

Abstract
Microglia represent 5 - 10% of all central nervous system (CNS) cells and are increasingly drawing attention due to their contributions during development, homeostasis, and disease. Although macrophages have been studied in detail for decades, specialized features of microglia, the tissue-resident macrophages of the CNS, have remained largely mysterious, in part due to limitations in the ability to recapitulate mature microglial properties in culture. Here, we illustrate a straightforward procedure for the rapid isolation of pure microglia from the mature rodent brain. We also describe serum-free culture conditions that support high levels of microglial viability over time. Microglia cultured under these defined-medium conditions exhibit elaborate ramified processes and dynamic surveillance behavior. We illustrate some effects of serum exposure on cultured microglia and discuss how these serum-free cultures compare to both serum-exposed cultures as well as microglia in vivo.
Keywords: Neuroscience, Issue 133, Microglia, Cell Culture, Defined Medium, Neuroscience, Macrophage, Serum-Free, Ramified, Immunology
Introduction
As macrophages of the CNS parenchyma, microglia interact with a vast array of neuronal circuitry and glial signaling networks. They play vital roles in development and homeostasis of the brain through synaptic pruning, apoptotic cell clearance, and transient interactions with neuronal processes1,2. Microglia are early responders to neurological injuries, extending their long, thin processes to lesion sites to coordinate inflammatory responses and limit bleeding3,4. Changes in microglial morphology and function are ubiquitous in both acute and chronic CNS injuries, and microglia exhibit altered morphology, localization, and expression of inflammatory mediators in a diverse range of disease states1. Human genetic studies indicate that mutations that alter risk for neurodegenerative diseases are often predominantly or exclusively expressed by microglia in the intact CNS, pointing to a critical role for microglia in disease pathogenesis or progression5. Given their prominence in injury and disease, furthering the understanding of microglial biology is a high priority for developing new therapeutic approaches.
Many critical advances to the understanding of microglial biology have arisen by extrapolating techniques and mechanisms discovered in studies of other macrophage populations including culture methods, gene expression profiles, and definitions of functional/morphological states. Although generalized macrophage functions often play out in surprising ways within the CNS landscape, microglia are themselves highly specialized, exhibiting a ramified morphology and a unique gene expression signature that sets them apart from other tissue macrophages6. Microglia have a lineage that is distinct from most other tissue macrophages; they colonize the CNS during an early embryonic wave of primitive hematopoiesis and self-renew throughout life, independent of contributions from definitive hematopoiesis7. The fully mature gene expression signature of adult microglia is not achieved until the second postnatal week8. Environmental cues from the surrounding tissue play a major role in dictating tissue-specific macrophage features6, which in the CNS includes limited exposure to blood-borne factors granted by the blood-brain barrier9.
One obstacle to fully understanding microglial contributions to CNS homeostasis and disease is the difficulty of recapitulating the specialized properties of mature microglia seen in vivo with purified cells in vitro. Many methods have been developed to isolate and culture intact microglia, but most approaches rely on serum to support cell survival. We have shown that addition of serum, which is an inherently variable reagent containing a vast array of bioactive molecules, is particularly problematic when working with microglia because it promotes an amoeboid morphology, increased proliferation, and increased phagocytosis9 often seen in vivo when microglia are exposed to blood borne factors after the disruption of the blood-brain barrier. By these metrics, serum-exposed cells resemble microglia in injury or disease states, but such alterations are reduced when microglia are cultured in defined growth medium containing CSF-1 (or IL-34), TGF-β, cholesterol, and selenite.
This protocol provides details for culturing juvenile rat microglia under serum-free conditions, related to recently published work9. This protocol has been streamlined for rats from postnatal day 21 - 30 (P21 - P30), but can be adapted to isolate microglia from rats and mice of any age, though yield and overall viability will vary depending on the species and age of the animal. Maximal yields and optimal viability is achieved when using slightly immature microglia (~P9), with yields and viability gradually tapering to somewhat lower levels in adult animals. Microglia can also be isolated from mice, but we have found that rat cells show significantly higher yields, viability, and complexity of ramified morphologies, when compared to mouse cells in serum-free cultures. Animals aged greater than P50 have not been evaluated with this protocol. This immunopanning isolation procedure has been optimized to minimize changes in microglial transcriptional profiles during isolation and to maximize downstream viability of the cells. Using these techniques and media formulations, high-viability primary cultures can be sustained for weeks. Microglia cultured under these conditions exhibit a highly ramified morphology involving rapid extension and retraction of processes and relatively low rates of proliferation. We highlight the significance of serum-exposure on these properties, and discuss strengths and weakness of this method relative to other approaches.
Protocol
All procedures involving rodents conformed to Stanford University guidelines, which comply with national and state laws and policies. All animal procedures were approved by Stanford University's Administrative Panel on Laboratory Animal Care.
NOTE: All solution and buffer compositions are provided in the Table of Materials.
1. Prepare a Petri Dish for CD11b Immunopanning (Day 0)
NOTE: Prepare 1 immunopanning dish for every 1 - 2 juvenile rat brains.
Add 25 mL of 50 mM Tris pH 9.5 solution to a 15-cm Petri dish.
Add goat anti-mouse IgG (H+L chains) to the dish for a final concentration of 6 µg/mL. Swirl plate to evenly distribute.
Incubate the dish for 1 - 3 h at 37 °C.
Rinse dishes three times with DPBS++ (phosphate-buffered saline (PBS) with Ca2+ and Mg2+), then replace with a solution of panning buffer containing 1 µg/mL OX42 antibody. Leave the dishes overnight at room temperature on a flat surface.
2. Tissue Collection (Day 1)
NOTE: This protocol should take ~3 - 4 h.
Before beginning, ensure all solutions are sterile and chilled on ice. Chill all instruments on ice and sterilize with ethanol prior to use.
Following appropriate regulatory procedures, sacrifice a juvenile laboratory rat by carbon dioxide asphyxiation. NOTE: Alternatively, younger animals may be sacrificed with a lethal dose of ketamine/xylazine (100 - 200 µL of 24 mg/mL ketamine, 2.4 mg/mL xylazine). Ketamine/xylazine must be used if animals are less than 14 days of age. If using multiple animals, extract the tissue from one animal and place it into pre-chilled DPBS++ on ice before proceeding to subsequent animals.
Pinch a hindpaw and ensure complete unresponsiveness from the animal before proceeding.
Transcardially perfuse the animal with 10 - 30 mL of ice-cold perfusion buffer using a 27½-G needle until buffer runs clear. NOTE: Volume of Perfusion buffer will vary depending on the size/age of the animal.
Immediately after perfusion remove the head of the animal. Coming in from the spinal cord cut the occipital condyle on each side with dissection scissors, be careful not to damage the brain. After cutting each side carefully, cut up one side along the parietal and frontal bone towards the nasal bone. With forceps carefully pull back the top of the skull, quickly remove the brain (or CNS structures of interest), and place into pre-chilled DPBS++ on ice.
Repeat for all remaining animals. NOTE: All proceeding steps should be performed in a laminar flow hood under proper sterile conditions.
3. Mechanical Dissociation (Day 1)
After all brains have been collected, chop one brain into 1 mm3 chunks in a Petri dish on ice with a cold scalpel blade, and transfer to an ice-cold dounce homogenizer with 5 - 7 mL ice-cold douncing buffer. Dissociate one brain at a time.
Dissociate the tissue using 10 - 20 gentle and incomplete strokes with a loose-fitting dounce homogenizer. Take care not to directly crush the tissue at the bottom of the homogenizer, but instead impel the tissue through the space between the sides of the piston and the homogenizer.
Carefully remove the piston to prevent introduction of air bubbles. Allow poorly dissociated tissue chunks to settle to the bottom of the homogenizer, and transfer supernatant to a new chilled 50-mL conical tube.
Replace the removed volume with fresh douncing buffer, and repeat steps 3.2 and 3.3 for a total of 3 - 4 rounds, or until all tissue has been dissociated. Repeat the procedure for each brain.
4. Myelin Removal (Day 1)
NOTE: Myelin removal is used for isolation of microglia from animals older than P12.
Measure the volume of the cell suspension in the 50-mL conical tube using a 25 mL pipette, then add ice-cold douncing buffer to adjust the total volume to 33.5 mL.
Add 10 mL of myelin separation buffer (MSB) to the cell suspension and mix thoroughly by inverting the tube several times. This will result in a 23% final concentration of MSB in a volume of 43.5 mL.
Centrifuge cells for 15 min at 500 x g at 4 °C with slow braking. NOTE: The centrifuge should take approximately 1.5 - 2 min to decelerate. This will generate an upper layer of myelin and dead cell debris, a somewhat murky supernatant, and a smaller pellet that is enriched for live cells.
Remove the top layer of myelin/debris and the supernatant with a pipette. Take care when removing the top layer to ensure as much of it is removed as possible.
Resuspend the cell pellet in 12 mL of panning buffer. Gently triturate the cell suspension to break up any clumps of cells that might remain.
5. Immunopanning (Day 1)
Pass the cell suspension through a 70-µm cell strainer to remove large debris or cell clumps.
Rinse the OX42-coated panning dish three times with DPBS++. Don't allow the plate to completely dry between washes.
Pour off the last DPBS++ wash and apply the filtered cell suspension to the panning dish. Gently swirl the plate to distribute the cells, then incubate the plate on a flat surface at room temperature for 20 min to allow cells to adhere. Do not incubate longer than 20 min or cells will become very difficult to recover from the dish.
Rinse the panning dish with DPBS++ 10 times to remove non-adherent cells. Microglia will be firmly attached to the plate, so swirl the plate with each rinse to ensure removal of other non-adherent cells.
Pour off the last DPBS++ wash and replace with 15 mL DPBS++ and 200 μL trypsin (1.25% stock solution).
Incubate the dish for ≤10 min at 37 °C with 10% CO2 to trypsinize. NOTE: Do not continue longer than 10 min or microglia will become difficult to remove.
After 10 min of trypsinization, microglia will still be stuck to plate. Pour off trypsin/DPBS++ and gently wash 2x with DPBS++ to remove trypsin, replace with 12 mL of ice-cold microglia growth medium (MGM).
Place panning dish on ice for 2 min to help weaken cell/substrate interaction, and make sure the dish is flat to prevent areas of the panning dish from drying out.
Pipette vigorously with a 10-mL pipette and pipet controller on high speed to recover cells from the panning dish. Draw a 16 x 16 grid with the stream of liquid from the pipette to try and remove all the cells.
Check cells under a microscope at 20X magnification to make sure cells have detached from plate. Mark spots on top of the dish where cells are still stuck, and repeat pipetting in those areas.
Collect cell suspension and aliquot 3 - 4 mL of supernatant per 15-mL conical tube. Spin for 15 min at 500 x g at 4 °C with slow braking. NOTE: Spinning microglia through a small volume allows for the maximum recovery of cells.
Aspirate the supernatant, leaving 0.5 mL of MGM with the cell pellet.
Resuspend each pellet in remaining MGM, and pool the cells from all the tubes.
Count cells with hemocytometer.
Plate cells as described in Steps 6 - 7 depending on the application.
Culture cells at 37 °C with 10% CO2. NOTE: Cells can be culture for up to 3 - 4 weeks with regular media changes or one week with no media changes.
6. Spot Coating Tissue Culture Plates/Coverslips (Day 1)
Plate 15 µL of collagen IV coating directly in the center of a 24-well anionic/cationic coated tissue culture plate (see Table of Materials for details) and incubate for 15 min at 37 °C with 10% CO2.
After counting cells with hemocytometer dilute cells to 2.3 x 105/mL in MGM. Aspirate collagen IV spot and immediately plate 15 µL of cell suspension to this spot; this will give 3.5 x 103 cells/spot. Incubate for 5 - 10 min at 37 °C with 10% CO2 to allow cells to adhere, add 500 µL of CO2-equilibrated TGF-β2/IL-34/cholesterol containing growth medium (TIC) gently to the well.
If plating on coverslips, first coat sterile glass coverslips with 10 µg/mL ploy-D-lysine (PDL) in H2O for 1 h at RT, wash 3 times with H2O, and let dry in a hood under UV light. Once coverslips are dry, proceed with spot plating on the coverslips as described in Step 6.2.
7. Plating for RNA/Protein Isolation
On day 1, coat the entire area of a 24-well anionic/cationic coated tissue culture plate with collagen IV coating and incubate for 10 min - 1 h at 37 °C with 10% CO2, then remove and immediately plate 3 - 5 x 104 cells/well in CO2 equilibrated TIC media.
On day 2, perform a 50% media change for each well the day after preparation. Dead cells tend to cluster in the center of the well, so take media from there.
Perform a 50% media change every 2 - 3 days to maintain the cultures. If media changes introduce an unwanted variable for the cultures, cells can survive for approximately 1 week without any media changes, as opposed to over 3 weeks with regular maintenance.
Representative Results
This protocol describes a method to culture high purity ramified microglia from juvenile rats. Similar results can be obtained using immature, perinatal and adult animals, as discussed below. Since cell isolations often include subtle nuances and many opportunities for cells to die, we used quality-control checkpoints to help determine success at various steps. We typically monitored cell suspensions using a hemocytometer at multiple steps in the isolation protocol (Figure 1A). Successful isolations should yield 2.5 - 5 x 105 cells/juvenile animal. Animals from P7 - P15 show yields closer to 5 x 105 cells/animal, whereas cells isolated from animals older than P30 show yields closer to 2.5 x 105 cells/animal. Beyond monitoring total cell counts throughout the protocol, it is important to determine the overall health of isolated cells, which can be difficult because microglia isolated in this manner have a rounded or bipolar morphology during the first few days in culture. Here, we have included phase images of P25 microglia in culture during the first 6 days after isolation in TIC and TIC containing 10% fetal calf serum (FCS) (Figure 1B).

Figure 1: Evaluation of cell health during the isolation protocol and over 6 days of culture. (A) Phase-contrast images illustrating quality-control checkpoints at the designated steps of the cell isolation procedure. Cellular suspensions were imaged by mixing 9 µL of the cellular suspension with 1 µL of 0.4% trypan blue before loading onto a hemocytometer. Other images show the panning dish itself, and all images are shown as one field at 20X magnification. Scale bar, 200 µm. MGM, microglia growth medium. (B) Time course of microglial morphological changes and proliferation over 6 days of culture in serum-free TIC growth medium or TIC plus 10% fetal calf serum (FCS). CD11b-immunopanned cells were spot plated on collagen-coated anionic/cationic coated tissue culture plate (see Table of Materials for details), and the same field was imaged by phase-contrast every 24 h. Scale bar, 100 µm. TIC, TGF-β2/IL-34/Cholesterol containing growth medium. Please click here to view a larger version of this figure.
Routine evaluation of the health and viability of isolated cells is important for reproducibility and the success of experiments. Here we show microglia isolated from a P21 animal cultured in MGM, TIC, or TIC supplemented with 10% FCS after 7 days in vitro (DIV) with a viability assay. If the isolation was successful, cultures show between 80 - 90% viability in TIC after 7 DIV. Cells cultured in the presence of serum eventually climb to near 100% viability by this measure, however this is artificially inflated both by rapid proliferation of serum-exposed cells and rapid phagocytosis of dead cells within these cultures (Figure 2A). In addition to robust survival, TIC cultured cells also show a ramified morphology when compared to FCS treated cells (Figure 2B, C).
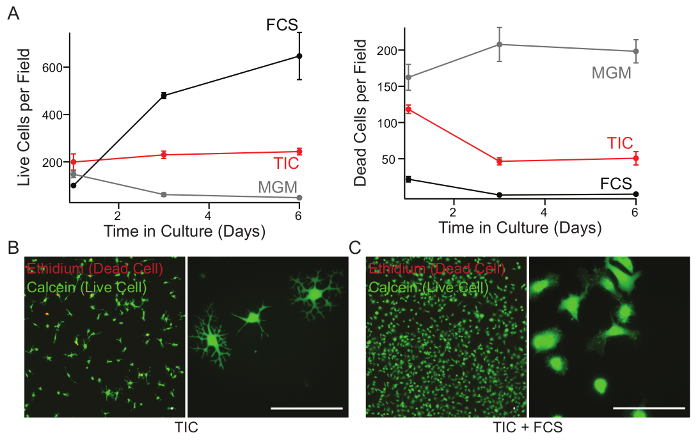
Figure 2: Survival of microglia in MGM, TIC or TIC + FCS over seven days in culture. (A) Cells were isolated from P21 rats and cultured in microglia growth medium (MGM), TGF-β2/IL-34/Cholesterol containing growth medium (TIC), or TIC + 10% fetal calf serum (FCS). At each time point, culture medium was supplemented with calcein AM (1.33 µM final) and ethidium homodimer (2.5 µM final) for 15 min to label live cells and dead cells respectively. Cells were counted in each field using an automated ImageJ script. (B and C) After 7 DIV, with no media changes, cells grown in TIC (B) or TIC supplemented with 10% FCS (C) were stained to label live and dead cells as above. Representative images were taken using a 10X (left) or 40X (right) objective. Scale bar, 40 µm. Error bars represent standard error of the mean (SEM). This figure was modified from Bohlen et al.9 and used with permission. Please click here to view a larger version of this figure.
In order to demonstrate successful isolation and culture of highly pure microglia, we isolated microglia from P21 rats and cultured these cells in TIC or TIC supplemented with 10% FCS for 8 days. Cells were then fixed and stained with microglial markers (Iba-1 and CD11b) and proliferation marker (Ki67, Figure 3A). In addition to staining for cell markers, RNA-seq on freshly isolated cells can provides a powerful and sensitive tool for evaluating the RNA profile of the cells, and can help inform the starting purity of cultures. CD11b immunopanning typically results in a small percentage of CD11b+ neutrophils/monocyte carryover in addition to CD11b+ microglia (Figure 3B). These cells will die in TIC after a few days, but can complicate analyses of earlier time points.
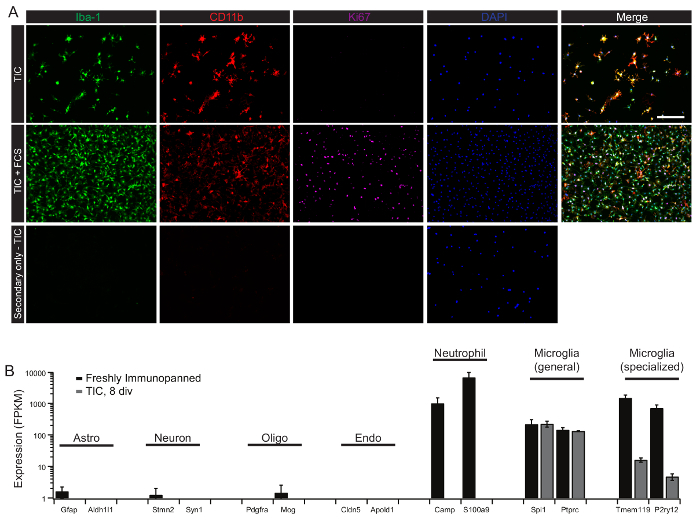
Figure 3: Purity of cultured cells as assessed by immunohistochemistry and RNA-seq. (A) Microglia were isolated from P21 rats, plated onto PDL/collagen coated glass coverslips and cultured in TGF-β2/IL-34/Cholesterol containing growth medium (TIC) for 8 days. Cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature and immune-stained with Iba-1 (1:500), CD11b (1:1500), and Ki67 (1:500). Briefly, fixed cells were blocked with 0.1% detergent (see methods table for details) and 10% normal donkey serum (NGS) for 1 h at room temperature. Cells were then incubated with primary antibodies in 0.1% detergent with 1% NGS overnight at 4 °C. Cells were washed three times for 5 min each in PBS. Cells were then incubated with secondary antibodies all 1:500 in 0.1% detergent with 1% NGS for 1 h at room temperature. Cells were again washed three times in PBS and mounted with DAPI-containing mounting medium. Representative images were taken at 40X magnification. Scale bar, 50 µm. (B) Expression of representative cell-type specific transcripts by freshly-isolated and cultured cells. Microglia were isolated from P21 rats and lysed directly from the immunopanning dish for RNA isolation and RNA-seq. Markers of other major CNS cell types (astrocytes, neurons, oligodendrocytes, and endothelial cells) show minimal contamination of CD11b-immunopanned cells. Neutrophil markers are detected in freshly-isolated cells (black), but are lost in serum-free cultures (gray). Cultured cells express high levels of common macrophage markers, but markers defining the specialized microglial signature are quickly downregulated by cultured cells. Error bars represent standard error of the mean (SEM). Data are modified from Bohlen et al.9 and used with permission. Please click here to view a larger version of this figure.
Staining for viability and cell markers allows for the determination of the survival and purity of the cells, but provides limited information on the dynamics of the culture. Here we have provided a time-lapse movie of P21 rat microglia after 14 DIV in TIC. After two weeks, serum-free cultures show dynamic surveillance behavior, with rapid process extensions and retractions throughout the dish (see Movie 1). Additionally, we have previously found that serum exposure transforms these resting state properties of microglia, resulting in lasting morphological and phagocytic changes. We have included a movie of microglia cultured for 5 days in TIC, then exposed to 10% FCS (see Movie 2).
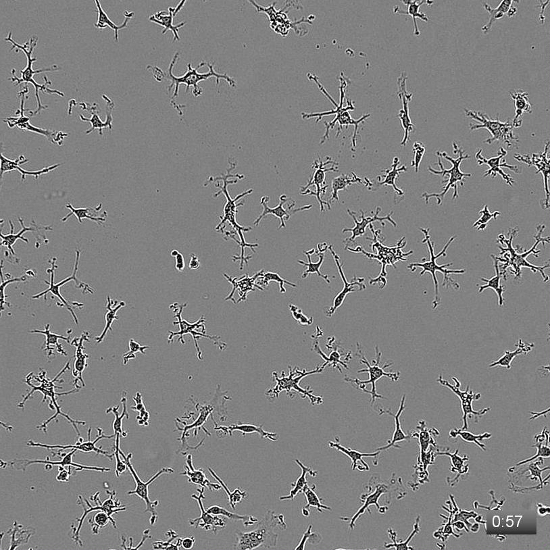 Movie 1: Defined-medium microglia cultures show ramified morphology with dynamic processes. Microglia were isolated from a P21 rat, spot plated onto collagen-coated anionic/cationic coated tissue culture plates, and cultured for 14 days in TIC. At 14 DIV, images were collected every 3 min. The movie plays at 6 frames/s, and the timestamp in the lower-right corner denotes the duration of imaging in h:min. Movie is reproduced from Bohlen et al.9 and used with permission. Please click here to view this video. (Right-click to download.)
Movie 1: Defined-medium microglia cultures show ramified morphology with dynamic processes. Microglia were isolated from a P21 rat, spot plated onto collagen-coated anionic/cationic coated tissue culture plates, and cultured for 14 days in TIC. At 14 DIV, images were collected every 3 min. The movie plays at 6 frames/s, and the timestamp in the lower-right corner denotes the duration of imaging in h:min. Movie is reproduced from Bohlen et al.9 and used with permission. Please click here to view this video. (Right-click to download.)
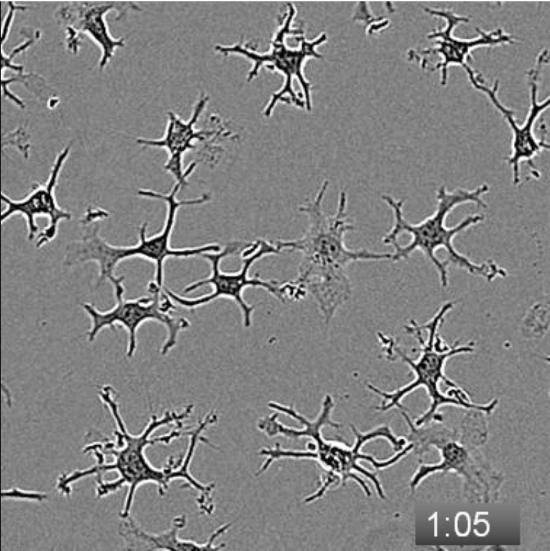 Movie 2: Serum exposure triggers a rapid morphological change in defined-medium microglial cultures. Microglia were isolated from a P21 rat, spot plated onto collagen-coated anionic/cationic coated tissue culture plates, and cultured for 5 days in TIC. At 5 DIV, images were collected every 5 min. Baseline motility/morphology was recorded, then at 2:55 (2 h, 55 min), a 50% media change was performed to add FCS to a final concentration of 10%. Cells were imaged for an additional 7 h. The movie plays at 6 frames/s, and the timestamp in the lower-right corner denotes the duration of imaging in h:min. Please click here to view this video. (Right-click to download.)
Movie 2: Serum exposure triggers a rapid morphological change in defined-medium microglial cultures. Microglia were isolated from a P21 rat, spot plated onto collagen-coated anionic/cationic coated tissue culture plates, and cultured for 5 days in TIC. At 5 DIV, images were collected every 5 min. Baseline motility/morphology was recorded, then at 2:55 (2 h, 55 min), a 50% media change was performed to add FCS to a final concentration of 10%. Cells were imaged for an additional 7 h. The movie plays at 6 frames/s, and the timestamp in the lower-right corner denotes the duration of imaging in h:min. Please click here to view this video. (Right-click to download.)
Discussion
Because microglia serve as sentinel immune cells of the CNS, they are highly responsive to environmental changes; therefore, great care is required to minimize inflammatory responses within the cells during their isolation and culture8. This is accomplished in this protocol through speed and temperature. Keeping the cells on ice or at 4 °C whenever possible greatly reduces activation, so all centrifugation steps take place at 4 °C, the animals are perfused with ice-cold perfusion buffer, and the protocol has been streamlined to minimize the time between tissue extraction and culturing of the cells. If assessing gene expression patterns of freshly-isolated cells, harvest cell lysates directly from the CD11b immunopanning dish prior to trypsinization to keep isolation-induced changes at a minimum. It should be noted that even carefully-isolated microglia execute an inflammatory response immediately upon being placed in warm temperatures, presumably due to recognition of damage-associated signals intrinsically associated with the isolation procedure. If the protocol was performed successfully, 2.5 - 5 x 105 cells/juvenile rat are expected.
The cell isolation protocol provided here is highly specific for isolating CD11b+ cells from the perfused CNS9. Although this cell population is comprised predominantly of microglia, it also contains low levels of other myeloid populations such as perivascular macrophages, meningeal macrophages, choroid plexus macrophages, monocytes, and neutrophils10. Choroid-associated myeloid cells can be essentially eliminated by recognition and removal of the choroid plexus prior to tissue dissociation. Meninges can also be removed to some extent, but complete elimination of all meningeal tissue is not practical within the timeframes targeted for rapid isolation described here. Therefore, to limit variability due to incomplete removal of meninges, we do not remove meninges. To minimize the number of circulating monocytes/neutrophils in the isolated material, it is also important to have a clean perfusion, and we typically discard tissue that is poorly perfused. Even with excellent perfusion, clearance of blood is not absolute, as will be evident by the presence of a small quantity of erythrocytes in cell pellets throughout the purification. Thus, small levels of monocytes and neutrophils are co-purified and contribute a distinct signature in transcriptomic profiles of freshly-isolated cells. However, circulating cells will quickly die in serum-free TIC media and do not represent persistent contaminants, although they can continue to survive and proliferate in serum containing cultures.
Another important point to achieving a healthy, viable culture is to take care during the mechanical dissociation steps. While douncing kills many neurons, glia, and other CNS cells, microglia survive this dissociation relatively unharmed (although microglia can also be killed by over-douncing). If done correctly, this protocol results in cell yields comparable to those obtained using proteolytic dissociation, whereas potentially confounding variables from tissue inflammatory responses at elevated temperatures are avoided. By performing consecutive incomplete rounds of mechanical tissue dissociation, single cells can be harvested from the suspension while minimizing the amount of mechanical stress on the overall culture.
The immunopanning protocol described here is a reliable and reproducible way to isolate microglia, but other comparable methods are available. We have obtained similar results using magnetic isolation with magnetic-activated cell sorting myelin depletion and CD11b selection. We have here focused on the immunopanning method because it has provided the highest-quality cultures to date, requires less specialized equipment, and does not require introduction of iron oxide-conjugated antibodies to the cells immediately prior to culture. Another approach commonly used for microglial isolation is flow cytometry, which has a major advantage over the current approach in that protocols exist for purification of bona fide microglia from other CD11b+ myeloid contaminants using surface antibodies against signature markers such as TEMEM1198. Though fluorescence-activated cell sorting results in an extremely pure cell population, it also imposes additional stresses to the cells and may result in lower viability cultures or reduced yields. Overall, we focus on antibody-based methods over label-free density centrifugation and shake-off preparations to maximize the reproducibility and purity of the cell population.
While this protocol is optimized for the culture of juvenile rat microglia, it can be used to culture microglia from various ages; total cell yields are maximal from 1 - 2-week-old rats, after the peak of microglial expansion has passed and before structural rearrangements that interfere with recovery of the cells from older tissue. Microglia can also be isolated and cultured from mouse and human tissue by replacing the CD11b antibody with an appropriate monoclonal antibody specific for mouse or human epitopes. While both mouse and human microglia survive under these culture conditions, they show morphological differences when compared to rat cells. Mouse cells retain a ramified morphology but have less elaborate processes, while human cells have an almost uniform amoeboid morphology when isolated and cultured according to this procedure.
Isolated microglia can be plated in different ways depending on the assays required. For optimal results (as illustrated in the live/dead assays and morphological movies shown here), 3.5 x 103 cells were "spot" plated in the center of the well of a 24-well anionic/cationic coated tissue culture plate after a 10-min coating of collagen IV as described in the above protocol. Alternatively, for experiments requiring large numbers of cells, such as RNA isolation, 3 - 5 x 104 cells can be plated per well of a 24-well plate after 10 min to 1 h of collagen IV coating. Cells plated at higher densities showed slightly lower viability and less complex morphology when compared to cells that were spot treated, possibly due to the impact of inflammatory signals secreted in response to the isolation. For immunohistochemistry or imaging requiring glass coverslips, cells exhibit the best survival and morphology when coverslips were first coated with PDL then coated in collagen IV as described above. For assays requiring a large number of conditions, cell viability is also high in 96-well or 384-well collagen-coated plates. In all conditions, the cells start out round or bipolar, begin to acquire processes around day 3 - 4, and take 5 - 10 days to exhibit the maximally ramified morphology illustrated in Figure 1 and Movie 1.
While this protocol results in a culture that exhibits dynamic surveillance behavior and morphology similar to resting microglia in vivo, these cells fail to fully retain the specialized gene expression signature of microglia in vivo. Many of the persistent gene changes observed in cultured cells reflect alterations typically seen in the embryonic or inflamed CNS9. As such, this protocol represents a step forward in the understanding of the conditions microglia require to survive in culture and serum-evoked changes, but further study is needed to understand the extrinsic signals provided by the CNS that fine-tune microglial properties and gene expression.
Disclosures
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Christopher and Dana Reeve Foundation International Research Consortium on Spinal Cord Injury, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the JPB Foundation, the Novartis Institute of Basic Research, generous contributions from Vincent and Stella Coates, and the Damon Runyon Cancer Research Foundation (DRG-2125-12).
References
- Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015;36(10):605–613. doi: 10.1016/j.it.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou N, et al. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A. 2016;113(4):1074–1079. doi: 10.1073/pnas.1520398113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9(12):1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Gosselin D, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017;356(6344) doi: 10.1126/science.aal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Ginhoux F. Ontogeny of Tissue-Resident Macrophages. Front Immunol. 2015;6:486. doi: 10.3389/fimmu.2015.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen CJ, et al. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron. 2017;94(4):759–773. doi: 10.1016/j.neuron.2017.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid Cells in the Central Nervous System. Immunity. 2017;46(6):943–956. doi: 10.1016/j.immuni.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]