

Abstract
As a promising genome editing platform, the CRISPR/Cas9 system has great potential for efficient genetic manipulation, especially for targeted integration of transgenes. However, due to the low efficiency of homologous recombination (HR) and various indel mutations of non-homologous end joining (NHEJ)-based strategies in non-dividing cells, in vivo genome editing remains a great challenge. Here, we describe a homology-mediated end joining (HMEJ)-based CRISPR/Cas9 system for efficient in vivo precise targeted integration. In this system, the targeted genome and the donor vector containing homology arms (~800 bp) flanked by single guide RNA (sgRNA) target sequences are cleaved by CRISPR/Cas9. This HMEJ-based strategy achieves efficient transgene integration in mouse zygotes, as well as in hepatocytes in vivo. Moreover, a HMEJ-based strategy offers an efficient approach for correction of fumarylacetoacetate hydrolase (Fah) mutation in the hepatocytes and rescues Fah-deficiency induced liver failure mice. Taken together, focusing on targeted integration, this HMEJ-based strategy provides a promising tool for a variety of applications, including generation of genetically modified animal models and targeted gene therapies.
Keywords: Genetics, Issue 133, CRISPR/Cas9, Targeted Integration, Homology-Mediated End Joining, In Vivo, Embryo, Genetically Modified Mice, Hydrodynamic Injection
Introduction
Precise, targeted genome editing is often required for producing genetically modified animal models and clinical therapies. Much effort has been made to develop various strategies for efficient targeted genome editing, such as zinc finger nuclease (ZFN), transcription activator-like effector nucleases (TALENs), and CRISPR/Cas9 systems. These strategies create targeted DNA double-strand breaks (DSB) in the genome, and take advantage of intrinsic DNA repair systems, such as homologous recombination (HR)1,2, microhomology-mediated end joining (MMEJ)3,4,5, and non-homologous end joining (NHEJ)6,7,8 to induce targeted integration of transgenes1,9. The HR-based strategy is currently the most commonly used genome editing approach, which is very efficient in cell lines, but not readily accessible to non-dividing cells due to its restricted occurrence in the late S/G2 phase. Thus, the HR-based strategy is not applicable for in vivo genome editing. Recently, the NHEJ-based strategy was developed for efficient gene knock-in in mouse tissues8. Nevertheless, the NHEJ-based method usually introduces indels at the junctions, making it difficult to generate precise genome editing, especially when trying to construct in-frame fusion genes8. MMEJ-based targeted integration is capable of precise genome editing. However, it only modestly increases the targeted integration efficiency in previous reports5. Therefore, improving the efficiency of precise targeted integration in vivo is urgently needed for broad therapeutic applications3.
In a recently published work, we demonstrated a homology-mediated end joining (HMEJ)-based strategy, which showed the highest targeted integration efficiency in all reported strategies both in vitro and in vivo10. Here, we describe a protocol for the establishment of the HMEJ system, and also the construction of the single-guide RNA (sgRNA) vectors targeting the gene of interest and the donor vectors harboring sgRNA target sites and ~800 bp of homology arms (Figure 1). In this protocol, we also describe the detailed steps for generation of DNA knock-in mice and brief steps for targeted integration in tissues in vivo. Moreover, a proof-of-concept study of the HMEJ-based strategy demonstrated its ability to correct Fah mutation and rescue Fah-/- liver failure mice, which further revealed its therapeutic potential.
Protocol
All procedures including animal subjects have been approved by the Biomedical Research Ethics Committee at the Shanghai Institutes for Biological Science (CAS).
1. Design of Donor Plasmids
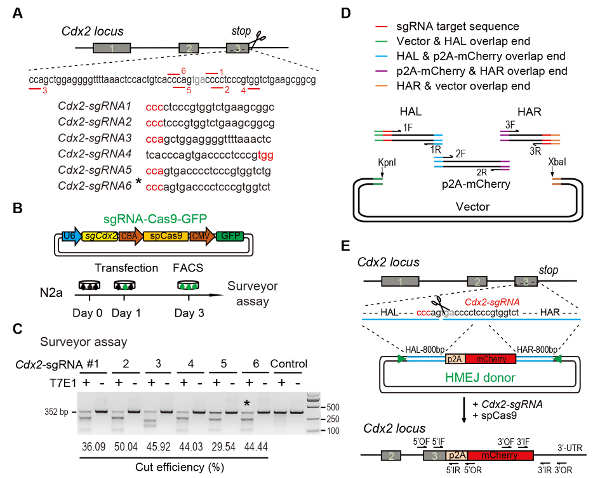
- Selection of sgRNA
- Linearize 2 µg of Cas9-CMV-EGFP expression vectors and sgRNA by BbsI digestion (1 µL of BbsI for 2 h at 37 °C at a final concentration of 1 U/µL in a volume of 20 µL). Then purify the product by gel purification kit with a 1% agarose gel in 1× TAE buffer.
- Mix a pair of sgRNA oligonucleotides in 10 µL of 1× T4 DNA ligase buffer to a final concentration of 50 µM. Incubate the oligo solution using a temperature gradient from 95 °C to 25 °C with a temperature change rate of 5 °C /5 min (95 °C for 5 min, then 90 °C for 5 min, 85 °C for 5 min, etc.), which will anneal the oligos.
- Mix 4 µL of annealed product, 2 µL of the linearized vector with 1 µL of T4 DNA ligase in 10 µL of 1× T4 DNA ligase buffer, and then ligate at 22 °C for 1-2 h (Figure 1B).
- Surveyor nuclease assay of sgRNA NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
- Transfect Cas9-sgRNA-EGFP expression vectors into N2a cell lines cultured in DMEM supplemented with 10% fetal bovine serum, 1% PSG, and 1% non-essential amino acid by the transfection kit (see Table of Materials). Incubate the transfected cells at 37 °C in 5% CO2.
- After 48 h of incubation, collect 5,000 transfected cells (GFP+) by fluorescence-activated cell sorting (FACS) using non-transfected cells as a control.
- Digest the collected cells in 2-5 µL oflysis buffer (0.1% Triton X-100, 0.1% Tween 20, and 100 µg/mL Proteinase K) at 56 °C for 30 min, and then heat inactivate proteinase K at 95 °C for 10 min.
- Amplify the sample by nested PCR (Table 1) using the manufacturer's protocol. The size of PCR products is set to 300-500 bp.
- Mix 1 µL of lysis product with DNA polymerase and a pair of outer primers recognizing the sequence around the sgRNA target site (0.1 µM, final concentration) (Table 1), and perform the primary PCR in a volume of 20 µL.
- Activate DNA polymerase at 95 °C for 5 min, and perform the primary PCR for 30 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 24 s (1 min/1 kb), with a final extension at 72 °C for 5 min.
- Perform the secondary PCR using 1 µL of primary PCR product and a pair of nested inner primers.
- Denature and re-anneal 300-600 ng of purified PCR product in 20 µL of 1× T7EI reaction buffer (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT pH 7.9) using a gradient of temperature from 95 °C to 25 °C with a rate of 5 °C /5 min.
- Add 1 µL of T7EI enzyme to the annealed PCR products and digest at 37 °C for 2 h. Then run the digestion product on 2% agarose gel in 1×TAE buffer at 120 V for 40 min until the fragments are separated (see Table of Materials).
- Construction of donor vector NOTE: To generate HMEJ donor vectors for Cdx2 gene, construct a donor DNA (800 bp HAL-p2A-mCherry-800 bp HAR) flanked with 23 nt Cdx2-sgRNAs targeting sequence at both ends (Figure 1D and Figure 1E). The PAM of target sequence was adjacent to the end of the homologous arm. Gibson assembly is recommended for HMEJ donor cloning.
- Amplify the 800 bp left homology arm (HAL) with forward primer-1F (containing 15-20 nt overlap sequence from vector, 23 nt Cdx2-sgRNA targeting sequence, and about 20 nt sequence from HAL) and reverse primer-1R (containing 15-20 bp overlap sequence from p2A-mCherry and about 20 nt sequence from HAL) at 0.1 µM final concentration using mouse genomic DNA at 200 ng/µL (Figure 1D, Table 1).
- Amplify the p2A-mCherry insertion fragment with forward primer-2F (containing 15-20 nt overlap sequence from HAL and about 20 nt sequence from insertion fragment) and reverse primer-2R (containing 15-20 nt overlap sequence from HAR and about 20 nt sequence from the insertion fragment) at 0.1 µM final concentration using genomic DNA or plasmid with reporter sequences at 100 ng/µL or 30 ng/µL (Figure 1D, Table 1).
- Amplify the 800 bp right homology arm (HAR) with forward primer-3F (containing 15-20 nt overlap sequence from vector, 23 nt Cdx2-sgRNA targeting sequence, and about 20 nt sequence from HAR) and reverse primer-3R (containing 15-20 nt overlap sequence from p2A-mCherry and about 20 nt sequence from HAR) at 0.1 µM final concentration using mouse genomic DNA at 200 ng/µL (Figure 1D, Table 1).
- Run all the PCR products on 1% agarose gel in 1×TAE buffer, and purify the PCR products of expected size by gel extraction kit, according to the manufacturer's instructions (Table 1).
- Digest 50-100 ng of a construct vector with by KpnI and XbaI. Mix 2 µL of the linearized vector at 30-40 ng/µL with three PCR amplified fragments (1 µL for each, 100-200 ng/µL) in 2x Gibson mix. Add H2O to adjust the final volume to 10 µL.Incubate the mix at 50 °C for 60 min.
- Transform competent E. coli cells with all the assembled product and extract the plasmid constructs by DNA extraction kit according to the manufacturer's instructions. Verify the HMEJ donor by DNA sequencing.
2. Genome Editing in Mouse Embryos Using the HMEJ-Based Method
- Production of Cas9 mRNA
- For Cas9 mRNA preparation, add the T7 promoter sequence to the Cas9 coding region by PCR amplification using the appropriate primer pair listed in Table 1. Add the primer Cas9 F/R at a final concentration of 0.1 µM and 20 ng of Cas9 expressing vector to 1× high fidelity DNA polymerase mix. Adjust the final volume to 50 µL with H2O.
- Activate DNA polymerase at 95 °C for 5 min, and perform the PCR for 36 cycles at 95 °C for 30 s, 60 °C for 30 s, and 68 °C for 4 min (1 min/1 kb), with a final extension at 68 °C for 10 min.
- Purify T7-Cas9 PCR product for in vitro transcription (IVT), and then transcribe 0.5-1 µg DNA by mRNA transcription kit at 37 °C for 8 h in a total volume of 20 µL, according to the manufacturer's instructions (see Table of Materials).
- Add 1 µL of DNase to the mixture to remove the DNA template at 37 °C for 15 min. Add a poly-A tail for 45 min at 37 °C and recover the Cas9 mRNA by RNA purification kit, according to the manufacturer's instructions (see Table of Materials).
- Production of sgRNA
- Generate the sgRNA template driven by a T7 promoter with high fidelity DNA polymerase as above. Choose an sgRNA scaffold containing vector as the template. The primers used are listed in Table 1.
- Purify the T7-sgRNA PCR product and use 0.5-1 µg DNA as the template for in vitro transcription of sgRNA using a short RNA transcription kit at 37 °C for 6 h in a total volume of 20 µL, according to the manufacturer's instructions (see Table of Materials).
- Add 1 µL of DNase to the mixture and continue the incubation at 37 °C for 15 min to remove the DNA template. Purify the sgRNAs by RNA purification kit, as above (see Table of Materials).
- Embryo collection, microinjection and in vitro culture
- Superovulate female B6D2F1 (C57BL/6 × DBA2J) mice (7-8 weeks old) by pregnant mare serum gonadotropin (PMSG), followed by human chorionic gonadotropin (hCG) 48 h later. After the hCG injection, house females with B6D2F1 males overnight.
- Sacrifice the females by CO2 anesthesia, 24 h after hCG injection. Collect the fertilized embryos from their oviducts (with 30 - 50 embryos for each female) in M2 medium.
- Place the fertilized embryos (about 300 eggs for one-day injection) into KSOM medium (5.55 g/L NaCl, 0.19 g/L KCl, 0.05 g/L KH2PO4, 0.05 g/L MgSO4•7H2O, 0.04 g/L Glucose, 1.12 g/L Sodium lactate, 2.1 g/L NaHCO3, 0.02 g/L Sodium pyruvate, 0.25 g/L CaCl2•2H2O, 0.004 g/L EDTA, 0.146 g/L L-glutamine, and 1 g/L Bovine serum albumin) at 37 °C in an incubator with 5% CO2.
- Mix Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL), and HMEJ donor vector (100 ng/µL), and add H2O to adjust the final volume to 10 µL. Put the mixture on ice.
- Pull capillary needles (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) using a Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300. See Table of Materials). Commercial needles would be an alternative substitution for the microinjection.
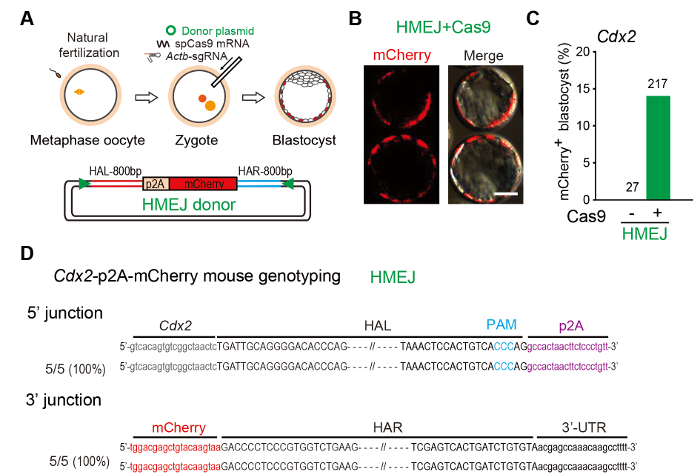
- Inject a probable volume of the mixture into the cytoplasm of zygotes with well-defined pronuclei in a droplet of HEPES-CZB medium containing 5 µg/mL cytochalasin B using a microinjector with constant flow settings (Figure 2A) (see Table of Materials)21. NOTE: Each group of zygotes should be injected within 20-30 min. Cytochalasin B could increase the viability of mouse zygote after injection. Alternatively, microinjection can be operated with the piezo system, as described previously22.
- Culture the injected zygotes in KSOM medium at 37 °C under 5% CO2 until the blastocyst stage after 3.5 days for fluorescence observation (Figures 2B and 2C).
- Embryo transfer and generation of mice
- Mate estrous ICR female mice with vasectomized ICR male mice on the same day as injection.
- Culture the injected zygotes into the 2-cell stage at 37 °C under 5% CO2, and transfer 25-30 2-cell embryos into oviducts of pseudopregnant ICR females at 0.5 day post coitum (dpc). Recipient mothers deliver pups at 19.5 dpc.
- Mouse genotyping
- Extract mouse genomic DNA from toe or tail samples using a DNA extraction kit, according to the manufacturer's instructions (see Table of Materials).
- Identify the 5' and 3' junction of knock-in events using 200-400 ng of genomic DNA measured by UV/vis spectrometry as a template to perform the PCR amplification.
- Activate DNA polymerase at 95 °C for 5 min, and perform the PCR for 38 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min (1 min/1 kb), with a final extension at 72 °C for 10 min. For the 5' junction, use the forward primer at the upstream of the HAL, with the reverse one on the knock-in fragment (p2A-mCherry). As to 3' junction, use the forward primer on the knock-in fragment (p2A-mCherry), with the reverse one on the downstream of the HAR (Table 1).
- Run 6 µL of the PCR product on 1% agarose gel in 1×TAE buffer and check for the expected fragment size. Then verify them by DNA sequencing (Figure 2D).
3. HMEJ-Based In Vivo Genome Editing in Hepatocytes
Place recipient C57BL/6J mouse (8 week) in a restraining device and put the tail through the slit.
Mix HMEJ donor vectors (30 µg) and spCas9 expression vectors (30 µg) in 2 mL of saline solution. For the control experiment, suspend HMEJ donor vectors (30 µg) in 2 mL of saline solution (Figure 3A).
Clean the mouse tail with 70% ethanol. Insert the needle into the tail vein and inject the plasmid DNA solution within 5-7 s. Remove the needle and release the mouse from the restraining device.
Sacrifice mice by CO2 anesthesia after 5-9 days after injection. Perfuse the mice transcardially with 0.9% saline, followed by 4% paraformaldehyde using a peristaltic pump, and fix the liver overnight at 4 °C.
Dehydrate the tissue using 30% sucrose overnight, until it sinks to the bottom of tube.
Section the frozen tissue at a thickness of 10 µm for liver samples.
Rinse the sections three times in 0.1 M phosphate-buffered (PB) and incubate them with primary antibody: Rabbit anti-mCherry (diluted in 5% NGS) overnight at 4 °C.
Wash sections three times in PB, and then incubate them with secondary antibody: Cy3-AffiniPure Goat Anti-Rabbit IgG for 2 h at room temperature on an orbital shaker.
Counterstain the sections with DAPI for 20 min and mount with glycerin on glass slides for further fluorescence observation (Figure 3B).
Representative Results
HMEJ-based genome editing in mouse embryos: To define the knock-in efficiency of the HMEJ-based method in mouse zygotes, we delivered Cas9 mRNA, sgRNA targeting the Cdx2 gene and the HMEJ donor into mouse zygotes, which was designed to fuse a p2A-mCherry reporter gene to the last codon of the Cdx2 gene (Figure 2A). The injected zygotes developed into blastocysts in the culture. To evaluate the knock-in efficiency, we analyzed the mCherry fluorescence with a fluorescent microscope, and we found that 12.9% of the blastocysts receiving HMEJ donors were positive for mCherry, which was strictly expressed in the trophectoderm (Figures 2B,2C). By sequencing the PCR positive mice, we also found that all examined integration events were precise in-frame integrations at both 5' and 3' junctions (Figure 2D).
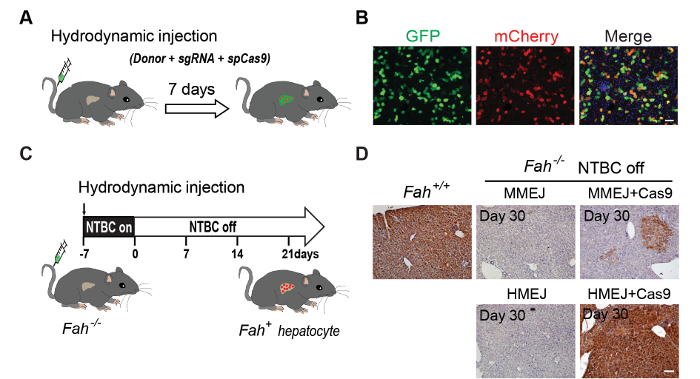
HMEJ-based genome editing in adult tissues and HMEJ-mediated gene therapy: To investigate whether HMEJ-based genome editing could be applied in adult tissues, we inserted the mCherry cassette right before the stop codon of Actb gene by transducing Actb-HMEJ constructs to C57/B6J mouse livers by tail-vein hydrodynamic injection (Figure 3A). After 7 days of injections, we found that nearly half of the transfected hepatocytes expressed mCherry as stained on the liver sections (Figure 3B).
To explore the possibility of using an HMEJ-based strategy for gene therapy, we employed fumarylacetoacetate hydrolase (Fah)-deficient mice. The Fah-/- mouse is a well-established hereditary tyrosinemia type I (HTI) mouse model, which harbors an insertion fragment in exon 5 of the Fah gene, causing frameshift mutations in the following sequence23. To maintain Fah-/- mice, we treated the Fah-/- mice with an inhibitor of the upstream of tyrosine catabolic pathway, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Here we set out to see whether MMEJ- and HMEJ-mediated gene correction could rescue Fah mutation in the Fah-/- mouse. We hydrodynamically injected Cas9 construct together with Fah-MMEJ or Fah-HMEJ constructs, designed to insert Fah cDNA of exon 5 to 14 into intron 4 of Fah gene, to Fah-/- mouse livers (Figure 3C). One week after injection, NTBC was withdrawn to induce liver damage (Figure 3C). After the withdrawal of NTBC, Fah-corrected hepatocytes of the Fah-/- mice receiving Fah-HMEJ and Cas9 constructs showed more effective proliferation than MMEJ-based method (Figure 3D).
Figure 1: HMEJ-mediated targeted integration in vitro. (A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
Figure 2: Genome editing in mouse embryos via HMEJ-mediated targeted integration (A) Experimental scheme of microinjection: A mixture of Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL), and donor plasmids (100 ng/µL) were injected into mouse zygotes. (B) Representative fluorescence images of mouse embryos edited by HMEJ strategy. Bar, 20 µm. (C) Knock-in efficiency indicated by percentage of mCherry+ blastocysts. Number above each bar, total blastocysts counted. (D) Sequence analysis of gene-edited mice at Cdx2 locus.PCR products amplified from 5' and 3' junction sites were sequenced. Upper, homology arm; purple, p2A; red, mCherry; HAR or HAL, right or left homologous arm. Dashed lines mark the region omitted for clarity. Figure modified from previous report10. Please click here to view a larger version of this figure.
Figure 3: HMEJ-mediated targeted integration in vivo. (A) Schematic overview of hydrodynamic tail vein injection. A mixture of plasmids expressing donor sequence and sgRNA, and plasmids expressing spCas9 were delivered to the liver via hydrodynamic tail vein injection. (B) Representative immunofluorescence images of hepatocytes. The liver sections were collected 7 days post injection. Scale bar, 50 µm. GFP, transfected cells. (C) Plasmids of either MMEJ- or HMEJ-mediated gene replacement strategy designed to insert Fah cDNA of exon 5 to 14 into intron 4 of Fah gene were delivered into Fah-/- mouse livers by hydrodynamic injection. NTBC on: Fah-/- mice were maintained on NTBC water; NTBC off: withdrawal of NTBC water (the first day of NTBC withdrawal was defined as day 0, which is the 7th day after injection). (D) Fah immunohistochemistry staining of liver sections from Fah-/- mice injected with MMEJ or HMEJ plasmids. Scale bar, 100 µm. Figure modified from previous reports5,10. Please click here to view a larger version of this figure.
Discussion
The most critical steps in the construction of HMEJ donor plasmids are: (1) selection of the sgRNA with high DNA cleavage efficiency and low distance between sgRNA cutting site and stop codon, and (2) proper construction of HMEJ donor. CRISPR/Cas9-mediated cleavage on both transgene donor vector (containing sgRNA target sites and ~800 bp homology arms) and targeted genome is necessary for efficient and precise targeted integration in vivo. The most critical steps of generation of knock-in mice using the HMEJ-based method are: (1) the preparation of high quality of Cas9 mRNA and sgRNA (No degeneration exists in Cas9 mRNA and sgRNA), and (2) the preparation of the high quality HMEJ donor plasmid. The plasmid shows no toxic effects on embryonic development.
Recently, an NHEJ-based method had also been reported for efficient in vivo genome editing8. Nevertheless, various types of indel mutations were usually induced at the junctions, as described in previous reports8, making it difficult to achieve precise integration. Here, the HMEJ-based strategy we described above showed precise targeted integration with hardly any indel mutations. Thus, an HMEJ-based strategy could be an ideal platform for replacing a mutated sequence (such as a point mutation) with the correct one, which is not applicable for NHEJ-based method.
Mosaicism is a major problem for gene editing in embryos. Injection of Cas9 protein instead of mRNA at an earlier embryonic stage may achieve transgene knock-in at one cell stage without mosaicism. For clinical applications, delivery of the CRISPR/Cas9 systems into adult tissues is still challenging.
There are many future potential uses of HMEJ-based genome editing. It can be used to generate genetically modified animal models. Considering its high knock-in efficiency in embryos, this method could significantly reduce the animal number needed for generating genetically modified animal models, and particularly opens up the possibility of generating non-human primate genetic models. HMEJ-based genome editing can lineage trace individual cell types in adult tissues, which is particularly useful for animal models, since there is a lack of available animal models, such as non-human primates. It can be used for targeted gene therapies: The most attractive application of an HMEJ-based strategy is gene therapy for clinic uses. In this study, we corrected the Fah mutation of hereditary tyrosinemia type I mice by hydrodynamic injection of the indicated vectors. However, delivery of the CRISPR/Cas9 system into adult tissues is still the major technical challenge for clinical use, as hydrodynamic injection is unlikely to be performed in patients. Currently, further improvement of the delivery strategy is urgently needed before translating this HMEJ-based method into the clinic.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by CAS Strategic Priority Research Program (XDB02050007, XDA01010409), the National Hightech R&D Program (863 Program; 2015AA020307), the National Natural Science Foundation of China (NSFC grants 31522037, 31500825, 31571509, 31522038), China Youth Thousand Talents Program (to HY), Break through project of Chinese Academy of Sciences, Shanghai City Committee of science and technology project (16JC1420202 to HY), the Ministry of Science and Technology of China (MOST; 2016YFA0100500).
References
- Yang H, et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 2013;154(6):1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 2011;29(8):731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S, et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 2014;5:5560. doi: 10.1038/ncomms6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano Y, et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 2015;5:8841. doi: 10.1038/srep08841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. 2017. [DOI] [PMC free article] [PubMed]
- Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 2014;24(1):142–153. doi: 10.1101/gr.161638.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca M, Lin VG, Guo N, Yang Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 2013;23(3):539–546. doi: 10.1101/gr.145441.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540(7631):144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 2017;27(6):801–814. doi: 10.1038/cr.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DW, et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 2011;13(1):66–71. doi: 10.1038/ncb2136. [DOI] [PubMed] [Google Scholar]
- Han DW, et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. 2012. [DOI] [PubMed]
- Ambasudhan R, et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 2011;9(2):113–118. doi: 10.1016/j.stem.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparman M, et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 2009;27(6):1255–1264. doi: 10.1002/stem.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatten G, Mitalipov S. Developmental biology: Transgenic primate offspring. Nature. 2009;459(7246):515–516. doi: 10.1038/459515a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadros RM, et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 2017;18(1):92. doi: 10.1186/s13059-017-1220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KE, et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 2016;217(6) doi: 10.3390/ijms17060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JW, et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 2015;33(11):1162–1164. doi: 10.1038/nbt.3389. [DOI] [PubMed] [Google Scholar]
- Harms DW, et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 2014;83:11–27. doi: 10.1002/0471142905.hg1507s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 2014;9(8):1956–1968. doi: 10.1038/nprot.2014.134. [DOI] [PubMed] [Google Scholar]
- Grompe M, et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 1993;7(12):2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- Paulk NK, et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 2010;51(4):1200–1208. doi: 10.1002/hep.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]