding of the fluorescently labeled receptor ligand and this will reduce the maximal binding signal (right panel, shown in a histogram representation). (C). A possible plate layout when performing the competitive binding assay in a 96-well plate format. In this case, six different compounds (cpd 1- 6) are tested in a 1/5 serial dilution series (in duplicate) ranging from 1,000 nM down to 0.32 nM. Please click here to view a larger version of this figure.
Figure 2: Binding of CXCL12AF647 to Jurkat cells. Jurkat cells were incubated with increasing amounts of CXCL12AF647, in absence of pre-incubation with compound. The mean fluorescence intensity (MFI, expressed in arbitrary light units) ± SD is shown (n = 2). Curve fitting was performed using nonlinear regression using a one-site total binding model following the equation
![]()
with Bmax (maximum specific binding) = 18,345; Kd = 92.8 ng/mL; NS (the slope of nonspecific binding) = 2.139; and Background = 332.6. Please click here to view a larger version of this figure.
Figure 3: Inhibition of the binding of CXCL12AF647 by active and inactive compounds. (A) Dose-dependent inhibition of CXCL12AF647 binding by pre-incubating cells with increasing concentrations of AMD3100, before addition of 25 ng/mL CXCL12AF647. Note that the maximal inhibited mean fluorescence intensity (MFI; expressed in arbitrary light units) is still slightly above the background (auto)fluorescence obtained for Jurkat cells (Mean ± SD (n = 3)). Curve fitting was performed using non-linear regression with a variable slope (four parameters) according to
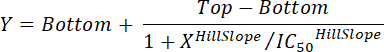
Here the HillSlope is 1.313 and the calculated IC50 is 18.12 ng/mL. (B). Dose-dependent effect of AMD11070 and maraviroc on CXCL12AF647 binding to CXCR4+ Jurkat cells (MFI ± SD (n = 3)). Curve fitting for the AMD11070 response was performed similarly with a HillSlope of 1.458 and a calculated IC50 value of 0.9052 ng/mL. For the maraviroc response, no fitting was performed given the lack of activity of this compound. Please click here to view a larger version of this figure.
Discussion
Compared to other types of binding assays (i.e., saturation binding and kinetic binding experiments), competition binding assays are best suited for screening purposes. Indeed, they allow evaluation of large batches of unlabeled compounds, for instance small molecules, by scoring their capability to interfere with the binding of a fixed amount of a labeled receptor ligand. Compounds that bind to other receptor sites than the labeled ligand might remain undetected in the assay. Although the competition binding experiment described herein focuses on the chemokine receptor CXCR4 in particular, it can likely serve as a blueprint for the design of competition binding assays for other GPCRs as well. For instance, the CXC chemokine receptor 7 (CXCR7) was described as an alternative receptor for CXCL12, capable of binding CXCL12 with high affinity29,30. Hence, the same labeled chemokine (CXCL12AF647) as used for the described CXCR4 competition binding assay can be used to set up CXCR7 binding experiments. Because CXCR4 and CXCR7 share CXCL12 as a common ligand, it would be important to work with cellular models in which only one of both receptors is expressed at the cell surface, in order to evaluate specific receptor binding. Our previous work has demonstrated that Jurkat cells do not show endogenous expression of CXCR717. Thus, in the assay described here for CXCR4, interference with binding to CXCR7 is ruled out.
Several key features of the competition binding assay allow it to be used as a fast, initial, screening tool to identify small molecules interfering with agonist binding to CXCR4. One of the assay's key benefits is the use of whole, living cells expressing the receptor of interest. This omits the need for labor-intensive cell membrane preparations. Also, the low level of background (auto)fluorescence detected with Jurkat cells is beneficial. Together with the large signal window obtained upon the addition of the defined amount of fluorescently labeled probe, it contributes to a favorable signal to background ratio. This facilitates the detection of potential binding inhibition by unlabeled compounds. In case this binding assay is performed with other CXCR4 expressing cells, we recommend to always evaluate these parameters (background fluorescence, signal window) upfront as they might differ from cell line to cell line. The fact that Jurkat cells endogenously express CXCR4 at a consistent level over long periods, makes it a convenient cellular model. The same consistent level of receptor expression might be obtained with stably transfected cell clones. In contrast, we do not recommend the use of transiently transfected cells because this will likely result into a much broader distribution of fluorescence intensities, fewer responsive cells in the assay and less consistency between experiments. Since no CXCR4 negative Jurkat cells exist, specificity of receptor binding to these cells was demonstrated by pre-incubation with well-established specific receptor antagonists (AMD3100 and AMD11070). These antagonists are structurally different from the labeled ligand. This is important because, when using the unlabeled variant of the ligand as competitor, both labeled and unlabeled ligand might also compete for binding to non-specific binding sites potentially present at the cell surface.
Apart from the cellular host, several other aspects of the assay described here need to be taken into account before starting to build a similar assay for other GPCRs. Fluorescently labeled probes that target GPCRs have been developed as alternatives for radioactive probes, but are currently only available for a limited number of GPCRs and they are relatively expensive. Moreover, modification of natural receptor ligands with fluorophores might alter the binding properties as compared to the unlabeled parent molecule, especially in case of very small ligands (e.g., amines) or small peptides31,32. For instance, in case of small ligands, a linker is generally required to separate the pharmacophore from the fluorophore to allow access to the receptor's orthosteric binding site31. Also, the stability of the receptor-fluorescent probe complex needs to be taken into account. In our experience, after 90 - 120 minutes (i.e., the time needed to run two consecutive 96-well plates), the mean fluorescent intensity for the positive control wells typically is reduced by ~5% compared to the average of all positive control wells on the plate, with occasional outliers between 5 and 10%.
Also, the intrinsic activity of the labeled ligand is of importance. Whereas receptor antagonists display no intrinsic activity on the receptor, labeled agonists likely induce receptor activation and, in consequence, receptor internalization. This compromises the reversible nature of the ligand-receptor interaction33. To minimize possible issues with receptor internalization, incubation with the labeled ligand (in case of a labeled agonist) need to be limited in time and should preferentially be performed at room temperature or lower. The fact that the procedure is performed at room temperature makes the assay also convenient and more compatible with screening efforts in plate format. Finally, a sufficient number of cells per sample (e.g., 250,000 per well of a 96-well plate) should be used in the assay to ultimately retain a sufficient number of cells (20,000 cells, "single events," based on the gating of the whole cell population; see Figure 1) for flow cytometry analysis. Using less cells would make it more difficult to consistently reach this number of analyzed cells, which might compromise the reliability of the read out. Using less cells per sample (per well) would also increase the time interval needed to analyze the sample by flow cytometry, and thus increases the overall time to analyze a whole 96-well plate. This compromises increased throughput. Due to the abovementioned reason, future miniaturization of the assay might thus turn out not to be a trivial task.
Taken together, a competition binding assay that is primarily used in our lab as a screening tool to identify small molecules that can compete with a fixed amount of labeled CXCL12 for binding to CXCR4 has been described in detail. These binding studies are of interest as they are a relatively fast and simple method to identify compounds capable of receptor binding. Although the identification of compounds with binding capacity is already valuable in itself, this type of assay does not provide information about the activity of the identified compounds. Hence, it remains necessary to further validate and characterize their potential agonistic or antagonistic activity in other pharmacological and functional receptor assays. Of interest, in the case of CXCR4 it has been shown that receptor antagonists that show increased activity in the binding assay (i.e., that generate low IC50 values for binding inhibition) perform relatively better in other functional CXCR4-related assays as well17.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank Eric Fonteyn for excellent technical assistance. This work has been supported by the KU Leuven (grant no. PF/10/018), Fonds voor Wetenschappelijk Onderzoek (FWO, grant no. G.485.08) and the Fondation Dormeur Vaduz.
References
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Milligan G, Kostenis E. Heterotrimeric G-proteins: A short history. Br J Pharmacol. 2006;147 Suppl 1:S46–S55. doi: 10.1038/sj.bjp.0706405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Tuteja N. Signaling through G protein coupled receptors. Plant Signal Behav. 2009;4:942–947. doi: 10.4161/psb.4.10.9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Rajagopal S. The beta-arrestins: Multifunctional regulators of G protein-coupled receptors. J Biol Chem. 2016;291:8969–8977. doi: 10.1074/jbc.R115.713313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Behnam Azad , Nimmagadda B, S The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- Flomenberg N, DiPersio J, Calandra G. Role of CXCR4 chemokine receptor blockade using AMD3100 for mobilization of autologous hematopoietic progenitor cells. Acta Haematol. 2005;114:198–205. doi: 10.1159/000088410. [DOI] [PubMed] [Google Scholar]
- Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, Walenkamp AM. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur J Cancer. 2013;49:219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Tsou LK, Huang YH, Song JS, Ke YY, Huang JK, Shia KS. Harnessing CXCR4 antagonists in stem cell mobilization, HIV infection, ischemic diseases, and oncology. Med Res Rev. 2017. [DOI] [PubMed]
- Schols D, Struyf S, Van Damme J, Este JA, Henson G, De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med. 1997;186:1383–1388. doi: 10.1084/jem.186.8.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hout A, D'Huys T, Oeyen M, Schols D, Van Loy T. Comparison of cell-based assays for the identification and evaluation of competitive CXCR4 inhibitors. PLoS One. 2017;12:e0176057. doi: 10.1371/journal.pone.0176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil) Biochem Pharmacol. 2009;77:1655–1664. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3:47–75. doi: 10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard LE, Nimmagadda S. CXCR4-based imaging agents. J Nucl Med. 2011;52:1665–1669. doi: 10.2967/jnumed.111.097733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xie Y, Oupicky D. Potential of CXCR4/CXCL12 Chemokine Axis in Cancer Drug Delivery. Curr Pharmacol Rep. 2016;2:1–10. doi: 10.1007/s40495-015-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmagadda S, Pullambhatla M, Stone K, Green G, Bhujwalla ZM, Pomper MG. Molecular imaging of CXCR4 receptor expression in human cancer xenografts with [64Cu]AMD3100 positron emission tomography. Cancer Res. 2010;70:3935–3944. doi: 10.1158/0008-5472.CAN-09-4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva RA, Peyre K, Pullambhatla M, Fox JJ, Pomper MG, Nimmagadda S. Imaging CXCR4 expression in human cancer xenografts: evaluation of monocyclam 64Cu-AMD3465. J Nucl Med. 2011;52:986–993. doi: 10.2967/jnumed.110.085613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatse S, Princen K, Liekens S, Vermeire K, De Clercq E, Schols D. Fluorescent CXCL12AF647 as a novel probe for nonradioactive CXCL12/CXCR4 cellular interaction studies. Cytometry A. 2004;61:178–188. doi: 10.1002/cyto.a.20070. [DOI] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- Louis KS, Siegel AC. Cell viability analysis using trypan blue: manual and automated methods. Methods Mol Biol. 2011;740:7–12. doi: 10.1007/978-1-61779-108-6_2. [DOI] [PubMed] [Google Scholar]
- Perry CM. Maraviroc: a review of its use in the management of CCR5-tropic HIV-1 infection. Drugs. 2010;70:1189–1213. doi: 10.2165/11203940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Moyle G, DeJesus E, Boffito M, Wong RS, Gibney C, Badel K, MacFarland R, Calandra G, Bridger G, Becker S. Proof of activity with AMD11070, an orally bioavailable inhibitor of CXCR4-tropic HIV type 1. Clin Infect Dis. 2009;48:798–805. doi: 10.1086/597097. [DOI] [PubMed] [Google Scholar]
- Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, Bachelerie F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart LA, Kilpatrick LE, Briddon SJ, Hill SJ. Probing the pharmacology of G protein-coupled receptors with fluorescent ligands. Neuropharmacology. 2015;98:48–57. doi: 10.1016/j.neuropharm.2015.04.033. [DOI] [PubMed] [Google Scholar]
- Vernall AJ, Hill SJ, Kellam B. The evolving small-molecule fluorescent-conjugate toolbox for Class A GPCRs. Br J Pharmacol. 2014;171:1073–1084. doi: 10.1111/bph.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart LA, White CW, Nguyen K, Hill SJ, Pfleger KD. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br J Pharmacol. 2016;173:3028–3037. doi: 10.1111/bph.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]