

Abstract
Visualizing the formation of multinucleated giant cells (MGCs) from living specimens has been challenging due to the fact that most live imaging techniques require propagation of light through glass, but on glass macrophage fusion is a rare event. This protocol presents the fabrication of several optical-quality glass surfaces where adsorption of compounds containing long-chain hydrocarbons transforms glass into a fusogenic surface. First, preparation of clean glass surfaces as starting material for surface modification is described. Second, a method is provided for the adsorption of compounds containing long-chain hydrocarbons to convert non-fusogenic glass into a fusogenic substrate. Third, this protocol describes fabrication of surface micropatterns that promote a high degree of spatiotemporal control over MGC formation. Finally, fabricating glass bottom dishes is described. Examples of use of this in vitro cell system as a model to study macrophage fusion and MGC formation are shown.
Keywords: Immunology and Infection, Issue 133, Macrophage fusion, multinucleated giant cell formation, live imaging, foreign body reaction, foreign body giant cell, inflammation, protocol
Introduction
The formation of MGCs accompanies a number of pathological states in the human body distinguished by chronic inflammation1. Despite agreement that mononucleated macrophages fuse to form MGCs2, surprisingly few studies have shown fusion in context with living specimens3,4. This is because clean glass surfaces that are required for most imaging techniques do not promote macrophage fusion when induced by inflammatory cytokines5. Indeed, if clean glass is used as a substrate for macrophage fusion, then low to intermediate magnification objectives (i.e., 10 - 20X) and more than 15 h of continuous imaging are often required to observe a single fusion event.
On the other hand, fusogenic plastic surfaces (e.g., permanox) or bacteriological grade plastic promote fusion2. However, imaging through plastic is problematic because the substrate is thick and scatters light. This complicates imaging since long working distance (LWD) objectives are required. However, LWD objectives usually have low light gathering capacity compared to their coverslip-corrected counterpart. Further, techniques that exploit changes in the polarity of light passing through the specimen such as differential interference contrast are impossible since plastic is birefringent. The barriers associated with the use of plastic are further underscored by the fact that it is impossible to predict where macrophage fusion/MGC formation will occur on the surface. Together, these limitations restrict the visualization of macrophage fusion to phase contrast optics, extended total imaging durations (>15 continuous hours), and low resolution.
We recently identified a highly fusogenic glass surface while conducting single-molecule super resolution microscopy with fixed macrophages/MGCs4. This observation was surprising because clean glass surfaces promote fusion at the very low rate of ~ 5% after 24 h in the presence of interleukin-4 (IL-4) as determined by the fusion index4. We found that the capacity to promote fusion was due to oleamide contamination. Adsorption of oleamide or other compounds that similarly contained long-chain hydrocarbons made the glass fusogenic. The most fusogenic compound (paraffin wax) was micropatterned, and it imparted a high degree of spatiotemporal control over macrophage fusion and a 2-fold increase in the number of fusion events observed within the same amount of time compared to permanox. These optical-quality surfaces provided the first glimpse into the morphological features and kinetics that govern the formation of MGCs in living specimens.
In this protocol we describe the fabrication of a variety of glass surfaces that can be used to monitor the formation of MGCs from living specimens. In addition, we show that these surfaces are amenable to far-field super-resolution techniques. Surface fabrication is dependent on the goal of the experiment, and each surface is described with related examples in the proceeding text.
Protocol
Procedures that utilize animals were approved by the Animal Care and Use Committees at Mayo Clinic, Janelia Research Campus, and Arizona State University.
1. Preparing Acid-cleaned Cover Glass
NOTE: Cover glass purchased from many manufacturers may not be as clean as expected. Consider routinely cleaning batches of cover glass before any procedure where microscopy is involved.
Purchase high stringency cover glass. Take special care to choose the correct thickness (0.15 or 0.17 mm). NOTE: The choice of cover glass thickness is dependent on the microscope objective and is listed directly on the objective barrel.
Incubate the cover glass in 12 M hydrochloric acid in a well-ventilated chemical fume hood for 1 h with sonication (42 kHz, 70 W). Repeat this step for two additional times with fresh 12 M HCl.
Fill a separate beaker with ultrapure water and add the cover glass. Sonicate the cover glass for 5 - 10 min in a well-ventilated chemical hood. Repeat this step ten times.
Incubate the cover glass in pure acetone in a well-ventilated chemical hood for 30 min with sonication. Repeat this step for two additional times.
Fill a separate beaker with sterile ultrapure water and add the cover glass. Sonicate the cover glass for 5 - 10 min in a well-ventilated chemical hood. Repeat this step ten times.
Incubate the cover glass in 100% ethyl alcohol in a chemical hood for 30 min with sonication. Repeat this step two additional times. NOTE: The purity of the ethyl alcohol is important. Contaminants in the form of dissolved solids will dry on the glass and can promote macrophage fusion.
For long-term storage, place the acid-cleaned cover glass in a container filled with pure ethyl alcohol. Alternatively, dry the cover glass with nitrogen gas and store in a vacuum desiccator.
2. Preparation of Fusogenic Optical-quality Surfaces
Dissolve DMSO-free high-melting point paraffin wax in ultrapure toluene. NOTE: Stock concentrations are made at 10 mg/mL, and are diluted 1:9 in ultrapure toluene to make a 1 mg/mL working solution. Caution: Toluene should be handled with care as a teratogen. Dispose of toluene according to the institutional chemical hygiene plan.
Apply the paraffin working solution to a dry acid-cleaned cover glass at a volume that evenly covers the glass surface. Decant excess solution and dry the cover glass with nitrogen gas or air. For bulk preparation, use a Coplin jar designed to accommodate the cover glass.
Polish the cover glass by 3 strokes in the x axis and next by 3 strokes in the y axis with a lint-free wipe (see Table of Materials).
Immediately before the experiment is conducted, wash the cover glass with sterile ultrapure water, and subsequently sterilize for 15 - 30 min with ultraviolet light in a biological safety hood. Alternatively, sterilize the cover glass by ethylene oxide or gamma irradiation.
3. Micropatterning Hydrocarbon-containing Compounds to Confine Fusion to Predetermined Regions
Dry the acid-cleaned cover glass and immobilize the glass on a flat surface.
Using forceps, carefully immerse a gold transmission electron microscopy finder grid in a working solution of the hydrocarbon compound(s). Choose a hydrocarbon compound that meets the end goal of the experiment (see Table 1). Wick away excess solution by gently tapping the grid on filter paper, and immediately place the grid in the center of the cover glass. Allow the toluene to dry for 2 min before proceeding to the next step.
Make sure the grid is bonded to the glass by gently inverting the glass. If the grid detaches repeat step 3.2 using a new cover glass. NOTE: If a plasma cleaner is not available, omit step 3.4.
Plasma clean the cover glass by treatment with vacuum gas plasma. NOTE: The finder grid acts as a mask to protect the underlying surface adsorbed with long-chain hydrocarbons from the plasma. The regions that are unprotected by the grid are rendered non-fusogenic by exposure to plasma. The amount of time the cover glass is expose to plasma should be determined empirically.
Using fine-tip forceps, carefully remove the grid from the glass surface to expose the micropattern.
4. Fabricating Glass Bottom Dishes
Drill a 6 - 10 mm circular hole in the bottom of a 35-mm plastic Petri dish using a step drill bit. NOTE: It is critical to use a step drill bit to create smooth edges. If edges are too rough, the cover glass will not bond flat to the bottom of the dish. Imperfectly flat surfaces make microscopy difficult.
Carefully mix and degas silicone elastomer according to the manufacturer's instructions (i.e., polydimethylsiloxane; PDMS).
Apply a thin coating of elastomer just proximate to the edge of (i.e., circumscribing) the hole. NOTE: The elastomer should appear as a continuous albeit thin band surrounding the hole.
Gently apply either the fusogenic cover glass (described in section 2), or the dry acid-cleaned cover glass (described in section 1) to the dish in order to cover the hole surrounded by elastomer. Ensure that the cover glass appears flush with the bottom of the dish and extends beyond the diameter of the hole so that a substantial portion of the glass is in contact with the plastic. Cure the elastomer by baking at 50 °C for 2 - 3 h.
If fusogenic glass is preferred, UV sterilize the dish and culture cells according to standard protocols. However, if a micropattern is preferred, proceed to step 3.2 for micropattern preparation (the gas plasma used during micropatterning sterilizes the dish and strongly couples PDMS to glass).
5. Collecting Thioglycollate-elicited Macrophages
Inject 8-week-old C57BL/6 mice with 0.5 mL of a sterile solution of 4% Brewer's thioglycollate as previously described3,4,6.
Seventy-two hours later, euthanize the animal according to approved animal care and use guidelines, and collect macrophages by peritoneal lavage with ice-cold phosphate-buffered saline supplemented with 5 mM ethylenediaminetetraacetate.
Centrifuge the macrophages at 300 x g for 3 min and resuspend in 1 mL of DMEM:F12 supplemented with 15 mM HEPES, 10% FBS, and 1% antibiotics (culture medium). NOTE: The cells are subsequently counted with a Neubauer hemocytometer. Macrophages are diluted to the appropriate concentration and applied to the surfaces. The number of cells to apply to a given surface should be carefully considered by the investigator for the purpose of the experimental question. Consult known standards in the primary literature.
After 30 min wash the surfaces with HBSS supplemented with 0.1% BSA to remove non-adherent cells, and replace with fresh culture medium. Return the cultures to the incubator (5% CO2 in air at 37 °C).
2 h later, aspirate the medium and replace with culture medium supplemented with 10 ng/mL of IL-4. Image the cells as described elsewhere4.
Representative Results
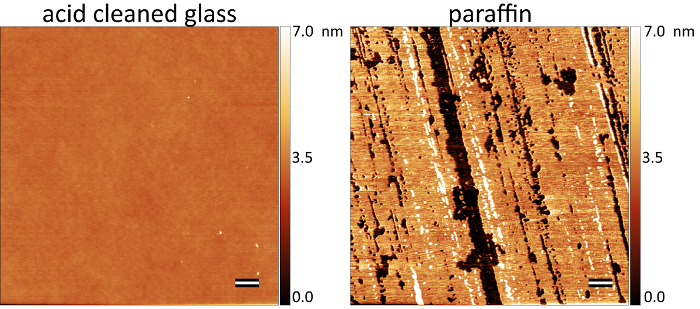
Physicochemical parameters of materials have dramatic effects on the extent of macrophage fusion7,8,9,10. Moreover, surface contaminants are known to promote macrophage fusion11. Therefore, it is important to start with clean cover glass as a negative control for macrophage fusion. When cleaned as described in protocol 1, the cover glass is exceptionally flat with no obvious surface features, whereas adsorption of long-chain hydrocarbons followed by surface polishing creates a degree of nanotopography (Figure 1).
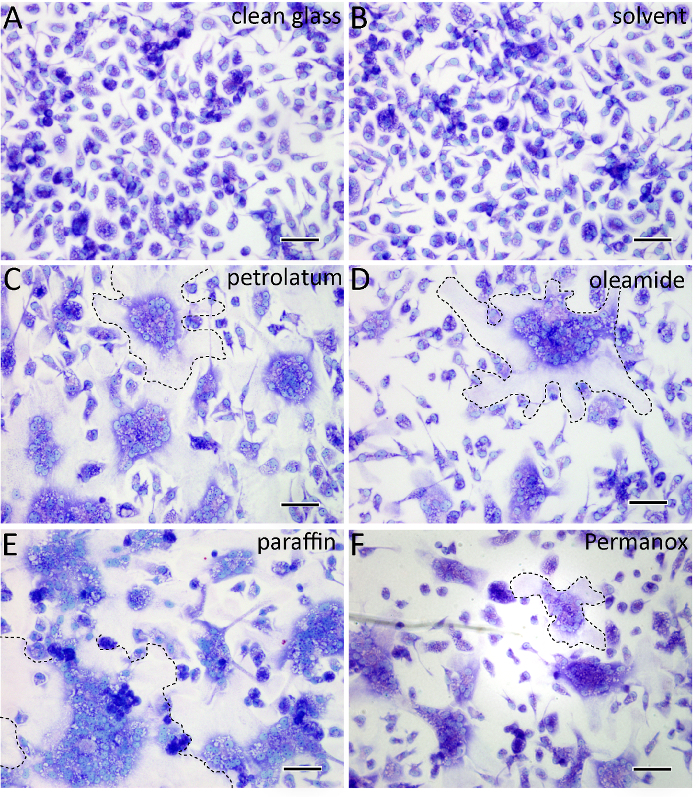
On clean cover glass surfaces, macrophages fuse at a very low rate (Figure 2). In contrast, after 24 h in the presence of IL-4, the macrophages applied to surfaces adsorbed with compounds containing long-chain hydrocarbons undergo robust fusion (Figure 2). Further, adsorption of solvent alone (e.g., toluene, isopropanol) does not make glass a fusogenic surface (Figure 2).
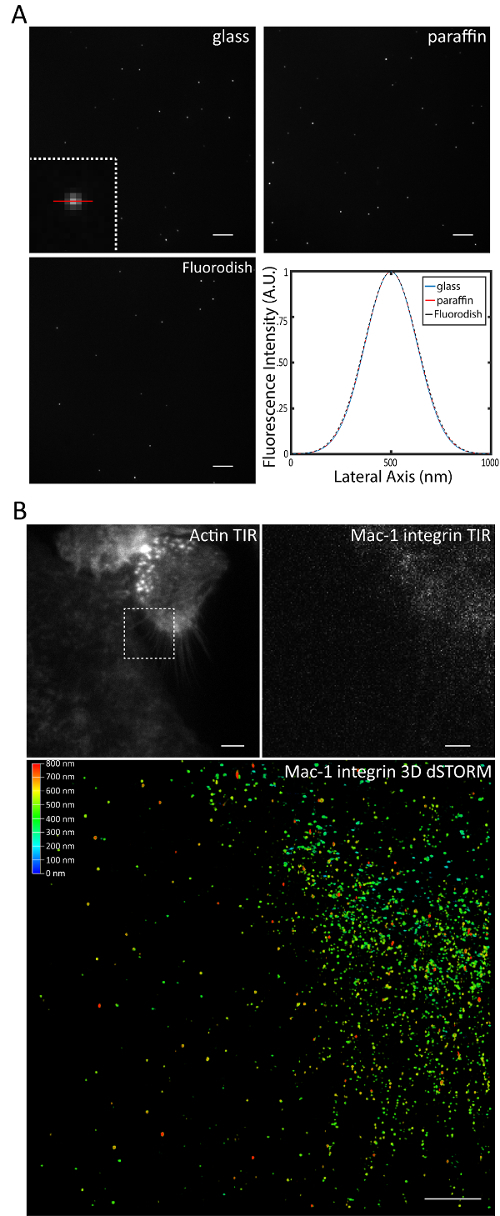
Although it is unlikely that a 10-nm layer of adsorbed hydrocarbons dramatically affects resolution, it was nevertheless important to quantitatively assess the potential difference in resolution among the various surfaces. Resolution is typically defined by the Rayleigh criterion. An alternative metric which is often used to approximate resolution is full-width at half maximum (FWHM) of structures smaller than the diffraction limit of light. Importantly, there is no apparent difference in FWHM of 100 nm fluorescent beads among the various surfaces (Figure 3A), suggesting that the characteristics of glass required for fluorescent imaging were sufficiently preserved. Furthermore, we were able to generate an evanescent field and perform 3D direct stochastic optical reconstruction microscopy of the macrophages applied to surfaces containing long-chain hydrocarbons (Figure 3B). A variety of live-imaging techniques are feasible when macrophages are applied to the surfaces (Figure 4, Supplemental Video 1, Supplemental Video 2).
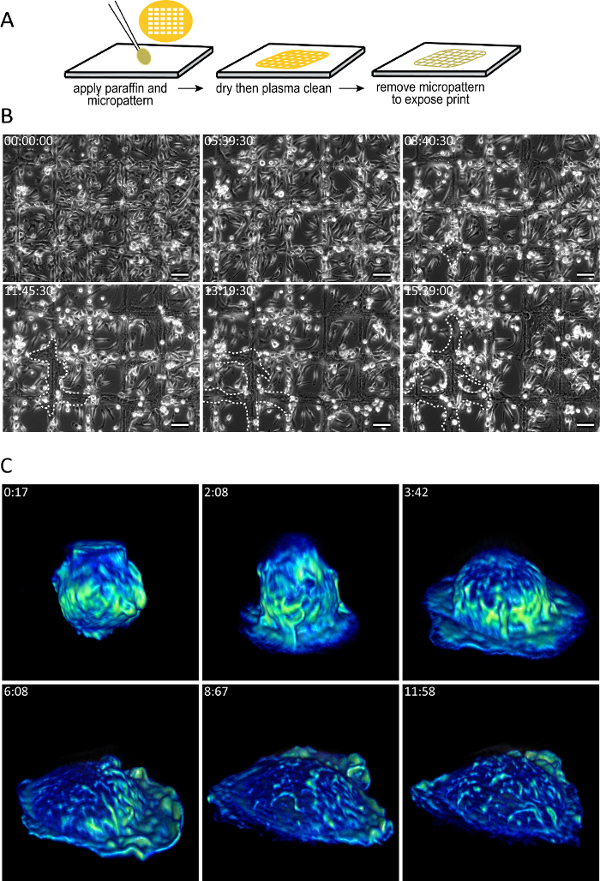
The advantage of each surface is described in Table 1. Note that the majority of advanced imaging techniques that require high NA oil immersion objectives are incompatible with most fusogenic plastic surfaces since they are thick, and have a degree of autofluorescence. It should be noted that surfaces adsorbed with long-chain hydrocarbons enable a large number of imaging techniques including12 PALM/dSTORM/GSDIM13,14,15,16,17, DIC, and others18,19,20,21 (Table 1). Furthermore, not only are a large number of imaging techniques possible (Table 1), but the use of a micropattern enables a high degree of spatiotemporal control over MGC formation (Figure 4B; Supplemental Video 1).
Figure 1: Paraffin adsorbed surfaces have a degree of nanoscale surface topography. AFM scans (5 x 5 µm) of glass surfaces show no obvious topographical features whereas paraffin-adsorbed surfaces contain arrays of material decorating the surface. The intensity scale on the right side of each micrograph shows height along the z-axis. The scale bars are 500 nm. Please click here to view a larger version of this figure.
Figure 2: Surfaces adsorbed with long-chain hydrocarbons promote macrophage fusion on cover glass. (A) In the absence of IL-4, there appears to be very few multinucleated cells on clean glass surfaces as determined by Wright's stain. (B) After 24 h in the presence of IL-4, surfaces adsorbed with solvent used to solubilize long chain hydrocarbons (e.g., toluene or isopropanol) have few multinucleated cells. (C-E) In contrast, surfaces adsorbed with long-chain hydrocarbons promote fusion and MGC formation. Paraffin adsorption results in the highest degree of IL-4 induced fusion. MGCs are outlined in black. (F) The degree of MGC formation on permanox plastic is shown for comparison. In each image, light blue shows nuclei and the cytoplasm appears purple. The scale bars are 100 µm. Please click here to view a larger version of this figure.
Figure 3: Surfaces adsorbed with long-chain hydrocarbons do not dramatically affect resolution. (A) Comparison of 100 nm fluorescent beads illuminated with Total Internal Reflection Fluorescence (TIRF) microscopy. The inset in the upper left panel is a magnified view a single fluorescent bead. The red line was superimposed to show an example line profile used to calculate FWHM. No apparent difference in FWHM of 100 nm beads was observed. The scale bars are 5 µm. (B) Comparison of the distribution of Mac-1 integrin as assessed by TIRF and 3D direct stochastic optical reconstruction microscopy. Macrophages were applied to an oleamide-containing surface and incubated with IL-4 for 24 h. The white inset in B highlights the region imaged in subsequent micrographs. The scale bars are 5 µm for the TIRF images and 2 µm for 3D STORM. Please click here to view a larger version of this figure.
Figure 4: Surfaces adsorbed with long-chain hydrocarbons enable time-resolved views of macrophage fusion and spreading. (A) The diagram shows the procedure used to generate a micropattern. (B) Micropatterned surfaces impart spatiotemporal control of macrophage fusion. Several hours after the application of IL-4, the macrophages were imaged with phase contrast. Note the migration of cells from the interior of the grid onto the micropattern. Fusion was observed only on the micropattern. The white dashes outline the expansion of a MGC. Time is shown in the upper left corner of each micrograph as hh:mm:ss. The scale bars are 50 µm. (C) Lattice Light Sheet microscopy20 reveals high spatiotemporal dynamics of a macrophage spreading on a paraffin-adsorbed surface. Color encodes the intensity distribution (yellow is most intense) of eGFP-LifeAct. Time is shown in the upper right corner of each micrograph as mm:ss. Please click here to view a larger version of this figure.
| Surface: | ||||||
| Technique | Paraffin | Oleamide | Petrolatum | MP paraffin | Permanox | Clean glass |
| phase contrast | √ | √ | √ | √ | √* | √ |
| brightfield | √ | √ | √ | √ | √* | √ |
| DIC | √ | √ | N/T | √ | x | √ |
| epifluorescence | √ | √ | √ | √ | √* | √ |
| confocal | √ | √ | √ | √ | √* | √ |
| TIRF | √ | √ | N/T | x | x | √ |
| PALM/dSTORM/GSDIM | √ | √ | N/T | x | x | √ |
| SIM | N/T | N/T | N/T | N/T | N/T | N/T |
| STED | N/T | √ | N/T | N/T | N/T | √ |
| LLSM | √ | N/T | N/T | √ | N/T | √ |
| Fusion Index | intermediate | intermediate | intermediate | high | intermediate | low |
| Fusion location | random | random | random | defined | random | random |
| MP-micropattern | ||||||
| DIC - differential interference contrast | ||||||
| TIRF - total internal reflection fluorescence | ||||||
| PALM - photoactivated localization microscopy | ||||||
| dSTORM - direct stochastic optical reconstruction microscopy | ||||||
| GSDIM - ground state depletion followed by individual molecule return | ||||||
| SIM - structured illumination | ||||||
| STED - stimulated emission depletion | ||||||
| LLSM - lattice light sheet microscopy | ||||||
| √ - compatible | ||||||
| x - incompatible | ||||||
| √* - possible only with LWD, low magnification, or immersion objectives | ||||||
| N/T - not tested |
Table 1: Potential uses of each surface. Note that the plastic surfaces preclude the use of most imaging techniques.
 Supplemental Video 1: Time-resolved phase contrast view of MGC formation. Note the apparent synchronicity of the MGC formation. Time is shown in hh:mm:ss in the upper left corner. The scale bar is 50 µm. Please click here to download this video.
Supplemental Video 1: Time-resolved phase contrast view of MGC formation. Note the apparent synchronicity of the MGC formation. Time is shown in hh:mm:ss in the upper left corner. The scale bar is 50 µm. Please click here to download this video.
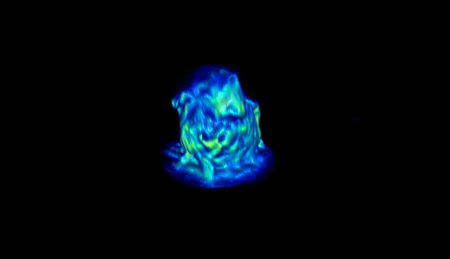 Supplemental Video 2: Lattice Light Sheet microscopy
20
of a thioglycollate-elicited eGFP-LifeAct macrophage spreading on a paraffin adsorbed surface.
Please click here to download this video.
Supplemental Video 2: Lattice Light Sheet microscopy
20
of a thioglycollate-elicited eGFP-LifeAct macrophage spreading on a paraffin adsorbed surface.
Please click here to download this video.
Discussion
The need to identify and subsequently develop optical-quality glass surfaces that promote macrophage fusion stemmed from the fact that until recently no published study directly visualized macrophage fusion in the context of living specimens3,4. This is due to the fact that fusogenic plastic surfaces that are commonly used require LWD objectives and are largely limited to phase contrast optics. These barriers were overcome by engineering an optical-quality glass surface that not only promoted extraordinary rates of macrophage fusion, but imparted a surprising degree of spatial control over the formation of MGCs.
Although this technology enables the use of advanced imaging techniques to monitor macrophage fusion, it does not come without limitation. Although the polished surfaces adsorbed with long-chain hydrocarbons (e.g., oleamide, paraffin wax, etc.) produce extraordinary rates of macrophage fusion compared to fusogenic plastic surfaces, fusion remains a spatially stochastic process (Figure 2). This limitation precludes the use of high magnification objectives to visualize fusion with living specimens since it is impossible to predict where fusion will occur. To overcome this limitation, we micropatterned paraffin to confine the fusion process to predetermined regions (Figure 4; Supplemental Video 1). Spatially confining macrophage fusion to the micropattern enabled us to predict where fusion would occur and therefore utilize higher magnification objectives to study this highly animate event.
What is the significance of using an optical-quality surface that imparts spatial control of macrophage fusion? First, since these surfaces promote high rates of fusion, it is possible to monitor fusion from living specimens within a reasonable timeframe. This is essential in the case of fluorescence microscopy since increased exposure of cells to light leads to photobleaching and phototoxicity. Second, these surfaces enable advanced imaging techniques based on the fluorescence or polarization to be employed (Figure 4; Table 1; Supplemental Video 1; Supplemental Video 2). Finally, the spatiotemporal control afforded by the micropatterned surfaces described in this protocol should expedite studies designed to identify the cellular and subcellular mechanisms that govern macrophage fusion.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We wish to thank members of the Ugarova laboratory and investigators in the Center for Metabolic and Vascular Biology for helpful discussion of this work. James Faust wishes to express his gratitude to the instructors at the European Molecular Biology Laboratory Super Resolution Microscopy course in 2015. We wish to thank Satya Khuon at Janelia for help with sample preparation for LLSM. During the review and filming portions of this work James Faust was supported by a T32 Fellowship (5T32DK007569-28). The Lattice Light Sheet component of this work was supported by HHMI and the Betty and Gordon Moore Foundation. T.U. is funded by NIH grant HL63199.
References
- McNally AK, Anderson JM. Interleukin-4 induces foreign body giant cells from human monocytes/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am J Pathol. 1995;147(5):1487. [PMC free article] [PubMed] [Google Scholar]
- Helming L, Gordon S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009;19(10):514–522. doi: 10.1016/j.tcb.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Podolnikova NP, Kushchayeva YS, Wu Y, Faust J, Ugarova TP. The Role of Integrins α M β 2 (Mac-1, CD11b/CD18) and α D β 2 (CD11d/CD18) in Macrophage Fusion. Am J Pathol. 2016;186(8):2105–2116. doi: 10.1016/j.ajpath.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust JJ, Christenson W, Doudrick K, Ros R, Ugarova TP. Development of fusogenic glass surfaces that impart spatiotemporal control over macrophage fusion: Direct visualization of multinucleated giant cell formation. Biomaterials. 2017;128:160–171. doi: 10.1016/j.biomaterials.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenney CR, Anderson JM. Alkylsilane-modified surfaces: Inhibition of human macrophage adhesion and foreign body giant cell formation. J Biomed Mater Res. 1999;46(1):11–21. doi: 10.1002/(sici)1097-4636(199907)46:1<11::aid-jbm2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Vignery A. Methods to fuse macrophages in vitro. Cell Fusion: Overviews Methods. 2008. pp. 383–395. [DOI] [PubMed]
- Zhao Q, et al. Foreign-body giant cells and polyurethane biostability: In vivo correlation of cell adhesion and surface cracking. J Biomed Mater Res. 1991;25(2):177–183. doi: 10.1002/jbm.820250205. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20(2):86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenney CR, Anderson JM. Effects of surface-coupled polyethylene oxide on human macrophage adhesion and foreign body giant cell formation in vitro. J Biomed Mater Res. 1999;44(2):206–216. doi: 10.1002/(sici)1097-4636(199902)44:2<206::aid-jbm11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- DeFife KM, Colton E, Nakayama Y, Matsuda T, Anderson JM. Spatial regulation and surface chemistry control of monocyte/macrophage adhesion and foreign body giant cell formation by photochemically micropatterned surfaces. J Biomed Mater Res. 1999;45(2):148–154. doi: 10.1002/(sici)1097-4636(199905)45:2<148::aid-jbm10>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Jenney CR, DeFife KM, Colton E, Anderson JM. Human monocyte/macrophage adhesion, macrophage motility, and IL-4-induced foreign body giant cell formation on silane-modified surfaces in vitro. J Biomed Mater Res. 1998;41(2):171–184. doi: 10.1002/(sici)1097-4636(199808)41:2<171::aid-jbm1>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19(11):780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- van de Linde S, et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protocols. 2011;6(7):991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- Fölling J, et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5(11):943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- Heilemann M, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Ange Chem Int Edit. 2008;47(33):6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Betzig E. Proposed method for molecular optical imaging. Optics Letters. 1995;20(3):237–239. doi: 10.1364/ol.20.000237. [DOI] [PubMed] [Google Scholar]
- Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Shtengel G, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. P Natl Acad Sci U S A. 2009;106(9):3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G, et al. Imaging cellular ultrastructure by PALM, iPALM, and correlative iPALM-EM. Method Cell Biol. 2013;123:273–294. doi: 10.1016/B978-0-12-420138-5.00015-X. [DOI] [PubMed] [Google Scholar]
- Chen BC, et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science. 2014;346(6208):1257998. doi: 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchon TA, et al. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat Methods. 2011;8(5):417–423. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]