

Abstract
Commensal bacteria are critical for physiological functions in the gut and dysbiosis in the gut may cause diseases. Here we report that mice deficient in cathelin-related antimicrobial peptide (CRAMP) were defective in the development of colon mucosa and highly sensitive to dextran-sulfate-sodium (DSS)-elicited colitis as well as azoxymethane-mediated carcinogenesis. Pretreatment of CRAMP−/− mice with antibiotics markedly reduced the severity of DSS-induced colitis, suggesting CRAMP as a limiting factor on dysbiosis in the colon. This was supported by observations that wide type (WT) mice co-housed with CRAMP−/− mice became highly sensitive to DSS-induced colitis and the composition of fecal microbiota was skewed by CRAMP-deficiency. In particular, several bacterial species that are typically found in oral microbiota, such as Mogibacterium neglectum, Desulfovibrio piger and Desulfomicrobium orale were increased in feces of CRAMP−/− mice and were transferred to WT mice during co-housing. When littermates from CRAMP+/− parents were examined, the composition of fecal microbiota of WT pups and heterozygous parents were similar to each other. In contrast, although the difference in fecal microbiota between CRAMP−/− and WT pups was small early on after weaning and single mouse housing, there was an increasing divergence with prolonged single housing. These results indicate that CRAMP is critical in maintaining colon microbiota balance and supports mucosal homeostasis, anti-inflammatory responses, and protection from carcinogenesis.
Introduction
Commensal bacteria are involved in various physiological functions in the gut and microbial imbalance (dysbiosis) is implicated for pathological changes (1). Microbiota in the colon is separated from the host compartment by a single layer of epithelial cells, which have the capacity to respond to and control the potential pathogenic threats from commensals (2, 3). In addition to the pattern recognition receptors, such as TLRs and related NOD proteins that recognize “microbe-associated molecular patterns (MAMPs)”, formylpeptide receptors (FPRs, Fprs for mouse), a subfamily of the 7-transmembrane G protein-coupled chemoattractant receptors, also interact with both pathogen- and damage-associated chemotactic molecules (4-6).
Both human FPRs and mouse Fprs are expressed by colonic epithelial cells and participate in important patho-physiological responses. Mice deficient in Fpr2 showed shortened colon crypt length with reduced epithelial cell proliferation, reduction in the severity of acute inflammatory responses to DSS but delayed mucosal restoration after injury, increased chemically-induced tumorigenesis and increased bacterial abundance in the colon (7). Thus, Fpr2 contributes to colonic epithelial homeostasis, inflammation and tumorigenesis in mice, presumably by interacting with commensal bacterial products. However, the capacity of Fpr2 (or human FPR2) to recognize endogenous ligands in the colon remains unclear.
One of the human FPR2 ligands, the antimicrobial peptide LL-37, is highly expressed by colon epithelia (8). The mouse orthologue of LL-37, cathelin-related antimicrobial peptide (CRAMP) (9) is expressed in epithelial cells of small intestine in the neonatal period (non-detectable by 21 day) and confers significant protection of the newborn from infection by enteric pathogen Listeria monocytogenes (10). Intrarectal administration of exogenous CRAMP alleviated DSS-induced colitis by preserving the mucous layer, reducing proinflammatory cytokine production and suppressing epithelial apoptosis (11). Oral administration of CRAMP-transformed Lactococcuslactis ameliorated DSS-induced colitis in mice (12). In addition, CRAMP mediates the migration of leukocytes and promotes innate and adaptive immune responses in mice by interacting with Fpr2 (9, 13).
Collectively, these results led us to hypothesize that endogenously produced CRAMP may actively participate in the patho-physiological processes in the colon by maintaining the homeostasis of microbiota. We tested this hypothesis by examining newly generated CRAMP−/− mice and demonstrate that CRAMP plays a critical role in colonic homeostasis, inflammation and carcinogenesis by regulating microbiota balance.
Materials and Methods
Mice
Wild type (WT) C57BL/6NCr mice were originally purchased from Charles River, Frederick, MD, and used as breeders. Systemic and CRAMPflox/flox mice were generated as detailed in Supplemental Figure 1. All mice used in this study were produced and maintained in the same room in the animal facility at the National Cancer Institute at Frederick, Frederick, MD. For colitis experiments, mice were transferred to another room on the same floor just before the experiment and kept there until the end. Six to 12 week-old male mice were used throughout the study. The experimental protocols of this study were approved by the Frederick National Laboratory for Cancer Research Animal Care and Use Committee, Frederick, MD, and all experiments were performed in accordance with relevant guidelines and regulations.
To generate littermates, CRAMP heterozygotes (CRAMP+/−) were generated by crossing CRAMP−/− mice to WT mice, and several cages of CRAMP+/− breeding pairs were set up. Pups were genotyped and weaned at 4 weeks. WT or CRAMP−/− pups were single housed for up to 13 weeks and feces were collected.
Induction of colitis and carcinogenesis
Colitis was induced by administration of mice with different concentrations (1.5 to 3%) of DSS (40,000-50,000 Da, Lot#: 4288597, Affymetrix, Cleveland, OH) in drinking water for 5 days. Mice were allowed to recover on normal drinking water for an additional 4 to 14 days. Disease activity index (DAI) was calculated as previously described (7, 14). Mice were euthanized when they lost 20% of the original weight or when they developed prolapse. In a few experiments, mice were euthanized when they lost 35% of the original weight.
To induce colitis-associated tumors, mice received intraperitoneal injection of 12.5 mg/kg azoxymethane (AOM, Product #: A5486, Sigma-Aldrich, St. Louis, MO) (15). Mice were exposed 7 days later to 1% DSS in drinking water for 1 week, followed by 2 weeks of normal drinking water. The DSS treatment was repeated for 3 cycles. All mice were euthanized 12 weeks after AOM injection when one of them developed prolapse. Colons were excised, flushed with PBS, and opened longitudinally. The size of each tumor was measured and the number of tumors was counted.
Antibiotic treatment and co-housing
Mice were given fresh drinking water containing a cocktail of antibiotics (ABX) (vancomycin, imipenem/cilastatin and neomycin) every two days for three weeks as previously described(16). DSS was dissolved in the ABX-containing drinking water. With regard to co-housing, several cages containing two WT and two CRAMP−/− mice were set up at the time of weaning at 4-weeks old and maintained for 4 weeks prior to use.
BrdU incorporation
To examine DNA synthesis, mice were intraperitoneally injected with 2 mg of BrdU (BD Biosciences). Mice were euthanized 2 or 24 hours after BrdU injection and the colons were harvested. The incorporation of BrdU was examined by immunohistochemistry (IHC) as described below.
Intestinal permeability
Intestinal permeability was determined by assessing the absorption of FITC-dextran as previously published (Laukoetter et al, 2007) and per manufacturer’s protocol. Mice were fasted for 4 hours, orally gavaged with 20ml/kg 40kDa dextran-FITC (Condrex, #4009; 25mg/ml). Following a further 3 hour fasting period, mice were euthanized and blood collected by cardiac puncture. Plasma was diluted 2-fold in PBS, placed in blacksided clear bottom 96 well plates (Corning Costar #3603) and assessed for fluorescence intensity at excitation of 490nm and emission 520 nm on a fluorescent plate reader. Concentration of plasma FITC-dextran was determined by comparison to a standard curve prepared with stock FITC-dextran diluted with normal mouse plasma. Five mice per group were used. Data was presented as mean ± SEM.
Histological analyses
Mouse colons were fixed in neutral buffered formalin and embedded in paraffin. Sections at 3 μm were prepared and stained with H&E. IHC for CD3, B220 and F4/80 to identify T cells, B cells and macrophages, respectively, was performed on LeicaBiosystems Bond Autostainer. Sections were blocked with 2% normal rabbit serum (Vector Laboratories) for 20 minutes and subsequently incubated for 30 minutes with anti-CD3 (AbDSerotec), anti-B220 (BD Biosciences) or anti-F4/80 (eBiosciences) Ab. Biotinylated secondary antibody (anti-rabbit IgG), diluted 1:100 in 1.5% normal rabbit serum, was applied to the slides for 30 minutes and subsequent detection was achieved with Leica Biosystems’ Intense R detection Kit. IHC for BrdU was performed manually using anti-BrdU antibody (Invitrogen) and the Vectastain Elite Standard ABC kit (Vector Laboratories).
RNA purification and cDNA synthesis
RNA was extracted from snap frozen distal colon with RNeasy mini kit (Qiagen) or from RNALater (Ambion)-treated distal colon with TRIzol (Invitrogen), as per instructions. Quality and yield were assessed by Nanodrop spectrophotometry. cDNA was synthesized using the QuantiTect reverse transcription kit (Qiagen, 205310) with inclusive genomic DNA wipeout buffer. Gene expression was assessed with TaqMan real-time PCR assays (Applied Biosystems) and TaqMan universal PCR mastermix (Applied Biosystems, 4304437) on Applied Biosystems 7300 real-time PCR system. Data was analyzed for relative expression by the ddCt method, normalized to HPRT endogenous control. To detect the expression of cytokines and chemokines, colons were placed in RNALater (Ambion). Total RNA was isolated from colon homogenates by using TRIzol (Invitrogen), precipitated, and then used.
DNA extraction, quantification and sequencing of fecal bacteria
Fecal DNA was prepared using Qiagen DNA Stool kit (Qiagen). Fecal samples were sequenced as described earlier (17). Briefly, V4 fragment of 16S rDNA using primers 515F-GTGCCAGCMGCCGCGGTAA and 806R-GGACTACHVGGGTWTCTAAT flanked by p5 and p7 Illumina Sequencing adaptors (p5 and p7), barcode (i5 and i7), pad (to optimize melting temperature) and a link sequence. PCR was done with 200 ng of fecal DNA using Accuprime High Fidelity Taq Polymerase (Invitrogen) with an initial step at 95°C for 2 min, followed by 25 cycles of 20 s at 95°C, 30 s at 55°C and 7 min at 72°C. PCR products were purified and normalized using SequalPrep Normalization Plate Kit (Invitrogen), resultant eluents were pooled together and quantified using Agilent Tapestation (High sensitivity DNA kits) and Qubit (DNA). Pooled DNA library from 96 samples was denatured, diluted and sequenced on Illumina MiSeq (using V2 500 cycle kit) according to the manufacturer’s protocol. Typically, we obtained around 100,000 high quality reads per sample. Sequence processing was performed using Mothur v1.30.0 as described in MiSeq 16S SOP protocol (17). Briefly, contigs were generated from the R1 and R2 sequences, followed by read assignment to corresponding barcodes; low quality sequences and chimeras were removed. Sequences were aligned to SILVA reference dataset, trimmed to the same length and clustered to produce operational taxonomic units (OTUs) with more than 97% similarity. Sequences were also classified using RDP and Green genes reference datasets. Both types of data, classified sequences and binned OTUs, were used in downstream analysis for comparisons between the groups. Beta-diversities (weighted and unweighted Unifrac) and alpha diversities (Chao and Inverse Simpson Index) were calculated using Mothur software. All sequence processing was done on Biowulf NIH cluster. OTUs and classified reads datasets were normalized to the geometric mean and analyzed using Partek Genomics Suite software, version 6.0, Partek Inc., St. Louis, MO, USA. Statistical analysis was performed using ANOVA and p-values were corrected for multiple comparisons using q-value test (< 0.1). Sequence data used in this study were deposited to the Short Read Archive (SRA) with the project number PRJNA429251 (SRA accession number: 128546; https://www.ncbi.nlm.nih.gov/sra/SRP128546).
Statistical analyses
All experiments were performed at least 3 times with comparable results. Statistical analysis was performed by using Student’s t-test or Log-rank (Mantel-Cox) test of the GraphPad Prism, Version 6, GraphPad Software, San Diego, CA. A value of p < 0.05 was considered to be statistically significant.
Results
CRAMP is critical for colon homeostasis
To examine whether endogenously produced CRAMP actively participate in the patho-physiological processes, in particular the homeostasis, in the colon, we generated CRAMP−/− mice (Supplement Fig. 1) and examined the colon of naïve mice. As shown in Figure 1A, the length of colonic crypts was significantly shorter in CRAMP−/− mice compared with those in WT mice. The shortening of the colon crypts in CRAMP−/− mice was associated with reduced proliferation of epithelial cells as demonstrated by significantly decreased BrdU incorporation (Fig. 1B). These results indicate that in the absence of CRAMP, naïve mouse colon epithelial cells are in a hypo-proliferative state.
Figure 1. CRAMP plays a role in colonic homeostasis.
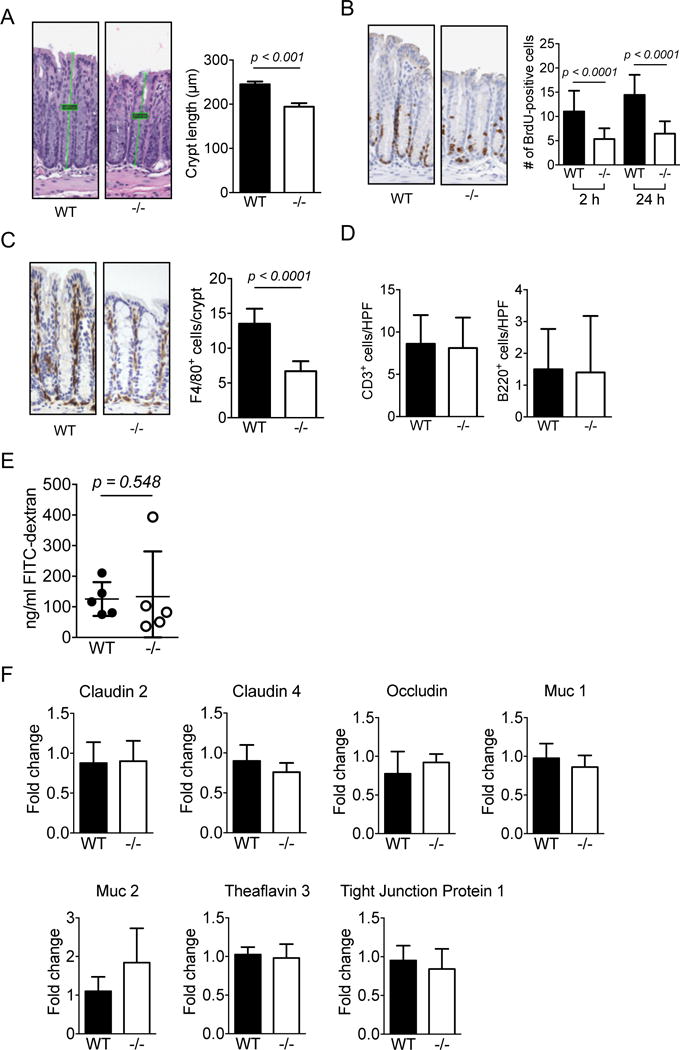
A. Distal sections of colons were obtained from 5 WT or CRAMP−/− mice, H&E sections were prepared and the length of colonic crypts (4 to 9 crypts from each colon) was measured. Data is presented as mean ± SEM of 5 mice in each group. B. BrdU was intraperitoneally injected into untreated WT or CRAMP−/− mouse. Colons were excised 2 or 24 hours after the injection and the incorporation of BrdU was examined by immunohistochemistry. BrdU-positive cells in 20 crypts per mouse were counted. Two mice per group were used for 2 hours and one mouse per group was used for 24 hours. Data is presented as mean ± SEM. C. The infiltration of macrophages (F4/80+) was examined by immunohistochemistry. Positive cells in 10 high power fields (HPF) were counted. Data is presented as mean ± SEM per HPF. D. The infiltration of T cells (CD3+) and B cells (B220+) was examined by immunohistochemistry. Positive cells in 10 high power fields (HPF) were counted. Data is presented as mean ± SEM per HPF. E. Mice were fasted overnight and then dextran-FITC was instilled into stomach. Blood was collected, serum was isolated and the concentration of FITC was measured. Data is presented as mean ± SD obtained from 5 mice per group. F. The expression of genes involved in the epithelial integrity, including cldn2, cldn4, occldn, muc1, muc2, TF3, TJP1, was examined. Data is presented as mean ± SD obtained from 5 mice per group.
Gut macrophages are normally located in the lamina propria in close proximity to luminal bacteria and regulate inflammatory responses to the bacteria that breach the epithelial layer to protect the mucosa (18). As shown in Figure 1C, F4/80+ macrophages were detected in subepithelial lamina propria of WT and CRAMP−/− mouse colons. However, the number of F4/80+ macrophages per crypt was significantly reduced in CRAMP−/− colons; whereas, the number of CD3+ T cells or B220+ B cells at the same location was similar between WT and CRAMP−/− mice (Fig. 1D). No active inflammation with neutrophil infiltration was detected in WT or CRAMP−/− mouse colons. There was also no sign of increased gut permeability, since the plasma concentration of gavaged FITC (Fig. 1E) and the expression levels of genes critical for the barrier functions, including claudin 2, claudin4, occludin, trefoil factor 3, MUC1, MUC2, and tight junction protein 1, were same between WT and CRAMP−/− mice (Fig. 1F). Thus, epithelial barrier integrity appears to be intact in naive CRAMP−/− mouse colons.
We next examined the number of isolated lymphoid aggregates (ILAs), which are randomly distributed in human and mouse colon and are a key element to mucosal immune regulation (19, 20). A similar number of ILAs was present in the colon of WT and CRAMP−/−mice (Fig. 2A, B). However, there was a significant increase in the number of larger ILAs, mainly composed of B220-positive B lymphocytes, in the colon of CRAMP−/− mice (Fig. 2A, B). These results suggest that CRAMP may play an important role in the immune response in the colon. We therefore examined the weight and length of colons of naïve WT and CRAMP−/− mice. Although there was no significant difference in the length of colons between WT and CRAMP−/− mice, the colon weight and the weight/length ratio were significantly increased in CRAMP−/− mice (Fig. 2C), suggesting the presence of potentially abnormal and low level inflammatory responses in the colon of CRAMP−/− mice.
Figure 2. The colon of naïve CRAMP−/− mice shows increased baseline inflammation.
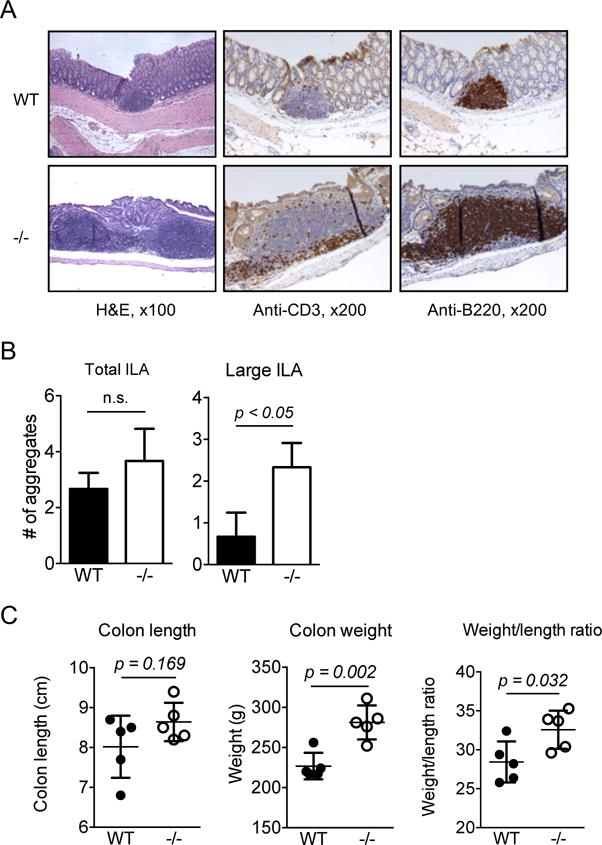
A. WT and CRAMP−/− colons were histologically examined for the presence of isolated lymph aggregates. n = 3. B. The number of total ILAs or large size ILAs in the entire length of colon was counted. Data is presented as mean ± SD of 3 mice. C. The weight and the length of the colons were measured and the weight/length ratio was calculated. Data is presented as mean ± SEM. Five mice were used for each group.
CRAMP−/−mice are highly sensitive to chemically induced colitis and carcinogenesis
We therefore induced acute colitis in WT and CRAMP −/− mice by administering 3% DSS in drinking water. WT mice showed clinical signs of acute colitis, including weight loss, rectal bleeding and diarrhea, around day 6 and 40% of the mice died by day 8. In contrast, all CRAMP−/− mice showed clinical signs of severe acute colitis by day 5 and 100% of mice died by day 8 (Fig. 3A, left panel). In response to a lower concentration of DSS (2%), all WT mice survived without showing significant signs of colitis, whereas all CRAMP−/− mice died by day 10 with signs of severe acute colitis (Fig. 3A, right panel). The length of CRAMP−/− mouse colons was significantly shortened by day 4 after 3% DSS intake (Fig. 3B), which was consistent with the signs of acute colitis.
Figure 3. CRAMP−/− mice are highly sensitive to chemically induced acute colitis.
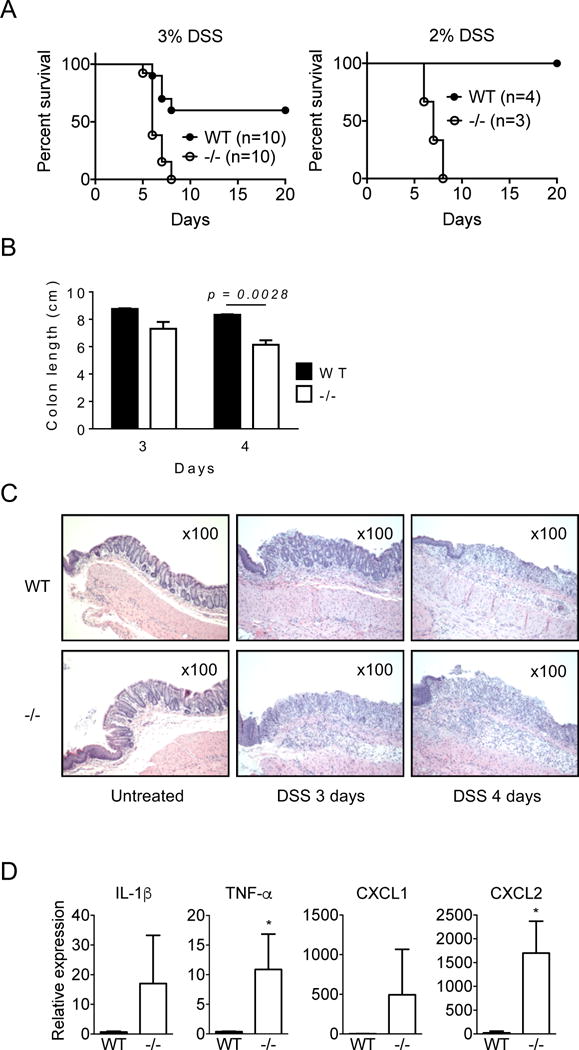
A. WT and CRAMP−/− mice were given 3% or 2% of DSS in drinking water and the survival of mice was examined. B. Colons were harvested from mice treated with 3% DSS on day 3 or day 4 and their length were measured. Two mice per group for day 3 and 3 mice per group for day 4 were used. Data is presented as the mean ± SEM. C. The distal section of colons from mice treated with 3% DSS was histologically examined by H&E staining. Original magnification was 200×. D. The expression of IL-1β, TNFα, CXCL1 and CXCL2 in the colon of mice treated with 3% DSS for 4 days was examined by real time RT-PCR. Data is presented as the mean ± SD. Three colons per group.
Histologically, the epithelia of CRAMP−/− mouse colon were markedly damaged and necrotic by day 3 and infiltrated by numerous leukocytes, including neutrophils. By comparison, WT mouse colon showed mostly intact epithelia (Fig. 3C). On day 4, the inflammatory responses, including leukocyte infiltration and tissue edema, significantly increased in CRAMP−/− mouse colon, as compared to WT mouse colon (Fig. 3C). We also detected increased expression of mRNAs encoding proinflammatory cytokines and chemokines, such as TNF-α and CXCL1, in the colon of CRAMP−/− mice 3 days after DSS treatment (Fig. 3D). These results confirm a higher sensitivity of CRAMP−/− mice to DSS-induced colitis and CRAMP is critical for protecting the colon from inflammatory insults.
We next examined the recovery of the colon mucosa following DSS exposure. Mice were given lower concentrations of DSS (1.5%) for 5 days and allowed to recover for an additional 15 days on regular water. As shown in Figure 4A, CRAMP−/− mice developed severe acute colitis even at this DSS concentration and recovered very slowly compared to WT mice. Their weight did not reach the original level even on day 37, and the colon length of CRAMP−/− mice was significantly shorter (Fig. 4B). Histologically, the mucosa of WT mouse colons showed little histological change compared to normal mucosa, whereas the mucosa of CRAMP−/− mouse colons were thickened with increased epithelial cell proliferation indicated by increased number of BrdU-positive cells and increased leukocyte infiltration, including neutrophils and mononuclear cells (Fig. 4C). When mice were treated with AOM plus three cycles of 1.5% DSS, CRAMP−/− mice developed more numerous tumors with larger sizes in the colon than WT mice (Fig. 4D, E, F). The length of colon was also markedly shortened in CRAMP−/− mice (Fig. 4D). Tumors formed in the colon of WT and CRAMP−/− mice were polypoid. All tumor sections we examined were histologically diagnosed as adenocarcinoma limited to the mucosa or submucosa (Fig. 4G). Thus, the colon of CRAMP−/− mice readily develops severe chronic colitis with increased epithelial cell proliferation, likely contributing to the development of colon cancer.
Figure 4. CRAMP−/− mice are highly sensitive to chemically induced chronic colitis and carcinogenesis.
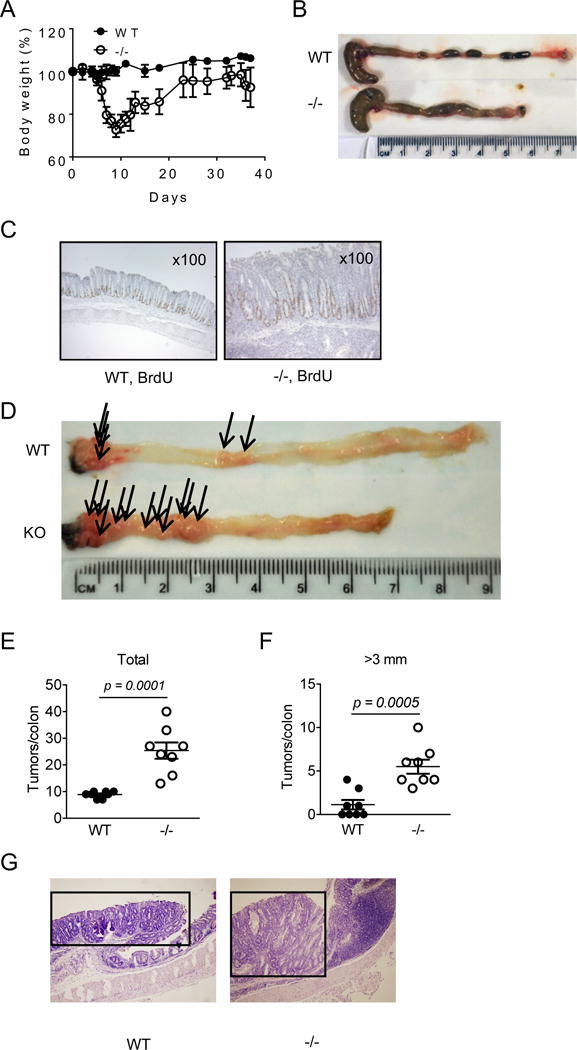
Mice were given 1.5% DSS for 5 days then were allowed to recover on regular water. On day 37, mice were euthanized and colons were harvested. A. Changes in body weight (%) are presented. B. Mice were subjected to 3 cycles of 1.5% DSS treatment and euthanized on day 50. Colons were harvested and the length was measured. C. On day 20, 2 mg of BrdU was intraperitoneally injected. Twenty-four hours later, mice were euthanized and the incorporation of BrdU into colon epithelial cells was examined by immunohistochemistry. D-F. The number and the size of tumors developed in the colon of 8 WT and 8 CRAMP−/− mice treated with AOM plus DSS were evaluated. Data is presented as the mean ± SEM. G. Tumors that developed in the colon of WT and CRAMP−/− mice were histologically examined by H&E staining. Tumors are indicated by an open square. Original magnification was 100×.
Pretreatment with antibiotics reduces the severity of DSS-induced colitis in CRAMP−/− mice
Intestinal microbiota and their products have been implicated in the pathogenesis of IBD and DSS-induced colitis in mice. It was previously demonstrated that mucosal destruction caused by DSS occurs without the involvement of intestinal microbiome which instead affects the susceptibility and responsiveness to DSS-induced damage (21). Since CRAMP is an antimicrobial peptide, the absence of CRAMP may alter the composition of microbiota, resulting in higher susceptibility of CRAMP−/− mice to DSS. To test the possibility, we pretreated WT and CRAMP−/− mice with a cocktail of ABX (16) for three weeks, followed by administration of 3% DSS in drinking water. As shown in Figure 5A, ABX treatment significantly rescued WT and CRAMP−/− mice from DSS-induced death. However, CRAMP−/− mice remained to show signs of mild acute colitis with a higher DAI and mild mucosal leukocyte infiltration on day 5 of DSS treatment as compared to WT mice (Fig. 5B). These results suggest that the increased susceptibility of CRAMP−/− mice to DSS may be dependent on the over growth of bacteria, but CRAMP may also protect mouse colon through additional mechanism(s) independent of its antimicrobial property.
Figure 5. Severe DSS-induced colitis in CRAMP−/− mice is dependent on bacteria and transferable.
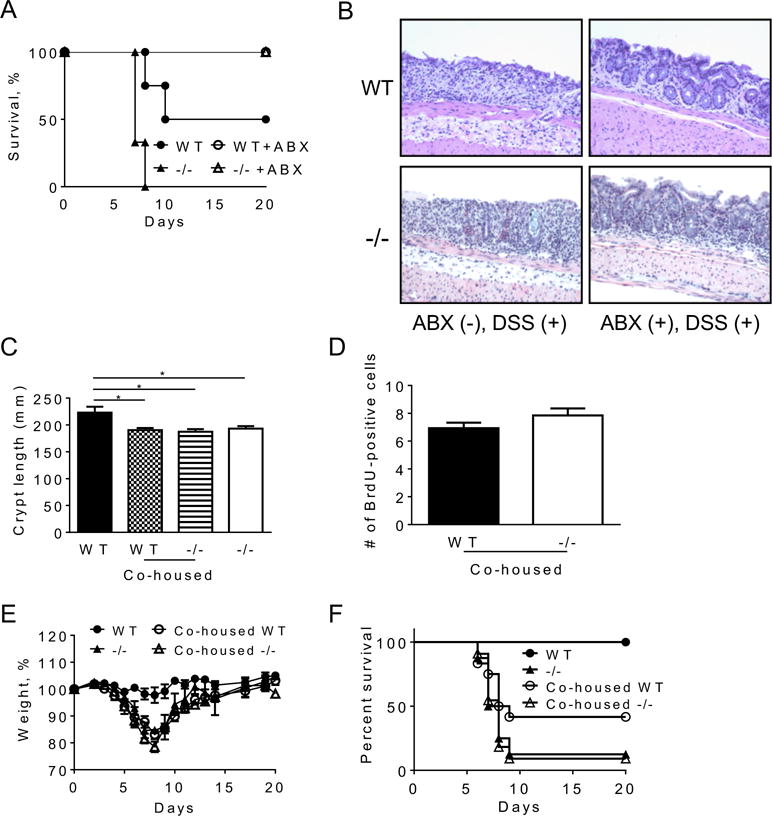
WT and CRAMP−/− were pretreated with a cocktail of ABX (vancomycin, imipenem/cilastatin and neomycin) for three weeks and then administered with 3% DSS in drinking water. A. The survival of mice was examined. Representative of 2 experiments with similar results was presented. ABX-treated 3 WT and 5 CRAMP−/−, and untreated 3 WT and 3 CRAMP−/− mice were used. B. One mouse from each group was euthanized and the histological changes were examined on a H&E section. C. WT mice and CRAMP−/− mice were co-housed for 4 weeks and the crypt length was examined on H&E section. Data is presented as mean ± SD. D. To examine the proliferation of epithelial cells, mice were intraperitoneally injected with 2 mg BrdU. Mice were euthanized 24 hours after the injection and the incorporation of BrdU was detected by immunohistochemistry. E, F. Mice were given 3% DSS for 5 days in drinking water and allowed to recover. The change in body weight (E) and the survival (F) were monitored. Representative data of 2 experiments was shown for body weight (5 non-co-housed WT and 5 non-co-housed CRAMP−/− mice, and 8 co-housed WT and 7 co-housed CRAMP−/− were examined). Data is presented as the mean ± SEM. The summary of 2 experiments is shown for survival (9 non-co-housed WT, 8 non-co-housed CRAMP−/− mice, and 12 co-housed WT and 10 co-housed CRAMP−/− mice were used).
The phenotype of CRAMP−/−mice is transferable to WT mice
To explore the possibility that CRAMP-deficiency may result in colonic dysbiosis, thereby enhance the sensitivity to DSS-induced acute colitis, we co-housed WT and CRAMP−/− mice for 4 weeks. As shown in Figure 5C, the crypt length of the colons of co-housed untreated WT mice was reduced to the level shown in untreated CRAMP−/− mice, with reduced proliferating epithelial cells (Fig. 5D). Administration of 2% DSS for 5 days caused only minor disease activity in non-co-housed WT mice, while non-cohoused CRAMP−/− mice suffered severe acute colitis with a high mortality, as observed in earlier experiments (shown in Fig. 2). Interestingly, WT mice co-housed with CRAMP−/− mice showed signs of severe acute colitis with significant weight loss and a 58% mortality rate (Fig. 5E, F). These results indicate that the high sensitivity of CRAMP−/− mice to DSS-induced colitis is transferable to WT mice by co-housing, presumably through the transfer of certain bacteria species that grew in the absence of CRAMP.
CRAMP-deficiency alters the composition of microbiota in the colon
To determine whether CRAMP-deficiency alters the composition of colonic microbiota in a naïve state and also during DSS-induced colitis, we sequenced DNA of microbiota in fecal pellets of mice. Figure 6A shows a significantly different microbiota composition between non-co-housed WT and non-co-housed CRAMP−/− mice. After 4 weeks of co-housing, the microbiota composition in the feces from WT mice markedly shifted toward that of CRAMP−/− mice. Treatment with DSS for 4 days revealed a clear difference in the microbiota composition between non-cohoused WT mice and the other groups. Similar difference persisted at 21 and 56 days after DSS treatment.
Figure 6. CRAMP-deficiency alters the composition of microbiota in the colon.

Fecal bacterial DNA was isolated from 5 uncohouse WT, 5 uncohoused CRAMP−/−, 8 co-housed WT and 6 co-housed CRAMP−/− mice before and 4, 21 and 56 days after DSS treatment. On day 21 and 56, DNA was isolated from only surviving mice. A. Principal-component analysis of unweighted Unifrac distances are visualized for different time points. B. 16S-sequence frequencies were analyzed by 16S amplicon high throughput sequencing of fecal microbiota. Data are shown as heat map of classified sequences (species level). Species were selected based on two-way ANOVA analysis using time and genotype as variables following by multiple tests correction (q<0.1).
A heatmap was generated to show 45 bacteria strains in mouse feces (Fig. 6B) that showed statistically significant differences (q < 0.1) between samples as determined by two-way ANOVA using time post-treatment and mouse genotypes as factors. The heatmap revealed that the majority of the changes in microbiota were caused by DSS treatment. Different species of bacteria were increased during acute period of colitis in non-co-housed WT and CRAMP−/− mice. In WT mice, the bacterial populations that were increased are Mucispirillum shaedleri, Clostridium populeti and Acetivibrio cellulosolven. Whereas in CRAMP−/− mice, we saw an increase in Odoribacter laneus, Ruminococcus lactaris, Desulfovibrio piger and Desulfomicrobium orale, Mogibacterium neglectum, Bacteroide sacidifaciens, and others. The same bacterial species that increased during the acute phase of DSS colitis in CRAMP−/− mice appear to be the ones that were transferred to WT mice during co-housing (Fig. 6B). This was evident by their significant increase in WT mice after co-housing with CRAMP−/− mice (Fig. 7). Among those species, there were bacteria typically found in oral microbiota, such as M. neglectum (22) and sulfate-reducing D. piger and D. orale. However, several known members of gut microbiota, such as O. laneus, R. lactaris, also increased. O. laneus increased in WT mice after co-housing with CRAMP−/− mice from almost undetectable levels to more than 10% of all the bacteria in the fecal sample (Fig. 7). Moreover, DSS caused severe and persistent dysbiosis in all mice tested, which was evident by a dramatic loss of bacterial species at different time points. Many bacterial species were markedly decreased already at day 4 post-DSS, including Lactobacillus intestinalis and Candidatus arthromitus (Segmented Filamentous Bacteria - SFB), and some, including Escherichia Coli, were lost at day 21 post-DSS treatment. Some bacterial species that were downregulated during colitis were mostly from Clostridium genus; however, their numbers recovered at later time points and returned to its pre-DSS levels.
Figure 7. CRAMP-deficiency alters the composition of certain bacteria in the colon.
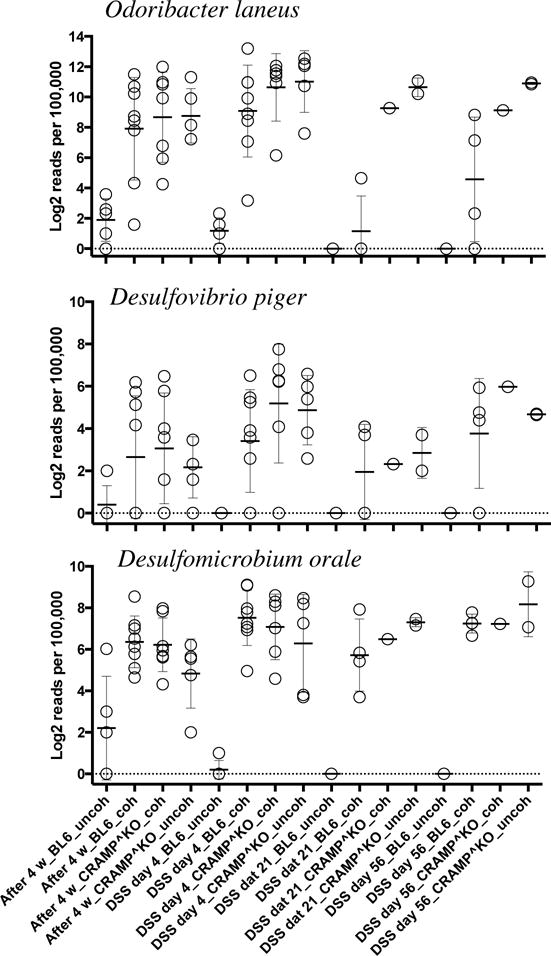
The composition of three genera, O. laneus, D. piger and D. orale, whose composition were significantly increased after co-housing of WT mice with CRAMP−/− mice is shown. X-axis represents groups and Y-axis represents Log2 transformed number of reads per 100,000 (typically sequenced per sample).
To obtain additional evidence supporting the role for CRAMP in the maintenance of microbiota in the colon, we generated WT and CRAMP−/− littermates by mating pairs of male and female heterozygous (CRAMP+/−) mice, and then single housed each littermate mouse, collected fecal pellets and analyzed their colonic microbiota. As shown in Figure 8A, the composition of fecal microbiota of WT pups and heterozygous parents were similar to each other. By contrast, although the difference in the composition between CRAMP−/− and WT pups was small early on after weaning and single mouse housing, there was an increasing divergence with the time (Fig. 8A). These results indicate a critical role of CRAMP in the maintenance of a balanced microbiota in the colon and the potential involvement of bacteria expanded in the absence of the anti-microbial CRAMP in the severity of DSS colitis. It should be noted that bacterial species identified in single housed WT and CRAMP−/− littermates (Fig. 8B) were different from those of earlier experiments (Fig. 6B), in which we focused on the species whose abundance changed after the induction of colitis in co-housed mice.
Figure 8. Colonic microbiota of CRAMP−/− mice diverges from that of littermate WT mice after single housing.
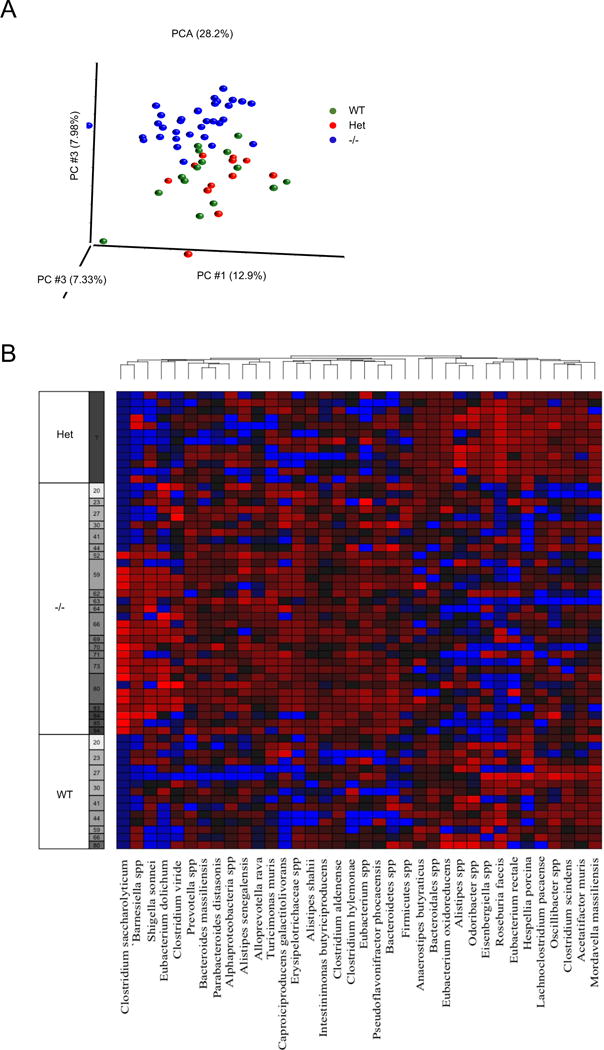
A. CRAMP heterozygotes (CRAMP+/−) were first generated by crossing CRAMP−/− mice to WT mice, and several cages of CRAMP+/− breeding pairs were set up. Pups were genotyped and weaned at 4 weeks. WT or CRAMP−/− pups were single housed for up to 13 weeks and feces were collected to analyze the bacterial composition. Principal-component analysis of unweighted Unifrac distances are visualized. B.16S-sequence frequencies were analyzed by 16S amplicon high throughput sequencing of fecal microbiota. Data are shown as heat map of classified sequences (species level). Species were selected based on two-way ANOVA analysis using time and genotype as variables following by multiple tests correction (q<0.1).
Discussion
Cathelicidins are a family of mammalian antimicrobial proteins that consist of an N-terminal putative signal peptide, a conserved cathelin-like domain and a C-terminal antimicrobial domain that varies remarkably in size (ranging from 12 to 97 amino acids). More than 40 members of the cathelicidin family have been identified in different species; however, humans and mice each produce only one cathelicidin, called human cationic antimicrobial protein 18 (hCAP18) and the mouse orthologue CRAMP, respectively. CRAMP−/− mice were previously demonstrated to develop more severe colitis than WT mice in response to DSS (23). However, the mechanistic basis of their observation remains unclear. In the present study, we revealed a novel role of CRAMP in colon mucosal homeostasis and provided a new mechanism by which CRAMP protects mice from DSS-induced colitis and associated carcinogenesis.
It is now widely accepted that commensal bacteria are involved in the regulation of intestinal epithelial cell turnover, promotion of epithelial restitution and reorganization of tight junctions, all of which are critical for fortifying barrier function; thus, microbial dysbiosis may cause pathological responses (1). In the present study, we revealed reduced proliferation of colon epithelial cells in CRAMP−/− mice, associated with increased susceptibility to DSS-induced colitis. We additionally showed that DSS-induced severe colitis in CRAMP−/− mice was partially preventable by antibiotics and the colon phenotype found in CRAMP−/− mice was transferable to WT mice after co-housing. These observations were corroborated by a clear difference in bacterial composition in the feces between non-cohoused WT and CRAMP−/− mice and co-housing of WT mice with CRAMP−/− mice dramatically shifted the bacterial composition in the feces of WT mice toward that of CRAMP−/− mice. Furthermore, colonic microbiota of CRAMP−/− mice diverged from that of littermate WT mice after single housing. Thus, CRAMP appears to play a critical role in the maintenance of healthy microbiota in the colon and in the prevention of certain bacteria outgrowth to cause severe colitis.
We noted an increase in several species of bacteria in WT mice post-co-housing with CRAMP−/− mice, including O.laneus, D. piger and D. orale, likely transferred from CRAMP−/− mice. Since M. neglectum, D. piger and D. orale are known components of oral microbiota, and cathelicidins are found in salivary glands, the increase in those species in the colon of CRAMP−/− mice could be due to their over-growth in the mouth. D. piger and D. oraleare are sulfate-reducing bacteria, and hydrogen sulfate has been implicated in the development of colitis (24). This emphasized the importance of CRAMP in the control of the balance of microbiota for disease prevention.
Cathelicidins are predominantly stored constitutively at high concentrations in the secondary granules of neutrophils, which, in the course of degranulation, release the C-terminal mature antimicrobial peptides (e.g. LL-37) by proteolytic cleavage. In addition to neutrophils, other leukocytes, including monocytes/macrophages, mast cells and epithelial cells, also generate cathelicidin, particularly in response to proinflammatory stimuli including cytokines, pathogen-associated molecular patterns (PAMPs) or tissue injury (13). Koon et al. previously demonstrated increased expression of CRAMP in the colon of mice with DSS-induced colitis (23). CRAMP expression was increased in the mucosal epithelium and also in colonic macrophages, which appeared to be regulated by the interaction between TLR9 and genomic DNA of bacteria present in the colon. CRAMP from BM-derived immune cells, especially macrophages, regulated DSS induction of colitis using mice transplanted by CRAMP−/− BM cells (23). We detected high level expression of CRAMP mRNA and protein in BM cells from WT mice (Supplemental Fig. 1B-C). In addition, myeloid cell-specific CRAMP−/− mice were more sensitive to DSS-induced colitis than control mice (Supplemental Fig. 2); thus, our results are consistent with the notion that myeloid cell-derived CRAMP is critical for control of colitis (23). However, deletion of CRAMP in intestinal epithelial cells also moderately increased the sensitivity to DSS, suggesting the participation of epithelium-derived CRAMP in colon homeostasis.
It is interesting to note that there are similarities in the phenotype between CRAMP−/− and Fpr2−/− mice in that they both show defects in colon mucosal development and repair (7). However, in contrast to Fpr2−/−mice, which showed decreased acute colitis syndrome upon intake of high dose DSS therefore with a reduced acute mortality, CRAMP−/−mice develop more severe colitis and rapidly became succumbed. Therefore, in addition to sustaining colon mucosal homeostasis, which may be dependent on interaction with the receptor Fpr2, the anti-microbial property of CRAMP actively participates in the control of pathogen overgrowth in the colon, which causes severe colitis. Further research is undergoing to more precisely clarify the role of CRAMP and Fpr2 interaction in the pathophysiological process in the colon.
In addition to the phenotypes described above, we also noted that CRAMP−/− mice consume more drinking water than WT mice in naïve state (data not shown), which is associated with more severe acute colitis observed in CRAMP−/− mice after inflammatory stimulation. Therefore, we adjusted the concentration of DSS based on their water consumption so that WT and CRAMP−/− mice would take in equivalent doses of DSS. All CRAMP−/− mice still exhibited severe colitis and died by day 9, whereas all WT mice survived (data not shown). This confirms that CRAMP−/− mice are more sensitive to DSS-induced colonic inflammation. Similar to systemic CRAMP−/− mice, myeloid cell-specific, but not IEC-specific CRAMP−/− mice, consumed higher volumes of drinking water than the control mice, an indication of their potential to suffer from more severe colitis.
In conclusion, CRAMP, among several antimicrobial peptides in the mammalian gut, plays a non-redundant role in mouse colon epithelial homeostasis and inflammation. In addition to its antimicrobial activity against a broad range of Gram-positive and Gram-negative bacteria, as well as fungi, in vitro (25), CRAMP appears to control the growth of certain pathological bacteria in the colon. Further studies are required to more precisely define the role of CRAMP in the maintenance of microbiota in the gut and in the overall host interactions with the microbiota.
Supplementary Material
Acknowledgments
The authors thank Dr. J. J. Oppenheim for reviewing the manuscript and Ms. Sharon Livingstone for secretarial assistance. The technical assistance provided by CIP Mouse Core and by Ms. Loretta Smith, NCI, and the staff of LASP, Leidos, Frederick National Laboratory for Cancer Research, is gratefully acknowledged.
This project was funded in part by federal funds from the NCI, NIH, under Contract No. HHSN261200800001E and by the Intramural Research Program of the NCI, NIH.
Abbreviations
- AOM
azoxymethane
- CRAMP
cathelin-related antimicrobial peptide
- DSS
dextran-sulfate-sodium
- FPRs
formylpeptide receptors
- MAMPs
microbe-associated molecular patterns
Footnotes
Disclosures
The authors declare no competing financial interests.
References
- 1.Yu LC, Wang JT, Wei SC, Ni YH. Host-microbial interactions and regulation of intestinal epithelial barrier function: From physiology to pathology. World J Gastrointest Pathophysiol. 2012;3:27–43. doi: 10.4291/wjgp.v3.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–363. doi: 10.1016/j.chom.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 4.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 5.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacological reviews. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Chen K, Xiang Y, Yoshimura T, Su S, Zhu J, Bian XW, Wang JM. New development in studies of formyl-peptide receptors: critical roles in host defense. J Leukoc Biol. 2016;99:425–435. doi: 10.1189/jlb.2RI0815-354RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen K, Liu M, Liu Y, Yoshimura T, Shen W, Le Y, Durum S, Gong W, Wang C, Gao JL, Murphy PM, Wang JM. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. The Journal of clinical investigation. 2013;123:1694–1704. doi: 10.1172/JCI65569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schauber J, Rieger D, Weiler F, Wehkamp J, Eck M, Fellermann K, Scheppach W, Gallo RL, Stange EF. Heterogeneous expression of human cathelicidin hCAP18/LL-37 in inflammatory bowel diseases. Eur J Gastroenterol Hepatol. 2006;18:615–621. doi: 10.1097/00042737-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–6265. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 10.Menard S, Forster V, Lotz M, Gutle D, Duerr CU, Gallo RL, Henriques-Normark B, Putsep K, Andersson M, Glocker EO, Hornef MW. Developmental switch of intestinal antimicrobial peptide expression. J Exp Med. 2008;205:183–193. doi: 10.1084/jem.20071022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tai EK, Wu WK, Wong HP, Lam EK, Yu L, Cho CH. A new role for cathelicidin in ulcerative colitis in mice. Exp Biol Med (Maywood) 2007;232:799–808. [PubMed] [Google Scholar]
- 12.Wong CC, Zhang L, Li ZJ, Wu WK, Ren SX, Chen YC, Ng TB, Cho CH. Protective effects of cathelicidin-encoding Lactococcus lactis in murine ulcerative colitis. J Gastroenterol Hepatol. 2012;27:1205–1212. doi: 10.1111/j.1440-1746.2012.07158.x. [DOI] [PubMed] [Google Scholar]
- 13.Yang D, de la Rosa G, Tewary P, Oppenheim JJ. Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009;30:531–537. doi: 10.1016/j.it.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 15.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2:1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 16.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai RM, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, Trinchieri G, Goldszmid RS. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. 2011;4:31–42. doi: 10.1038/mi.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cesta MF. Normal structure, function, and histology of the spleen. Toxicol Pathol. 2006;34:455–465. doi: 10.1080/01926230600867743. [DOI] [PubMed] [Google Scholar]
- 20.Yeung MM, Melgar S, Baranov V, Oberg A, Danielsson A, Hammarstrom S, Hammarstrom ML. Characterisation of mucosal lymphoid aggregates in ulcerative colitis: immune cell phenotype and TcR-gammadelta expression. Gut. 2000;47:215–227. doi: 10.1136/gut.47.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakazawa F, Poco SE, Jr, Sato M, Ikeda T, Kalfas S, Sundqvist G, Hoshino E. Taxonomic characterization of Mogibacterium diversum sp. nov. and Mogibacterium neglectum sp. nov., isolated from human oral cavities. Int J Syst Evol Microbiol. 2002;52:115–122. doi: 10.1099/00207713-52-1-115. [DOI] [PubMed] [Google Scholar]
- 23.Koon HW, Shih DQ, Chen J, Bakirtzi K, Hing TC, Law I, Ho S, Ichikawa R, Zhao D, Xu H, Gallo R, Dempsey P, Cheng G, Targan SR, Pothoulakis C. Cathelicidin signaling via the Toll-like receptor protects against colitis in mice. Gastroenterology. 2011;141:1852–1863. doi: 10.1053/j.gastro.2011.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rowan FE, Docherty NG, Coffey JC, O’Connell PR. Sulphate-reducing bacteria and hydrogen sulphide in the aetiology of ulcerative colitis. Br J Surg. 2009;96:151–158. doi: 10.1002/bjs.6454. [DOI] [PubMed] [Google Scholar]
- 25.Travis SM, Anderson NN, Forsyth WR, Espiritu C, Conway BD, Greenberg EP, McCray PB, Jr, Lehrer RI, Welsh MJ, Tack BF. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect Immun. 2000;68:2748–2755. doi: 10.1128/iai.68.5.2748-2755.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.