

Environmental DNA reveals unsuspected shark diversity and calls for monitoring and protection of residual populations.
Abstract
In the era of “Anthropocene defaunation,” large species are often no longer detected in habitats where they formerly occurred. However, it is unclear whether this apparent missing, or “dark,” diversity of megafauna results from local species extirpations or from failure to detect elusive remaining individuals. We find that despite two orders of magnitude less sampling effort, environmental DNA (eDNA) detects 44% more shark species than traditional underwater visual censuses and baited videos across the New Caledonian archipelago (south-western Pacific). Furthermore, eDNA analysis reveals the presence of previously unobserved shark species in human-impacted areas. Overall, our results highlight a greater prevalence of sharks than described by traditional survey methods in both impacted and wilderness areas. This indicates an urgent need for large-scale eDNA assessments to improve monitoring of threatened and elusive megafauna. Finally, our findings emphasize the need for conservation efforts specifically geared toward the protection of elusive, residual populations.
INTRODUCTION
Human activities are largely responsible for the ongoing defaunation of ecosystems worldwide, causing massive population declines and local species extirpations (1, 2). This global wave of defaunation may dramatically increase local “dark diversity,” defined as the suite of species that should be present within a certain region, based on their habitat requirements and dispersal ability, yet are absent (3). That is, dark diversity encompasses the diversity of locally absent species, although biogeographic history, as well as ecological and environmental conditions, suggests their presence (4). High dark diversity not only may imperil ecosystem functioning (5) but also represents potential for recovery of purportedly absent species (3, 6). A substantial portion of megafaunal dark diversity is composed of mobile, rare, elusive, and threatened species that are highly challenging to detect (1). Accordingly, an ongoing concern is whether this megafaunal dark diversity has been correctly measured or overestimated as a result of nondetection of remaining individuals by traditional sampling methods. The answer to this question has significant implications in terms of management and conservation because the presence of previously undetected individuals may require immediate action to prevent extirpation of remnant individuals, whereas the confirmed absence of species requires different management considerations (6).
Detecting species occurrences and extirpations is more challenging in the ocean than on land because most habitats remain hardly accessible and therefore poorly investigated (7). Similarly, accurate assessment of dark diversity is particularly problematic for low-density, mobile species such as sharks. Sharks are one of the most threatened marine taxa (8). They often have a high intrinsic vulnerability to fishing due to slow population growth (9), and with shark products such as dried fins reaching high commercial value (up to $1697 kg−1), they have a high exposure to international trade (10). Throughout the Pacific, the density of reef sharks has declined to 3 to 10% of prehuman levels (11), and even the most well-managed marine protected areas appear inadequate in maintaining healthy shark populations (8, 12).
It is unclear, however, whether reported levels of dark diversity of sharks are due to local extirpations or to a failure to detect remaining animals. Similar to most terrestrial vertebrates, sharks exhibit learning abilities linked to avoidance behavior, and repeated exposure to negative anthropogenic interactions may increase their elusiveness (13). This raises the possibility that sharks’ prevalence in marine habitats, even close to humans, may be greater than previously thought, with individuals being less detectable, therefore overinflating the apparent level of dark diversity.
Environmental DNA (eDNA) analysis is based on the retrieval of genetic material naturally released by organisms in their environments, and it is emerging as a noninvasive method to detect and identify even rare and elusive species in a wide range of ecosystems (14), including marine waters (15, 16). Here, we assessed the potential of eDNA metabarcoding in providing a more accurate estimate of the dark diversity of sharks on coral reefs of the New Caledonian archipelago by contrasting eDNA analysis with traditional underwater visual census (UVC) and baited remote underwater video station (BRUVS) survey methods.
RESULTS AND DISCUSSION
Lower dark diversity than previously estimated
Of the 26 historically present species in the regional pool (17), only 9 species of sharks were detected in 2758 UVCs and 385 BRUVS (Fig. 1, A to C). The dark diversity of sharks was thus initially estimated at 65% of the regional pool (that is, 17 species were not detected) using traditional survey methods. Despite two orders of magnitude less sampling effort, eDNA detected 44% more species than UVC or BRUVS; with only 22 samples, 13 shark species were detected, reducing the previously estimated dark diversity to 50% of the regional pool (13 undetected species). Six species were only detected by eDNA, three species were only detected by UVC and BRUVS (of which, one species only by the BRUVS), whereas six species were detected by all three methods (Fig. 2).
Fig. 1. Sampling design and analyses of surveys across the New Caledonian archipelago, southwestern Pacific.
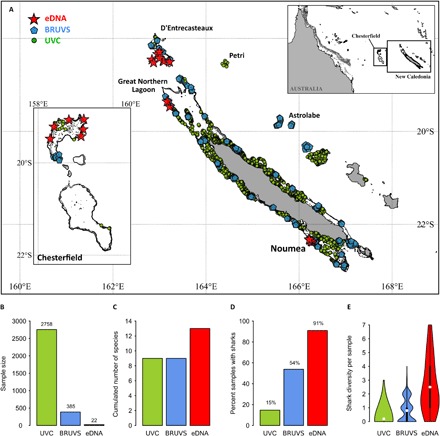
(A) Sampling design in the New Caledonian archipelago (red stars, eDNA; blue pentagons, BRUVS; green dots, UVC). (B) Sample size (UVC, n = 2758; BRUVS, n = 385; eDNA, n = 22). (C) Cumulated number of shark species detected. (D) Frequency of samples with sharks detected. (E) Violin plot showing detected shark species richness, significantly different between techniques (P < 0.001, Kruskal-Wallis test), with eDNA detecting more shark species (2.5 ± 1.9) compared to BRUVS (0.8 ± 0.8) and UVC (0.2 ± 0.5) (P < 0.001, Dunn’s tests). White dots are mean values; thick black bars correspond to interquartile ranges; thin black lines are 95% confidence intervals.
Fig. 2. Detection of shark species with different sampling methods.
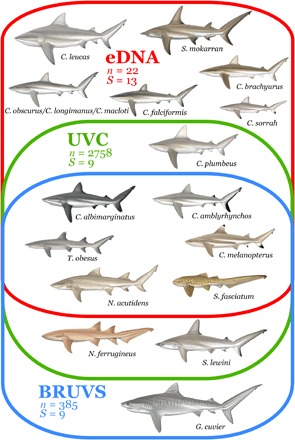
Venn diagram showing the species detected by eDNA (n = 22 samples, S = 13 species), UVC (n = 2758 samples, S = 9 species), and BRUVS (n = 385 samples, S = 9 species). Scientific drawings courtesy of M. Dando.
Sharks were observed in only 15% of UVCs (n = 405 of 2758) and 54% of BRUVS (n = 207 of 385; Fig. 1D). Furthermore, shark diversity was low for each sample, with only 3% of UVCs and 23% of BRUVS recording more than one species. When excluding the two most common species, the gray reef shark (Carcharhinus amblyrhynchos) and the whitetip reef shark (Triaenodon obesus), other shark species were observed in only 2% of UVCs and 13% of BRUVS.
In contrast, sharks were detected in 91% of eDNA samples (Fig. 1D), with 64% of samples revealing at least two species. No sharks were detected in the negative controls (see Materials and Methods). Even after excluding gray reef and whitetip reef sharks, 68% of eDNA samples revealed one or more shark species. The mean shark diversity per sample was significantly different between techniques (P < 0.001, Kruskal-Wallis test), with eDNA detecting at least three times more species (2.5 ± 1.9; P < 0.001, Dunn’s tests; Fig. 1E) than BRUVS (0.8 ± 0.8) and UVC (0.2 ± 0.5). These results suggest that the level of dark diversity of sharks on New Caledonian coral reefs is much lower than previously estimated with traditional techniques.
For any given time interval, eDNA detects biodiversity at a greater spatiotemporal scale than the traditional methods used in this study; this may de facto increase the number of species detected by eDNA analysis (18). For example, UVCs sample limited visual areas (<500 m2), within specific habitats, over short temporal periods (<2 hours) (19). Similarly, although the bait plume from BRUVS can attract sharks from surrounding habitats, their detection capabilities are constrained by both visual range (<50 m) and operation time (a few hours) (12). In contrast, eDNA may detect species at a greater temporal scale (a few hours to a few days) due to the persistence of cellular material in the water (20, 21). Moreover, as water masses are in constant movement (particularly along the outer slopes of coral reefs), eDNA transported from different habitats (for example, open ocean) could potentially result in an overestimation of species richness in a given habitat. However, apart from occasional coral reef transients (for example, the great hammerhead, Sphyrna mokarran), all shark species detected by eDNA are regularly observed in coral reef habitats (Fig. 2) (22). Hence, it is unlikely that our diversity estimate has been inflated by oceanic inputs of external eDNA. Moreover, recent studies have indicated that eDNA analysis is powerful enough to distinguish species assemblages separated by small distances, even when comparing inshore and offshore habitats (23, 24).
Persisting shark populations in human-impacted areas
Scientific literature has repeatedly highlighted the footprint of anthropogenic activities on shark populations worldwide [for example, see the studies of Edgar et al. (25) and Robbins et al. (26)]. Similarly, BRUVS and UVC surveys suggest that sharks are quasi-absent near Nouméa, the capital city of New Caledonia (19). Human-induced behavioral changes have been observed in both terrestrial (27) and marine vertebrates (28), resulting in an overestimation of fish densities in marine reserves and an underestimation in impacted areas due to differential responses to diver presence (29). Little is known about the extent of similar behavioral sampling bias in sharks (13). However, our results suggest that this bias may play a role in shark detection, particularly near densely populated areas, where eDNA detected a significantly greater diversity of sharks compared to UVC and BRUVS (P < 0.001, Kruskal-Wallis test; P < 0.001, Dunn’s tests; Fig. 3A). Failure of traditional methods to detect comparative levels of shark diversity around human-populated areas may be a reflection not only of low shark densities but also of avoidance behavior in the remaining individuals. Conversely, as sharks in relatively undisturbed areas may display curiosity or naivety (29), we would expect BRUVS and UVC to reveal high shark diversity in “wilderness” areas located more than 20 hours travel time from the main regional city (19, 30). However, eDNA detected three times more species in these areas (3.1 ± 2.0) than BRUVS (1.3 ± 0.8) and UVC (0.9 ± 0.8; P < 0.001, Kruskal-Wallis test; P = 0.001, Dunn’s tests; Fig. 3B), demonstrating that eDNA appears more effective at estimating dark diversity, even when animal behavior may bias direct observations positively or negatively. Because of spatial heterogeneity of our sampling design between the three techniques (Fig. 1A), we performed the same analysis for overlapping collection sites and found consistent results (fig. S1).
Fig. 3. Number of shark species per sample in contrast to human impacts.
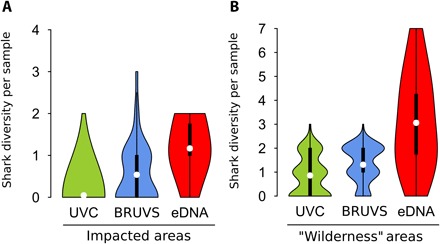
Violin plot showing detected shark species richness by the different methods in (A) impacted areas (Nouméa, the capital city) and (B) wilderness areas (Chesterfield, D’Entrecasteaux, Great Northern Lagoon, Petri, and Astrolabe). White dots are mean values; thick black bars correspond to interquartile ranges; thin black lines are 95% confidence intervals. Differences between methods are highly significant for both types of areas (P < 0.001, Kruskal-Wallis tests), with eDNA detecting more species per sample than BRUVS and UVC (P < 0.001, Dunn’s tests).
Increased species detectability revealed by rarefaction curves
Regional species diversity may be assessed by rarefaction curves linking the number of detected species to sampling effort (31). Rarefaction curves associated with the three methods (UVC, BRUVS, and eDNA) were fitted using six models with contrasted features (asymptotic versus nonasymptotic; two versus three parameters). The best model was nonasymptotic (power) for UVC-based survey and asymptotic for BRUVS and eDNA surveys (negative exponential and rational function, respectively; Table 1). The rarefaction curve for BRUVS reached nine species for the New Caledonian archipelago after 385 samples (Fig. 4, B and C). We then fitted the same asymptotic model (rational function) to the three rarefaction curves to compare their asymptotes. We show that doubling BRUVS sampling effort would result in the detection of only a single additional species (Table 1 and Fig. 4D). Meanwhile, the rarefaction curve for UVC attains nine species after 2758 UVCs (Fig. 4, B and C). According to the common model, doubling the number of UVCs would be required to detect one additional shark species (Table 1 and Fig. 4D), and both BRUVS and UVC rarefaction curves plateau at 10 species. In contrast, a few hundred eDNA samples could provide an accurate assessment of regional shark diversity (Fig. 4, B to D, and Table 1), requiring much less time and equipment than traditional survey methods. Furthermore, this would rapidly reveal a considerable proportion of shark species unseen by traditional methods that tend to overlook rare and elusive species in regional inventories (Fig. 4A). In addition, eDNA sampling was only conducted during 3 months in 2015, whereas BRUVS were deployed over a 3-year period (2012 to 2014) and UVC surveys were conducted between 1986 and 2014, reinforcing the potential of eDNA metabarcoding to detect species. Our results are very conservative in the sense that traditional methods were carried out over a large spatiotemporal scale; thus, they are more likely to sample unique events like rare species appearance or migration.
Table 1. Models fitted for species rarefaction curves obtained from UVC, BRUVS, and eDNA using the nls function in the stats package and the AICc for small sample bias (package AICmodavg).
The best-fitting model for each sampling technique is in bold, whereas the overall best-fitting model (all three techniques) is underlined.
| Model | Formula | Asymptotic | Number of parameters | AICc | ||
| UVC | BRUVS | eDNA | ||||
| Power | S = aXb | No | 2 | −4345 | 528 | 14 |
| Exponential | S = a + b log(X) | No | 2 | −191 | 210 | 23 |
| Negative exponential | S = a(1 − e− bX) | Yes | 2 | 4940 | 281 | 24 |
| Negative exponential | S = a + (b − a)e− cX | Yes | 3 | −609 | −675 | −19 |
| Monod | S = a/(1 + bX− 1) | Yes | 2 | 2321 | −185 | −5 |
| Rational function | S = (a + bX)/(1 + cX) | Yes | 3 | −2562 | −421 | −49 |
Fig. 4. Sample-based rarefaction curves.
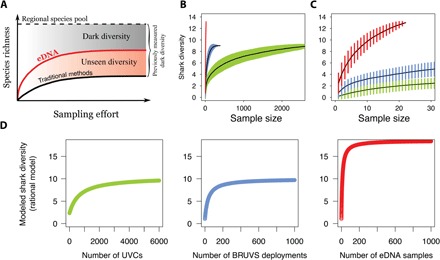
(A) Theoretical illustration of dark diversity measured by traditional methods, simply unseen but illuminated by eDNA, revealing a lower amount of dark diversity (absent species). Rarefaction curves showing accumulated sampled shark diversity measured by the different techniques (green, UVC; blue, BRUVS; red, eDNA). (B) Based on all samples. (C) Zoomed in to 30 samples. Error bars indicate SD. (D) Estimated rarefaction curves for UVC, BRUVS, and eDNA when increasing sampling effort, based on the best common model (rational function).
Limits and uncertainty of species detection
Using the 127–base pair (bp) cytochrome c oxidase subunit I (COI) fragment, we did not find an unequivocal correspondence between molecular operational taxonomic units (MOTUs) and species because some MOTUs had 100% sequence identity matches with multiple species in the Barcode of Life Data System (BOLD) database (32). This issue mainly concerns the Carcharhinus genus, which is recognized as taxonomically problematic and polyphyletic (33). Therefore, although 16 different shark MOTUs were identified, we opted for the conservative approach of merging MOTUs to present the minimum species richness (13 species). With a trade-off between primer universality and taxonomic resolution (16), the imperfect nature of currently available metabarcoding primers introduces a degree of uncertainty regarding the identification of certain species (34).
Only three species (tiger shark, scalloped hammerhead shark, and nurse shark) that were observed by UVC and/or BRUVS were not detected by eDNA (Fig. 2). The primers used in this study have already been shown to be capable of amplifying eDNA from both Sphyrnidae (hammerhead sharks) and Galeocerdo cuvier (tiger shark), but not Ginglymostomatidae (nurse sharks), explaining the absence of Nebrius ferrugineus (tawny nurse shark) from eDNA detections (32). Previous studies have indicated that hammerhead and tiger sharks only occur at very low densities in New Caledonia (12). Although our results show the power and potential of eDNA for the detection of shark diversity, our sampling effort was insufficient for an exhaustive survey not only because of stochasticity in polymerase chain reaction (PCR) and sequencing but also because scalloped hammerhead and tiger sharks may not have been present at the specific location and time of sampling. It is thus very likely that increasing the eDNA sampling effort would detect them in New Caledonia because the rarefaction curves show that eDNA can outperform the other methods in terms of species detectability (12), but additional work on refining the design of primers is needed to cement eDNA as a standard tool for the study of dark diversity of sharks.
There are also important caveats associated with eDNA detection, and traditional survey methods still have a number of advantages over eDNA methodologies. Foremost, eDNA-based methods cannot provide information on size, condition, developmental stage (eggs, larvae, juveniles, and adults), sex, behavior, and movement patterns of the target organism. Furthermore, when using typically maternally inherited mitochondrial markers, it remains impossible to distinguish hybrids, which may be the result of breeding between native and invasive species, from their maternal species. Finally, inferring species abundance from eDNA still remains a challenging but promising avenue and is a key area for further research (32, 35, 36).
New light for megafauna conservation
Here, we highlight the potential of eDNA metabarcoding for the rapid assessment of elusive megafauna species richness and, for the first time, for the determination of the extent of purported local species extirpations. We call for the introduction of eDNA assessments to complement traditional survey methods for the improvement of species detection and, hence, more efficient conservation strategies for threatened and elusive megafauna.
First, eDNA allows the reappraisal of previous estimates of species occurrences, which are used to define Criteria B of International Union for Conservation of Nature (IUCN) Red List Categories. With 46.8% of chondrichthyan species data deficient in IUCN assessments (37), this knowledge may markedly aid Red List classifications and the design and implementation of future conservation measures. For instance, a species is classified as Vulnerable if its extent of occurrence, that is, its continuous geographic range, is less than 20,000 km2 and declining. This kind of assessment is hardly achievable, trustable, and repeatable with traditional sampling methods owing to low detectability. However, eDNA provides an affordable, powerful, and standardized tool to assess large-scale occurrences, even for elusive megafauna. IUCN classification can also be based on population size (Criteria C), and although much more research is needed in this field, quantitative methods to infer eDNA concentration offer promise toward good estimates of relative abundance (36, 38). Alternative approaches include detecting and distinguishing individuals of a given population as a measure of abundance. Differentiating populations from a single shark species has recently been achieved using eDNA (39). This strategy, even in its infancy, opens a new era in the field of population genetics using eDNA and in population size assessment, potentially fueling IUCN Criteria C. Monitoring shark populations of New Caledonia may greatly benefit from these methodological advances because the effectiveness of conservation efforts is still under scrutiny for megafauna (12, 40).
In addition, increased knowledge of dark diversity may guide the direction of conservation-based decision-making because we can learn a lot from absent species (6). To halt biodiversity loss, it is imperative to understand why some species are missing from areas where biogeographic history, as well as current ecological and environmental conditions, predicts their presence. Seeking common characteristics (for example, ecological needs, dispersal ability, and body size) among species constituting the dark diversity can help identify key determinants of vulnerability, decline, or extirpation and guide appropriate management strategies (4). High proportions of dark diversity in a given region or area indicate the need for widespread conservation efforts across multiple species, whereas low proportions of dark diversity suggest that more tailored solutions are required to reduce pressures on specific species apparently missing. Areas showing a high observed diversity and low dark diversity can be considered as refugia and thus potential sources for recolonization. Consequently, such areas deserve high conservation priority (6, 41). By contrast, areas with relatively high dark diversity compared to the observed species richness need restoration efforts focusing on mitigation of species threats and increasing connectivity. In New Caledonia, mapping and monitoring the dark diversity related to the observed diversity of sharks along human gradients would provide a relevant indicator of wildlife status to inform management.
Large-scale efforts to restore local species pools are more feasible in terrestrial than in marine environments. For example, creating habitat connectivity to promote species reestablishment has previously been shown to be successful [for example, see the study of Di Minin et al. (42)]. However, this requires alleviation of the original stressors (through, for instance, protected areas) to avoid affecting newly reconnected populations (42). Assisted reintroduction is another potential solution in terrestrial habitats, albeit costly and controversial (43, 44). Because there are fewer manipulative solutions for the marine environment, the discovery of remaining individuals close to human-impacted areas requires more immediate, alternative actions, such as the establishment of marine reserve networks and connecting suitable habitats, to preserve the remainder of the species and increase population densities by decreasing threats. eDNA will most likely prove progressively useful in marine conservation and hence will be playing an increasingly important role in the formulation of policies to aid species conservation.
MATERIALS AND METHODS
Study sites
The New Caledonian archipelago is located in the southwestern Pacific Ocean (Fig. 1A). It is composed of “Grande Terre” surrounded by one of the largest barrier reefs in the world and numerous isolated islands and remote reefs. Sampling occurred across the New Caledonian Archipelago, including waters from the Coral Sea Marine Park. Study areas encompassed a gradient of human density, from high population density (near the capital, Nouméa) to wilderness reefs at >20 hours travel time from the main regional city [Chesterfield Reefs, D’Entrecasteaux Reefs, Astrolabe Reefs, Petri, and Great Northern Lagoon (12, 19, 30)]. The regional pool of sharks in New Caledonia is inventoried at 49 species, including 26 shallow-water species (17).
UVC and BRUVS data sets
UVC and BRUVS protocols used in this study are described in detail by D’Agata et al. (19) and Juhel et al. (12). Here, 2758 UVCs were conducted by day in various coral reef habitats at depths of 1 to 15 m from 1986 to 2014 (Fig. 1A). Three hundred eighty-five BRUVS (45) were deployed by day at a mean depth of 16 m (±10 m SD; range, 3 to 48 m) in different coral reef habitats between September 2012 and October 2014. BRUVS are video systems that record for 1 hour in the presence of standardized bait (1-kg pilchards). Shark occurrence was measured through video analysis, and species identification was double-checked by trained operators.
eDNA collection and sample processing
eDNA samples were collected during September to November 2015, on coral reef external slopes, with a reef topography ranging from 20 to 40 m. Each 4-liter water sample consisted of 2 liters sampled at a depth of 5 m and 2 liters sampled at a depth of 20 m, collected with a Niskin water sampler. After collection, water samples were individually covered and stored on ice before filtration. Water was subsequently filtered using sterile mixed cellulose esters filters (Merck Millipore; diameter, 47 mm; pore size, 0.45 μm) and then stored at −20°C in 2.0-ml screw-cap microcentrifuge tubes containing silica beads, drying out the filters and preventing DNA degradation (32). DNA was extracted from the filters using the DNeasy PowerSoil DNA Isolation Kit (Qiagen), following the manufacturer’s protocol. Purified extracts were assessed for DNA concentration in a Qubit fluorometer (Thermo Fisher Scientific).
Strict adherence to contamination control was followed at all field and laboratory stages to prevent the occurrence of contamination, including the use of disposable gloves and single-use sterile collection bottles and filtration equipment, and the bleaching (50% bleach) of sampling devices, laboratory equipment, and surfaces. In addition, a dedicated controlled eDNA laboratory at the University of Salford, with separate rooms designated for the physical separation of eDNA extraction, pre-PCR preparations, and post-PCR procedures, was used for all laboratory work. To identify any potential contamination, negative control DNA extraction blanks (elution buffer from extraction kit) and PCR blanks were also ran (32).
Library preparation and sequencing
An elasmobranch-specific COI primer set was used for the amplification of eDNA metabarcoding markers. The previously published primer set consisted of a novel reverse primer (“Shark COI-MINIR”, 5′-AAGATTACAAAAGCGTGGGC-3′) (46) and two universal fish barcoding forward primers (FishF2, 5′-TCGACTAATCATAAAGATATCGGCAC-3′; FishF1, 5′-TCAACCAACCACAAAGACATTGGCAC-3′) (46, 47), yielding an amplicon of 127 bp. Samples were sequenced in a single multiplexed Illumina MiSeq run, along with samples from a related project, which are not included in this study, for a total of 96 samples including two negative controls, using four sets of 24 primers with attached eight-base sample-specific oligo-tags differing in at least three bases (table S1) (48). To increase variability of the amplicon sequences, a variable number (two, three, or four) of fully degenerate positions (Ns) was added at the beginning of each primer (49). The full, sequenced PCR product consisted of 195 bp, including the amplicon, primers, sample tags, and leading Ns.
For PCR amplification, a single-step protocol was used, directly attaching the eight-base tagged primers. The PCR mix recipe was as follows: A total volume of 20 μl included 2 μl of 10× buffer (BioLine), 0.6 μl of 50 mM MgCl (BioLine), 0.5 μl each of the 5 μM forward primers (Eurofins), 1 μl of the 5 μM reverse primer, 0.2 μl of 10 mM deoxynucleotide triphosphate mix (BioLine), 0.2 μl of BioTaq DNA polymerase (5 U/μl; BioLine), a standardized amount (10 ng) of the filter-extracted eDNA template, and 13 μl of sterile water. The PCR profile included an initial denaturing step of 95°C for 15 min, 35 cycles of 94°C for 1 min, 52°C for 1 min, and 72°C for 1 min and a final extension step of 72°C for 5 min. The quality of all amplifications was assessed by electrophoresis, running the products through a 1.5% agarose gel stained with GelRed (Cambridge Bioscience), and visualized on an ultraviolet light platform. All PCR products (including two PCR-negative controls) were pooled by marker into four multiplex sample pools and purified using MinElute columns (Qiagen). Four Illumina libraries were subsequently built from the four pools, using a NextFlex PCR-free library preparation kit (Bioo Scientific). The libraries were quantified using a NEBNext qPCR quantification kit (New England Biolabs) and pooled in equimolar concentrations along with 1% PhiX (v3, Illumina) serving as a positive sequencing quality control. The libraries with a final molarity of 8 pM were sequenced on an Illumina MiSeq platform in a single MiSeq flow cell using v2 chemistry (2 × 150–bp paired ends).
Bioinformatics analyses
The bioinformatic analysis was based on an OBITools metabarcoding software suite (50). The pipeline used for data analysis is summarized in table S2. Quality of the reads was assessed using FastQC. Paired-end reads were aligned using illuminapairedend, and alignments with quality scores >40 were kept. The aligned data set was demultiplexed using ngsfilter. A length filter (obigrep) was applied to the aligned reads (120 to 135 bp), and reads containing ambiguous bases were removed. The reads were then dereplicated using obiuniq, and a chimera removal step was performed using the uchime-denovo algorithm (51) implemented in vsearch (52). The MOTUs were delimited using the sumaclust algorithm (50) with a constant similarity threshold of 99%. Taxonomic assignment of the representative sequences for each MOTU was performed using the ecotag algorithm (50). We built a bespoke elasmobranch reference database using a custom R script for retrieving all COI elasmobranch sequences available from the BOLD database (53), subsequently selecting those that included our 127-bp target fragment. The custom R script is available from http://github.com/metabarpark/R_scripts_querying_databases. To add homologous sequences from other non-elasmobranch taxa, an in silico PCR was performed against release R117 of the EMBL-EBI database using ecoPCR (54). Subsequently, the obtained reference sequences were added to the elasmobranch sequences obtained from BOLD. These additional reference sequences were added to our elasmobranch database to avoid the incorrect assignment of amplified sequences, belonging to other taxa, to elasmobranchs. This combined reference database is available from http://github.com/metabarpark/Reference-databases. The final refining of the data set included taxonomy clustering of MOTUs assigned to the same species.
The risk of contamination adds to the challenges associated with eDNA metabarcoding (18, 55), with the possibility of introducing false-positive results. It is likely to detect a species represented by a single sequence read in a sample, but the possibility of contamination or sequencing error cannot be excluded as the potential cause of MOTU detection. We subsequently adopted a conservative approach to our analyses and removed single-read MOTUs from our samples to avoid potential false positives.
Statistical analyses
Given the violation of the normality assumption and the unbalanced design with different number of samples depending on the techniques, Kruskal-Wallis tests followed by Dunn’s tests were performed to test for differences in shark diversity per sample among techniques. The vegan package was used for rarefaction analyses, followed by model fitting using the nls function in the stats package and the Akaike information criterion corrected for small sample bias (AICc, package AICmodavg). Models were fitted for the three methods independently (UVC, BRUVS, and eDNA; Table 1); subsequently, a common model was selected by comparing AICc for the three methods simultaneously. Statistical analyses were performed in the R program environment (R Development Core Team 2012, version 3.4.0).
Supplementary Material
Acknowledgments
We thank the crew of the research vessels Amborella (Government of New Caledonia), Alis Institute of Research for Development, and Plan B (Waitt Institute) for their help with data collection during oceanographic campaigns. We thank G. Mou-Tham and many others for UVC collection and BRUVS analyses. We would also like to thank M. Dando for the scientific drawings used in the manuscript. Funding: This work is an outcome of the Pristine, APEX and e(lasmo)DNA projects co-funded by the Total Foundation, the Pew Charitable Trusts, the Government of New Caledonia, and the University of Salford R&E strategy funding. Author contributions: G.B., S. Mariani, D.M., and L.V. designed the study with inputs from all coauthors. J.B., O.S.W., and S. Mariani designed and carried out the eDNA laboratory procedures and bioinformatics pipeline. M.K., L.V., J.-B.J., G.B., W.D.R., and many others collected the UVC, BRUVS, and eDNA samples. G.B., J.B., O.S.W., L.V., and D.M. analyzed the data and drafted the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from L.V. (laurent.vigliola@ird.fr).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaap9661/DC1
fig. S1. Number of shark species per sample in overlapping collection sites.
table S1. Full sequences of the 24 tagged primer sets used.
table S2. Metabarcoding pipeline for COI Elasmobranchii Fields et al. primers.
REFERENCES AND NOTES
- 1.Dirzo R., Young H. S., Galetti M., Ceballos G., Isaac N. J. B., Collen B., Defaunation in the Anthropocene. Science 345, 401–406 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Young H. S., McCauley D. J., Galetti M., Dirzo R., Patterns, causes, and consequences of anthropocene defaunation. Annu. Rev. Ecol. Evol. Syst. 47, 333–358 (2016). [Google Scholar]
- 3.Pärtel M., Szava-Kovats R., Zobel M., Dark diversity: Shedding light on absent species. Trends Ecol. Evol. 26, 124–128 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Moeslund J. E., Brunbjerg A. K., Clausen K. K., Dalby L., Fløjgaard C., Juel A., Lenoir J., Using dark diversity and plant characteristics to guide conservation and restoration. J. Appl. Ecol. 54, 1730–1741 (2017). [Google Scholar]
- 5.Soliveres S., van der Plas F., Manning P., Prati D., Gossner M. M., Renner S. C., Alt F., Arndt H., Baumgartner V., Binkenstein J., Birkhofer K., Blaser S., Blüthgen N., Boch S., Böhm S., Börschig C., Buscot F., Diekötter T., Heinze J., Hölzel N., Jung K., Klaus V. H., Kleinebecker T., Klemmer S., Krauss J., Lange M., Morris E. K., Müller J., Oelmann Y., Overmann J., Pašalić E., Rillig M. C., Schaefer H. M., Schloter M., Schmitt B., Schöning I., Schrumpf M., Sikorski J., Socher S. A., Solly E. F., Sonnemann I., Sorkau E., Steckel J., Steffan-Dewenter I., Stempfhuber B., Tschapka M., Türke M., Venter P. C., Weiner C. N., Weisser W. W., Werner M., Westphal C., Wilcke W., Wolters V., Wubet T., Wurst S., Fischer M., Allan E., Biodiversity at multiple trophic levels is needed for ecosystem multifunctionality. Nature 536, 456–459 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lewis R. J., de Bello F., Bennett J. A., Fibich P., Finerty G. E., Götzenberger L., Hiiesalu I., Kasari L., Lepš J., Májeková M., Mudrák O., Riibak K., Ronk A., Rychtecká T., Vitová A., Pärtel M., Applying the dark diversity concept to nature conservation. Conserv. Biol. 31, 40–47 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Webb T. J., Mindel B. L., Global patterns of extinction risk in marine and non-marine systems. Curr. Biol. 25, 506–511 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Davidson L. N. K., Dulvy N. K., Global marine protected areas to prevent extinctions. Nat. Ecol. Evol. 1, 0040 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Hobday A. J., Smith A. D. M., Stobutzki I. C., Bulman C., Daley R., Dambacher J. M., Deng R. A., Dowdney J., Fuller M., Furlani D., Griffiths S. P., Johnson D., Kenyon R., Knuckey I. A., Ling S. D., Pitcher R., Sainsbury K. J., Sporcic M., Smith T., Turnbull C., Walker T. I., Wayte S. E., Webb H., Williams A., Wise B. S., Zhou S., Ecological risk assessment for the effects of fishing. Fish. Res. 108, 372–384 (2011). [Google Scholar]
- 10.McClenachan L., Cooper A. B., Dulvy N. K., Rethinking trade-driven extinction risk in marine and terrestrial megafauna. Curr. Biol. 26, 1640–1646 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Nadon M. O., Baum J. K., Williams I. D., McPherson J. M., Zgliczynski B. J., Richards B. L., Schroeder R. E., Brainard R. E., Re-creating missing population baselines for pacific reef sharks. Conserv. Biol. 26, 493–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juhel J.-B., Vigliola L., Mouillot D., Kulbicki M., Letessier T. B., Meeuwig J. J., Wantiez L., Reef accessibility impairs the protection of sharks. J. Appl. Ecol. 55, 673–683 (2018). [Google Scholar]
- 13.Mourier J., Brown C., Planes S., Learning and robustness to catch-and-release fishing in a shark social network. Biol. Lett. 13, 20160824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohmann K., Evans A., Gilbert M. T. P., Carvalho G. R., Creer S., Knapp M., Yu D. W., de Bruyn M., Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 29, 358–367 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Miya M., Sato Y., Fukunaga T., Sado T., Poulsen J. Y., Sato K., Minamoto T., Yamamoto S., Yamanaka H., Araki H., Kondoh M., Iwasaki W., MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomsen P. F., Møller P. R., Sigsgaard E. E., Knudsen S. W., Jørgensen O. A., Willerslev E., Environmental DNA from seawater samples correlate with trawl catches of subarctic, deepwater fishes. PLOS ONE 11, e0165252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.P. Tirard, Requins du Caillou (2011). [Google Scholar]
- 18.Goldberg C. S., Turner C. R., Deiner K., Klymus K. E., Thomsen P. F., Murphy M. A., Spear S. F., McKee A., Oyler-McCance S. J., Cornman R. S., Laramie M. B., Mahon A. R., Lance R. F., Pilliod D. S., Strickler K. M., Waits L. P., Fremier A. K., Takahara T., Herder J. E., Taberlet P., Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307 (2016). [Google Scholar]
- 19.D’Agata S., Mouillot D., Wantiez L., Friedlander A. M., Kulbicki M., Vigliola L., Marine reserves lag behind wilderness in the conservation of key functional roles. Nat. Commun. 7, 12000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minamoto T., Fukuda M., Katsuhara K. R., Fujiwara A., Hidaka S., Yamamoto S., Takahashi K., Masuda R., Environmental DNA reflects spatial and temporal jellyfish distribution. PLOS ONE 12, e0173073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomsen P. F., Kielgast J., Iversen L. L., Møller P. R., Rasmussen M., Willerslev E., Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLOS ONE 7, e41732 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D. A. Ebert, S. Fowler, L. Compagno, Sharks of the World (Wild Nature Press, 2013). [Google Scholar]
- 23.Port J. A., O’Donnell J. L., Romero-Maraccini O. C., Leary P. R., Litvin S. Y., Nickols K. J., Yamahara K. M., Kelly R. P., Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 25, 527–541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Donnell J. L., Kelly R. P., Shelton A. O., Samhouri J. F., Lowell N. C., Williams G. D., Spatial distribution of environmental DNA in a nearshore marine habitat. PeerJ. 5, e3044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar G. J., Stuart-Smith R. D., Willis T. J., Kininmonth S., Baker S. C., Banks S., Barrett N. S., Becerro M. A., Bernard A. T. F., Berkhout J., Buxton C. D., Campbell S. J., Cooper A. T., Davey M., Edgar S. C., Försterra G., Galván D. E., Irigoyen A. J., Kushner D. J., Moura R., Parnell P. E., Shears N. T., Soler G., Strain E. M. A., Thomson R. J., Global conservation outcomes depend on marine protected areas with five key features. Nature 506, 216–220 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Robbins W. D., Hisano M., Connolly S. R., Choat J. H., Ongoing collapse of coral-reef shark populations. Curr. Biol. 16, 2314–2319 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Frid A., Dill L. M., Human-caused disturbance stimuli as a form of predation risk. Ecol. Soc. 6, 11 (2002). [Google Scholar]
- 28.French S. S., González-Suárez M., Young J. K., Durham S., Gerber L. R., Human disturbance influences reproductive success and growth rate in California sea lions (Zalophus californianus). PLOS ONE 6, e17686 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goetze J., Januchowski-Hartley F. A., Claudet J., Langlois T. J., Wilson S. K., Jupiter S. D., Fish wariness is a more sensitive indicator to changes in fishing pressure than abundance, length or biomass. Ecol. Appl. 27, 1178–1189 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Maire E., Cinner J., Velez L., Huchery C., Mora C., Dagata S., Vigliola L., Wantiez L., Kulbicki M., Mouillot D., How accessible are coral reefs to people? A global assessment based on travel time. Ecol. Lett. 19, 351–360 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Gotelli N. J., Colwell R. K., Quantifying biodiversity: Procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 4, 379–391 (2001). [Google Scholar]
- 32.Bakker J., Wangensteen O. S., Chapman D. D., Boussarie G., Buddo D., Guttridge T. L., Hertler H., Mouillot D., Vigliola L., Mariani S., Environmental DNA reveals tropical shark diversity in contrasting levels of anthropogenic impact. Sci. Rep. 16886 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sorenson L., Santini F., Alfaro M. E., The effect of habitat on modern shark diversification. J. Evol. Biol. 27, 1536–1548 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Stat M., Huggett M. J., Bernasconi R., DiBattista J. D., Berry T. E., Newman S. J., Harvey E. S., Bunce M., Ecosystem biomonitoring with eDNA: Metabarcoding across the tree of life in a tropical marine environment. Sci. Rep. 7, 12240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jo T., Murakami H., Masuda R., Sakata M. K., Yamamoto S., Minamoto T., Rapid degradation of longer DNA fragments enables the improved estimation of distribution and biomass using environmental DNA. Mol. Ecol. Resour. 17, e25–e33 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto S., Minami K., Fukaya K., Takahashi K., Sawada H., Murakami H., Tsuji S., Hashizume H., Kubonaga S., Horiuchi T., Hongo M., Nishida J., Okugawa Y., Fujiwara A., Fukuda M., Hidaka S., Suzuki K. W., Miya M., Araki H., Yamanaka H., Maruyama A., Miyashita K., Masuda R., Minamoto T., Kondoh M., Environmental DNA as a ‘snapshot’ of fish distribution: A case study of Japanese jack mackerel in Maizuru Bay, Sea of Japan. PLOS ONE 11, e0149786 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dulvy N. K., Fowler S. L., Musick J. A., Cavanagh R. D., Kyne P. M., Harrison L. R., Carlson J. K., Davidson L. N. K., Fordham S. V., Francis M. P., Pollock C. M., Simpfendorfer C. A., Burgess G. H., Carpenter K. E., Compagno L. J. V., Ebert D. A., Gibson C., Heupel M. R., Livingstone S. R., Sanciangco J. C., Stevens J. D., Valenti S., White W. T., Extinction risk and conservation of the world’s sharks and rays. eLife 3, e00590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lacoursière-Roussel A., Côté G., Leclerc V., Bernatchez L., Quantifying relative fish abundance with eDNA: A promising tool for fisheries management. J. Appl. Ecol. 53, 1148–1157 (2016). [Google Scholar]
- 39.Sigsgaard E. E., Nielsen I. B., Bach S. S., Lorenzen E. D., Robinson D. P., Knudsen S. W., Pedersen M. W., Jaidah M. A., Orlando L., Willerslev E., Møller P. R., Thomsen P. F., Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nat. Ecol. Evol. 1, 0004 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Bauer H., Chapron G., Nowell K., Henschel P., Funston P., Hunter L. T. B., Macdonald D. W., Packer C., Lion (Panthera leo) populations are declining rapidly across Africa, except in intensively managed areas. Proc. Natl. Acad. Sci. U.S.A. 112, 14894–14899 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pärtel M., Szava-Kovats R., Zobel M., Community completeness: Linking local and dark diversity within the species pool concept. Folia Geobot. 48, 307–317 (2013). [Google Scholar]
- 42.Di Minin E., Hunter L. T. B., Balme G. A., Smith R. J., Goodman P. S., Slotow R., Creating larger and better connected protected areas enhances the persistence of big game species in the Maputaland-Pondoland-Albany biodiversity hotspot. PLOS ONE 8, e71788 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seddon P. J., Griffiths C. J., Soorae P. S., Armstrong D. P., Reversing defaunation: Restoring species in a changing world. Science 345, 406–412 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Weise F. J., Stratford K. J., van Vuuren R. J., Financial costs of large carnivore translocations—Accounting for conservation. PLOS ONE 9, e105042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harvey E. S., Cappo M., Butler J. J., Hall N., Kendrick G. A., Bait attraction affects the performance of remote underwater video stations in assessment of demersal fish community structure. Mar. Ecol. Prog. Ser. 350, 245–254 (2007). [Google Scholar]
- 46.Fields A. T., Abercrombie D. L., Eng R., Feldheim K., Chapman D. D., A novel mini-DNA barcoding assay to identify processed fins from internationally protected shark species. PLOS ONE 10, e0114844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward R. D., Zemlak T. S., Innes B. H., Last P. R., Hebert P. D. N., DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1847–1857 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guardiola M., Uriz M. J., Taberlet P., Coissac E., Wangensteen O. S., Turon X., Deep-sea, deep-sequencing: Metabarcoding extracellular DNA from sediments of marine canyons. PLOS ONE 10, e0139633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O. S. Wangensteen, X. Turon, Metabarcoding techniques for assessing biodiversity of marine animal forests, in Marine Animal Forests, S. Rossi, L. Bramanti, A. Gori, C. Orejas, Eds. (Springer, 2015), pp. 1–26. [Google Scholar]
- 50.Boyer F., Mercier C., Bonin A., Le Bras Y., Taberlet P., Coissac E., obitools: A unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 16, 176–182 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R., UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rognes T., Flouri T., Nichols B., Quince C., Mahé F., VSEARCH: A versatile open source tool for metagenomics. PeerJ. 4, e2584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ratnasingham S., Hebert P. D. N., BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ficetola G. F., Coissac E., Zundel S., Riaz T., Shehzad W., Bessière J., Taberlet P., Pompanon F., An in silico approach for the evaluation of DNA barcodes. BMC Genomics 11, 434 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomsen P. F., Willerslev E., Environmental DNA—An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaap9661/DC1
fig. S1. Number of shark species per sample in overlapping collection sites.
table S1. Full sequences of the 24 tagged primer sets used.
table S2. Metabarcoding pipeline for COI Elasmobranchii Fields et al. primers.