

Deubiquitinase family proteins are broadly involved in modulating antiviral response through dynamic and diverse mechanisms.
Abstract
The innate immune response conferred by type I interferons is essential for host defense against viral infection but needs to be tightly controlled to avoid immunopathology. We performed a systematic functional screening by CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) knockout and overexpression to investigate the roles of the deubiquitinating enzyme (DUB) family in regulating antiviral immunity. We demonstrated that the expression of a large fraction of DUBs underwent complex temporal alteration, suggesting a dynamic program of feedback regulation. Moreover, we identified previously unrecognized roles of a subset of DUBs, including USP5, USP14, USP22, USP48, USP52, COPS5, and BRCC3, in inhibiting antiviral immunity at various levels. We explored an unexpected mechanism where multiple DUBs, such as USP5 and USP22, form diverse signalosomes with E3 ligases or DUBs to alter the substrates’ ubiquitination state instead of directly cleaving the ubiquitin chains on substrates via their protease activity. Altogether, our study has revealed a panoramic view of the broad and dynamic involvement of DUB family proteins in regulating antiviral responses.
INTRODUCTION
The innate sensing of and responses to microbial threats are mediated by pattern-recognition receptors (PRRs). Upon the detection of virus-specific pathogen-associated molecular patterns, effective immune responses against invading RNA and DNA viruses depend on the induction of type I interferons (IFNs) (1, 2). Viral RNA and DNA can be recognized by a variety of PRRs, including Toll-like receptors (TLRs), retinoic acid–inducible gene-I (RIG-I)–like receptors (RLRs), NOD-like receptors (NLRs), and several additional DNA sensors (3–6). Many TLRs such as TLR3 and TLR7/9 in an endosome-associated compartment trigger a signaling pathway mediated by TRIF (TIR-domain-containing adapter-inducing interferon-β) or MyD88 (myeloid differentiation primary response 88) (3), whereas RLRs, including the RNA helicase RIG-I, MDA5 (melanoma differentiation-associated protein 5), and LGP2 (laboratory of genetics and physiology 2), are responsible for sensing intracellular viral RNAs (7). Upon RNA recognition, RLRs mediate the recruitment of the mitochondrial signaling adaptor MAVS (mitochondrial antiviral signaling protein) to activate downstream interferon regulatory factor 3/7 (IRF3/7) and nuclear factor κB (NF-κB) signaling pathways (8). Recent studies have shown that a number of viral DNA sensors, including DEAD-box helicase 41 (9), IFI16 (interferon gamma inducible protein 16) (10), and cyclic GMP-AMP synthase (cGAS) (11), can initiate type I IFN production through another membrane-associated adaptor, STING (stimulator of interferon genes). The key adaptors (MAVS and STING) of both RNA and DNA sensors need TANK (TRAF family member-associated NF-κB activator)–binding kinase 1 (TBK1) to activate the downstream transcription factors IRF3/7 and NF-κB, which lead to the transcriptional activation of type I IFNs (12). Although type I IFN is critically required for antiviral immunity, if unchecked, excessive production of type I IFN can lead to immunopathology. Thus, to ensure a balanced immune response that is effective against viruses but not self-injurious, the type I IFN signaling pathway is subjected to stringent regulation.
It has been well documented that posttranslational modifications, especially ubiquitination, play critical roles in activation and regulation of type I IFN signaling (13). Because ubiquitination is a dynamic and reversible process, several specialized families of proteases and deubiquitinating enzymes (DUBs) cleave poly-ubiquitin chains, thereby reversing the process of ubiquitin ligases (14, 15). A few members of DUBs have been identified to play critical regulatory roles through their deubiquitinase activity in type I IFN pathways. For example, the tumor suppressor cylindromatosis (CYLD) negatively regulates type I IFN signaling by removing K63-linked ubiquitination of RIG-I (16). A20, another well-elucidated DUB, negatively regulates virus-induced type I IFN activation by cleaving K63-linked poly-ubiquitin chains from TBK1 and IRF7 (17, 18). MYSM1 (Myb-like, SWIRM and MPN domain-containing protein 1) impairs antiviral signaling activation by deubiquitinating TNF receptor–associated factor 3 (TRAF3) and TRAF6 (19). Ubiquitin-specific protease 3 and 21 (USP3 and USP21, respectively) are also involved in the negative regulation of RIG-I–mediated type I IFN signaling through their deubiquitinase activity (20, 21).
Despite the importance of several reported DUBs, the functions of a large number of other DUBs during antiviral innate immune responses remain unknown, and a systematic inquiry of their involvement in type I IFN signaling has not been carried out. Here, we perform a CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9)–based functional screening of DUBs that reveals the broad and diverse roles of DUBs in the activation and regulation of type I IFN pathways. On the basis of the ubiquitin-modulating functions of DUBs, we demonstrate six different modes of action of DUBs in type I IFN regulation, two of which involve novel mechanisms, completely independent of their catalytic activities. Altogether, our findings have revealed previously unidentified roles of a large portion of DUBs in controlling antiviral signaling and provide new insights into molecular mechanisms used by DUBs in controlling ubiquitin editing of their target proteins.
RESULTS
DUB proteins are differentially expressed upon virus infection and IFN stimulation
The human genome encodes approximately 100 putative DUBs, which can be grouped into at least five subfamilies: the ubiquitin-specific proteases/ubiquitin-specific processing proteases (UBPs), the ubiquitin C-terminal hydrolases (UCHs), the ovarian tumor proteases (OTUs), the Josephin or Machado-Joseph disease protein domain proteases (MJDs), and the Jab1/MPN domain-associated metalloisopeptidase (JAMM) domain proteins (14). To have a better and more comprehensive understanding of the function of DUB proteins, we classified each subfamily into several subgroups (Fig. 1A), which were based on the composition in relation to the DUB catalytic motif. Furthermore, we determined whether individual DUB genes encoded isoforms that lack a functional domain using SpliceMiner (22). The analysis reveals that more than half of all DUBs (67%) have more than one splice variant (Fig. 1B). Fifty-two percent of the DUBs have splicing variants that lacked a DUB catalytic domain, and 24% of the DUBs have isoforms with an incomplete interaction domain, such as the ubiquitin-binding domain or the zinc finger domain, which may perform as negative regulators to compete with the given full-length DUB proteins.
Fig. 1. A large number of DUBs are regulated by virus-induced type I IFN signaling at the transcriptional level.
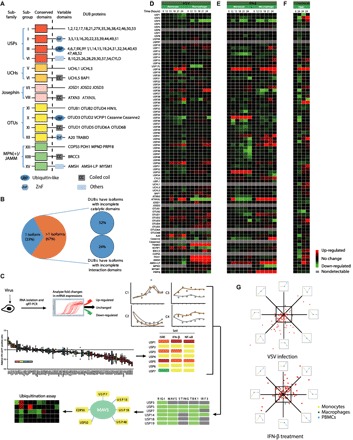
(A) Domain organization of DUB subfamilies [USPs, UCHs, Josephins, OTUs, and MPN(+)/JAMMs]. (B) DUB splice variants were mapped according to the UniProt database. Isoforms with incomplete catalytic domains or interaction domains were analyzed. (C) Schematic diagram illustrating the screening approach. (D) qRT-PCR analysis of the mRNA level of individual DUBs in THP-1 monocytes (left, lanes 1 to 6) or THP-1–derived macrophages (right, lanes 7 to 12) with VSV (MOI, 0.01) infection for the indicated time points. Data were normalized to the mRNA levels in untreated cells (0 hours) and were presented in a heat map. (E) qRT-PCR analysis of mRNA levels of individual DUBs in THP-1 monocytes (left, lanes 1 to 6) or THP-1–derived macrophages (right, lanes 7 to 12) treated with IFN-β (1000 U/ml) for the indicated time points. Data were normalized to the basal mRNA levels and were presented in a heat map. (F) qRT-PCR analysis of mRNA levels of the indicated DUBs in VSV-infected (MOI, 0.01) (lane 2), HSV-1–infected (MOI, 0.1) (lane 3), or IFN-β–treated (1000 U/ml) (lane 4) PBMCs. Data were normalized to the basal mRNA levels. (G) Distribution of different expression tendencies of DUBs between different cell types upon VSV infection or IFN-β stimulation. Each point represents the difference of a certain DUB expression among different cell types, plotted by the natural logarithm of expression difference ratio between THP-1 monocytes and THP-1–derived macrophages on the horizontal axis and the ratio between PBMCs and THP-1–derived macrophages on the vertical axis. Data are representative of three independent experiments (D to F).
Because several recent studies have reported the great potential of DUB proteins in regulating virus-induced IFN pathway activation, the following strategies were designed to obtain a global view of their regulatory functions. First, we checked the dynamic expression patterns of DUBs after virus infection and IFN activation. Second, 170 small guiding RNAs (sgRNAs) targeting 85 individual DUB family genes were designed and constructed into the lentiviral vectors expressing Cas9. Human embryonic kidney–293T (HEK293T) cells were infected with lentiviruses delivering Cas9 and sgRNA to generate sg_DUB 293T cells. The knockdown efficiency in sg_DUB 293T cells was confirmed by real-time quantitative polymerase chain reaction (qRT-PCR); the sgRNAs with better knockdown efficiency were used for the subsequent experiments. All sg_DUB 293T cells were tested for their ability to restrict vesicular stomatitis virus (VSV) infection, as well as their ability to affect the IFN-β, NF-κB, and ISRE (IFN-stimulated response element) activation in reporter assays using different stimulations. Next, protein-protein interaction assays and in vivo protein ubiquitination assays were carried out to further investigate the function of the candidate DUB proteins (Fig. 1C).
Because THP-1 monocytes or THP-1–derived macrophages, resembling innate immune cells, play a critical role in host immune responses to viral infection, we used THP-1 cells (a human monocyte cell line) to examine DUB engagements during antiviral responses (23, 24). We systematically analyzed the expression of 85 DUB family members in monocyte-like cells (THP-1) and THP-1–derived macrophages after VSV infection or IFN-β treatment. As expected, a portion of the DUBs were differentially expressed after viral infection or IFN treatment. Screening data were translated into a heat map representing the change in relative expression of DUBs at different time points (0, 12, 15, 18, 21, and 24 hours) (Fig. 1, D and E, and table S1). The expression patterns of DUBs can be divided into six different main clusters (C1 to C6) (table S2). During VSV infection, nine DUB genes (such as A20 and USP18) were up-regulated in both THP-1 and macrophages (C1), whereas two DUB genes (USP35 and Cezanne) were down-regulated in these cells. Thirty-two DUB genes remained unchanged throughout the experiment (C3). For the IFN-β treatment, eight DUB genes were up-regulated, and three DUB genes were down-regulated in both cell types. On the other hand, the rest of the DUBs (C4 to C6, 50% after VSV infection and 53% after IFN activation) were differentially regulated between THP-1 monocytes and macrophages (table S2). Similarly, we also analyzed the expression of DUB genes in human peripheral blood mononuclear cells (PBMCs) by VSV and HSV-1 (herpes simplex virus 1) infection or IFN-β treatment (Fig. 1F and table S1). More than half of the DUB genes in PBMCs (62.5% after VSV infection and 58.8% after IFN activation) followed a similar expression pattern to THP-1 monocytes and macrophages (Fig. 1G). Together, the different expression patterns of DUBs after virus infection and IFN treatment reveal that DUB proteins might have great potential in regulating virus-induced IFN pathway by forming feedback loops.
A large portion of DUBs modulate the type I IFN-mediated antiviral response
To further investigate the roles of DUBs in antiviral immunity, we screened the antiviral functions of a panel of DUBs, using CRISPR/Cas9-mediated gene-editing technology. One hundred seventy sgRNAs targeting 85 individual DUB family genes were designed and constructed into the lentiviral vectors expressing Cas9. HEK293T cells, which have been widely used in functional studies for DUB genes and various innate immune responses (15, 23, 24), were infected with lentiviruses delivering Cas9 and sgRNA to generate sg_DUB 293T cells. The knockdown efficiency in sg_DUB 293T cells was confirmed by qRT-PCR; the sgRNAs with better knockdown efficiency were used for the succeeding experiments (fig. S1). The sg_DUB cells were infected with VSV–green fluorescent protein (eGFP) and went through fluorescence-activated cell sorting (FACS) analysis. The percentage of infected (GFP-positive) cells was determined by fluorescence microscopy or flow cytometry (Fig. 2A and fig. S2A). We confirmed the feasibility of the system by testing several molecules whose function in antiviral immunity has been elucidated, including USP19 (25), USP21 (21), and TBK1 (12). The replication of VSV-eGFP was restricted in sg_USP19 and sg_USP21 cells, but was augmented in sg_TBK1 cells as expected (fig. S2, B and C). By screening 85 DUBs, we found that a large proportion of DUB-targeted sgRNAs can modulate virus replication. The relative infection ratios of sg_DUBs cells in accordance with nontargeting control sg_NT cells are displayed from high to low, and range from 30 to 141% (fig. S2, D and E). Another sgRNA targeting mCherry was also used as a negative control to ensure the feasibility of the system. To test the possibility that DUBs govern antiviral immunity through the modulation of the type I IFN pathway, we determined the activity of IFN-β–, NF-κB–, or ISRE-luc in the presence of limiting amounts of RIG-I [two caspase activation and recruitment domains (2CARD)] or cGAS/STING or after SeV (Sendai virus) infection in all sg_DUB 293T cells, respectively (Fig. 2A). The degree of increase in induction of sg_NT samples indicated the extent of reporter activation by transfection or infection (fig. S2F). All data from each subset of the screens were normalized by sg_NT samples (as 1) and clustered into a heat map (fig. S2, G to M). Overall, more than 30 sg_DUBs cells (36% of total) exhibited increased activation of two or more reporters upon stimulation of RIG-I (2CARD) or after SeV infection. Similarly, 32 sg_DUB cells (38% of total) exhibited elevated activation of two or more reporters in the presence of cGAS and STING.
Fig. 2. Identification of DUBs that modulate antiviral response.
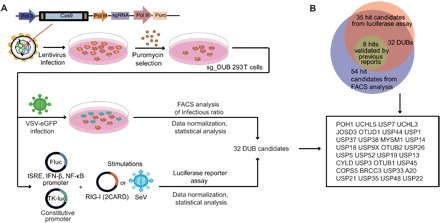
(A) Schematic overview of assay. 293T cells were transduced with lentiviruses expressing Cas9 and sgRNAs targeting individual DUBs. After puromycin selection, sg_DUB 293T cells were infected with VSV-eGFP (MOI, 0.01) for 18 hours. VSV-eGFP positive ratios were determined by FACS analysis, and the infection ratios were normalized to cells transduced with nontargeting control (NT) sgRNA. On the other hand, sg_DUB 293T cells were transfected with ISRE, IFN-β, or NF-κB luciferase reporters and then transfected with RIG-I (2CARD) or infected with SeV. The luciferase reporter induction was measured and normalized to NT sgRNA-expressing cells. (B) Demonstration of the hit candidate selection by Venn diagrams.
We next identified the hit candidates through two criteria: (i) The P value of the difference between the sg_NT and the sg_DUB group is less than 0.05, and (ii) relative VSV-eGFP infection ratio is above 110% or below 90% in FACS analysis, and at least two reporters’ relative induction of the sg_DUB group is above average or below 50% of the average in a luciferase reporter assay. These candidate-selection methods were followed by the analytical strategy used in a similar study (24). Fifty-four hit candidates were identified by analyzing VSV-eGFP infection ratios, and the reporter induction assay yielded 35 hit candidates. The 32 genes identified both by infection ratio and reporter induction assays were selected as DUB candidates for further validations (Fig. 2B) and most of them were negative regulators. USP3, USP19, USP21, USP38, OTUB1/2 (OTU deubiquitinase, ubiquitin aldehyde binding 1/2), A20, and MYSM1, which were negative regulators previously reported by us and other groups (17, 19–21, 25–27), were identified as hit candidates in the screen, thus validating the experimental approach.
To further validate our screening approach, we subsequently transfected candidate functional DUBs along with several previously characterized regulatory DUBs into 293T cells and then infected them with SeV to determine the activation of three different reporters (fig. S3, A and B). As expected, enforced expression of most of the tested DUBs (such as USP3, USP5, USP13, USP14, USP18, USP22, and USP38) inhibited the activation of IFN-β, NF-κB, and ISRE reporters upon SeV infection. However, there was one exception where overexpression of USP7 also inhibited signaling activation. We assumed that USP7 may exert different functions with different abundances and distributions (28, 29).
To further confirm the function of identified DUBs in type I IFN production, we transfected 293T cells with RIG-I (2CARD) plasmids, together with individual DUB plasmids, and transferred the supernatant to Vero cells 36 hours after transfection. The Vero cells were deficient in IFN production themselves, so any antiviral effect observed in Vero cells originated from the cytokines in the supernatant that we transferred. Next, the Vero cells were infected with VSV-eGFP and followed by flow cytometry analysis (fig. S3C). As expected, overexpression of USP52, USP38, CYLD, USP22, OTUB2, USP25, USP21, USP48, USP5, USP19, and USP14 inhibited the production of antiviral cytokines, resulting in higher levels of infection of Vero cells. On the contrary, DUBs such as UCHL3 (ubiquitin C-terminal hydrolase L3), UCHL5, and POH1 [PSMD14 (proteasome 26S subunit, non-ATPase 14)] induced an antiviral effect conferred by IFNs during VSV-eGFP infection (fig. S3, D and E). Altogether, these results suggest that a large portion of DUBs function as regulators in RLR-mediated and DNA sensor–mediated type I IFN signaling.
To further validate our screening results from the CRISPR/Cas9 system, we used several DUB-specific small interfering RNA (siRNA) to knock down the expression of DUBs, which showed inhibitory function in antiviral immunity. All siRNA efficiently inhibited the expressions of endogenous indicated DUBs in 293T cells (fig. S3F). We next assessed the effects of DUB knockdown by siRNA on VSV-eGFP replication. Knockdown of the indicated DUBs resulted in reduced viral replication (fig. S3, G and H). Similarly, we assessed the effects of DUB knockdown by siRNA on IFN-β induction and obtained similar results to those found in the CRISPR/Cas9 system (fig. S3I).
DUBs function at different levels of the type I IFN signaling pathway
We next sought to identify at which level in the type I IFN signaling pathway do individual DUBs function as regulators. To this end, we transfected plasmids of DUB candidates in the presence of the established signaling molecules RIG-I (2CARD), MAVS, cGAS/STING, TBK1, or IRF3 (5D), which function sequentially in the RLR-mediated or DNA sensor–mediated type I IFN pathway, and measured the IFN-β luciferase activity. We found that RIG-I (2CARD)–induced IFN-β induction was down-regulated by 16 DUBs and up-regulated by 4 DUBs (Fig. 3A and fig. S4A). The observed functions of these DUBs here were consistent with our previous screening results. To get an intuitional view of where individual DUBs function, the results were summarized in a heat map (Fig. 3B). The last molecule in the pathway at which each individual DUB still inhibits or enhances the signaling provides an indication of the level at which each DUB functions. USP3 only inhibited RIG-I (2CARD)–induced signaling activation, which was consistent with our previous study (20). Four DUBs (USP5, USP14, USP22, and POH1) acted at relative downstream of the pathway to affect IRF3-induced signaling. Meanwhile, a lot of these DUBs exerted their function up to the TBK1 level. Unexpected “gaps” were found in the pathway mapping for USP14 and USP18. The inhibition of TBK1-activated signaling but not cGAS/STING-induced activation by USP14 and USP18 might be attributed to their manifold roles in the signaling pathway. We and others also showed that USP14 inhibited cGAS degradation, and USP18 promoted the stabilization of STING, which promoted the cGAS-STING signaling (30, 31). In such cases, the heterogeneity of these genes that function at different levels of signaling pathway may be neutralized.
Fig. 3. DUB proteins act at different levels of type I IFN pathways.
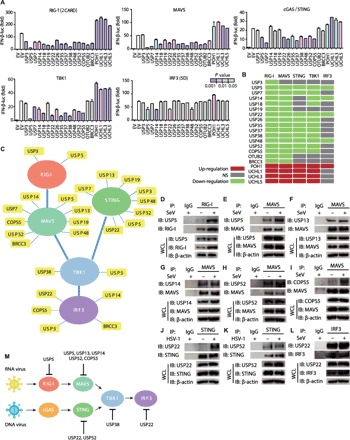
(A) Luciferase activity in 293T cells transfected with an IFN-β luciferase reporter, together with vectors for RIG-I (2CARD), MAVS, cGAS/STING, TBK1, and IRF3 (5D), along with an empty vector or with expression vectors for the indicated DUBs. Columns are colored by significance relative to EV (empty vector) control (t test); color scale is given in the figure. (B) Heat map summary of (A). Negative regulations are marked green, positive regulations are marked red, and nonsignificant (NS) changes are marked gray. (C) Summary of interactions between DUBs and key molecules in type I IFN signaling pathways. (D to I) Coimmunoprecipitation and immunoassay of extracts of A549 cells infected with or without SeV (MOI, 0.1) for 12 hours with the indicated antibodies. IgG, immunoglobulin G. (J and K) Coimmunoprecipitation and immunoassay of extracts of A549 cells infected with or without HSV-1 (MOI, 0.1) for 12 hours with the indicated antibodies. (L) Coimmunoprecipitation and immunoassay of extracts of A549 cells infected with or without SeV (MOI, 0.1) for 12 hours with the indicated antibodies. (M) Summary of multilayered regulations of DUBs, according to the combination of inhibitory function levels and endogenous protein interactions. Data are representative of three independent experiments [mean and SEM in (A)]. IP, immunoprecipitate; IB, immunoblot; WCL, whole cell lysate.
To further investigate the connections between DUBs and signaling molecules, we performed coimmunoprecipitation experiments to identify signaling molecules associated with DUBs. Extensive interactions between the adaptor proteins (MAVS and STING) and DUBs were observed. For example, USP13, USP19, and USP52 associated with both MAVS and STING (Fig. 3C and fig. S4, B to H). Besides, USP22 interacted not only with the adaptor STING but also with the transcription factor IRF3 and inhibited the signaling up to the IRF3 level. To have a better understanding of the dynamic interactions between DUBs and their target after viral infection, we examined the interaction of endogenous DUBs and the key molecules (Fig. 3, D to L). COPS5 (COP9 signalosome subunit 5) and USP22 associated with MAVS or STING only after viral infection. Meanwhile, SeV enhanced the interaction of USP5 and USP13 with MAVS. The multilayered regulations of DUBs, whose interactions were validated endogenously, from upstream to downstream, are summarized in Fig. 3M, including our previous finding that USP38 can interact with TBK1 (27). Collectively, these results demonstrate that DUBs regulate type I IFN signaling at different levels.
DUBs regulate the expression of antiviral genes upon viral infection
To better understand the roles of DUB candidates in antiviral responses, we generated A549 cells with diminished relevant DUB expression by expressing specific sgRNAs targeting individual DUBs. A549 is a lung epithelial cell line and is commonly used in the study of antiviral response (15, 23, 24). To ensure the efficient knockdown of individual DUBs by sgRNAs, primers were designed for individual DUBs, which crossed the supposed Cas9 cleave site near the PAM (protospacer adjacent motif) regions. The sgRNA targeting efficiency among different DUBs ranged from ~50 to 90% (fig. S5A). The cell lines were infected with VSV, SeV, or HSV-1. To analyze the replication of viruses, mRNAs of viruses were determined by qRT-PCR (figs. S4I and S5B). We observed higher levels of VSV, SeV, and HSV-1 mRNAs within cells expressing USP7, and POH1 targeted sgRNAs upon viral infection. The replication of VSV, SeV, and HSV-1 was restrained when we knocked down USP5, USP14, USP22, USP35, USP38, USP48, and USP52. However, OTUB1 and BRCC3 seemed to have no influence on HSV-1 replication, which suggested that they may target immune responses against RNA viruses. Consistently, the absence of USP7 and POH1 diminished IFN-β and IFIT2 gene expressions after viral infection, whereas USP5, USP14, USP22, USP35, USP38, USP48, and USP52 exerted opposite functions, which resulted in higher IFN-β and IFIT2 mRNA expression (figs. S4J and S5C). Overall, these data confirm the initial results from screening assays and strongly support the regulatory roles of DUBs in antiviral innate immune responses.
To further investigate the functions of these DUBs under physiological functions in primary immune cells, we deleted the relevant DUBs by the Cas9/sgRNA systems with high efficiency (37 to 67%) in human PBMCs from two independent donors (fig. S5D). Despite the inter-donor variations, decreased expression of USP5, USP14, USP22, USP38, USP48, USP52, COPS5, and BRCC3 consistently elevated IFN-β induction by at least 11% in both donors after SeV infection. The expression of two ISGs, IFIT2 and IFIT1, was also enhanced as expected (fig. S5E and fig. S5G). The effect of DUB gene targeting on the expression of pro-inflammatory cytokines after viral infection was also measured. In general, the induction of TNFα and IL-6 mRNAs was up-regulated, although some exceptions among two donors were observed (fig. S5F and fig. S5H). We further showed that knockdown of these DUBs in PBMCs also augmented the secretion of IFN-β and interleukin-6 (IL-6; fig. S5, I and J). The cell lysates of PBMCs were analyzed to ensure equal cell numbers in all samples and the knockout (KO) efficiency of individual DUB by the used sgRNAs at protein levels (fig. S5K). Overall, these results indicate that the majority of the tested DUBs are negative regulators of antiviral responses not only in cell lines but also in primary immune cells.
DUBs regulate the ubiquitination of the key molecules in the type I IFN signaling pathway
The above studies indicated a regulatory role for DUBs in type I IFN production. Because the general function of DUBs is to cleave the poly-ubiquitin chains (14), we wondered whether these regulatory DUBs can remove the poly-ubiquitin chains on the key proteins in type I IFN signaling pathways. Coimmunoprecipitation and immunoblot analysis were used to examine the regulatory functions of several potent DUBs on the ubiquitination of the key molecules, including RIG-I, MAVS, STING, TBK1, and IRF3, in type I IFN signaling. The ubiquitination levels of the target molecules were quantified and summarized in a heat map (Fig. 4A and fig. S6 A to G). The ubiquitination of RIG-I was down-regulated by USP3, which was consistent with our previous report (20). In addition, the ubiquitination of STING was impaired by USP5, USP13, and USP22. To our surprise, we also observed that several DUBs, including USP5 and USP13, can up-regulate the ubiquitination of the key molecules, such as RIG-I and MAVS. The presence of seven lysines (K6, K11, K27, K29, K33, K48, and K63) creates a large variety of poly-ubiquitin chains with different conformations, which then serve as signatures for substrates to distinct fates (13). We wondered whether DUBs affected specific linkages of ubiquitination that contribute to the total alteration. We next set out to examine the K6-, K11-, K27-, K29-, K33-, K48-, and K63-linked ubiquitin modifications of the indicated signaling molecules mediated by each DUB. The regulatory roles of these DUBs on the ubiquitination level of each key molecule with different linkages were analyzed and summarized (Fig. 4B and fig. S6, H to P). The ubiquitination level was reduced significantly (twofold) in 21 out of 315 samples while there were significant enhancements (twofold) in 21 out of 315 samples. Among them, USP5 increased K11- and K48-linked ubiquitination of RIG-I after SeV infection, which was in agreement with enhanced total ubiquitination of RIG-I. Meanwhile, the increasing K11- and K48-linked ubiquitination level may lead to the degradation of RIG-I, because a reduction in the abundance of RIG-I was observed (fig. S6H). Similarly, the enhanced K11-linked ubiquitination of MAVS caused by USP13 led to the increase of total ubiquitination. On the other hand, the reduction of K63-linked ubiquitination of MAVS and TBK1 was caused by BRCC3, which is well known as a K63-specific DUB (32). More DUBs (USP13, USP38, and COPS5) were considered to enhance one type of ubiquitination while inhibiting other types of ubiquitination on a single molecule. To validate our findings, we next assessed the effect of DUB knockdown on the endogenous ubiquitination of the targets and obtained consistent results in our overexpression system (fig. S6 Q to Z). Together, these results demonstrate that DUBs can both accelerate and restrain the ubiquitination of the key proteins in type I IFN signaling pathways.
Fig. 4. DUBs regulate type I IFN signaling by altering ubiquitination in both catalytic activity–dependent and –independent manners.
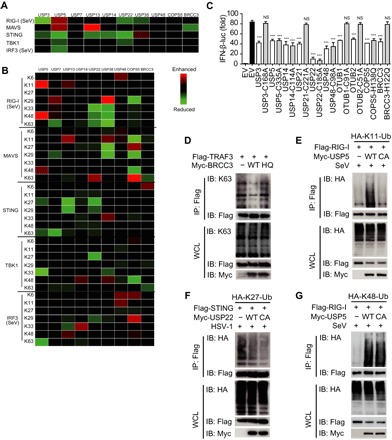
(A) Heat map summary of regulatory roles of DUBs on total ubiquitination of key molecules in the type I IFN signaling pathway. (B) Heat map summary of regulatory roles of DUBs on ubiquitination of key molecules with different linkages in the type I IFN signaling pathway. Data are obtained by band intensity analysis (IP-HA/IP-Flag) and are normalized to the control sample. (C) Luciferase activity in 293T cells transfected with an IFN-β luciferase reporter, together with vectors for RIG-I (2CARD), along with an empty vector or with expression vectors for the indicated WT DUB or catalytically inactive mutants. (D) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-TRAF3, Myc-BRCC3, and Myc-BRCC3-H122Q with the indicated antibodies. (E) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-RIG-I and HA-K11-Ub together with Myc-USP5 or Myc-USP5-C335A followed by SeV infection with the indicated antibodies. (F) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-STING and HA-K27-Ub together with Myc-USP22 or Myc-USP22-C185A followed by HSV-1 infection with the indicated antibodies. (G) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-RIG-I and HA-K48-Ub together with Myc-USP5 or Myc-USP5-C335A with the indicated antibodies. Data are representative of three independent experiments [mean and SEM in (C)]. NS, nonsignificant (P > 0.05); ***P < 0.001.
DUBs regulate type I IFN signaling in both catalytic activity–dependent and –independent manners
Because the enhancement of ubiquitination caused by DUBs was observed, we suspect that the role of DUBs in type I IFN signaling might be beyond their protease activity. To address this question, we generated catalytically inactive mutants of DUB candidates and found that the mutants of USP3, USP21, OTUB1/2, and BRCC3 (BRCA1/BRCA2-containing complex subunit 3) abolished their inhibitory functions, whereas the mutants of several other DUBs still inhibit IFN-β activation (Fig. 4C). To further confirm the functions of DUB protease activity, we also detected the influences of DUB mutations on modifying the ubiquitin chains of their substrate. The enzymatically inactive mutant of BRCC3 (BRCC3-H122Q) abolished its function on cleaving the K63-linked ubiquitination of TRAF3 (Fig. 4D), which suggested that the enzyme activity of BRCC3 was required for this cleavage process. The enzymatically inactive mutant of USP5 (USP5-C335A) failed to enhance the K11-linked ubiquitination chains on RIG-I after SeV infection (Fig. 4E), which indicated that the enhancement of ubiquitination was also dependent on its enzyme activity of DUBs in several cases. However, the detailed mechanism used by USP5 in enhancing K11-linked ubiquitin chains of RIG-I and the functions of K11-linked ubiquitination chains of RIG-I in the signaling pathway still need to be further elucidated. On the contrary, the enzyme activity of several candidate DUBs was not required to reduce ubiquitination on their target. For instance, both wild-type (WT) USP22 and its enzymatically inactive mutant (USP22-C185A) decreased the K27-linked ubiquitination on STING (Fig. 4F), which suggested that the catalytic activity of USP22 was not responsible for the cleavage of the K27-linked ubiquitination of STING. Moreover, both USP5 and USP5-C335A could increase the K48-linked poly-ubiquitin chains on RIG-I after SeV infection (Fig. 4G), which indicated that USP5 might enhance the ubiquitination on RIG-I independently of the enzyme activities. Together, our findings reveal the diverse mechanisms used by DUBs in regulating type I IFN signaling and modifying the ubiquitination chains on their targets, which were either dependent of their protease activity or not.
DUBs altered substrate ubiquitination through complex and dynamic mechanisms
It has been reported that the protease activity of several DUBs, such as USP3 and MYSM1 (19, 20), was critical in modulating the ubiquitination chains on their substrate and regulating IFN signaling. Our findings reveal that the catalytic activity of several DUBs is not necessary in controlling the type I IFN signaling pathway, which implies novel strategies used by DUBs. To further confirm this finding, we reconstituted Cas9-resistant WT DUBs or enzyme-deficient mutants that include at least two point mutations in the C terminus of a guide RNA (gRNA) recognized zone in sg_DUB 293T cells. We found that all the mutants of the candidate DUBs exerted the same function in regulating IFN-β induction as WT ones (Fig. 5A). To further explore how DUBs implement their function without their protease activity, USP22 and USP5 were selected because the catalytic activity of both was dispensable for the modification of the targets’ ubiquitination. To detect how USP22 reduced K27-linked ubiquitination of STING independent of its catalytic activity, we performed a coimmunoprecipitation screening for USP22. Coimmunoprecipitation and immunoblot analyses revealed that USP22 could interact with USP13, which could specifically cleave K27-linked ubiquitin chains on STING (33), and the interaction between USP22 and USP13 became stronger after HSV-1 stimulation (Fig. 5B). Moreover, USP22 significantly promoted the interaction between USP13 and STING (Fig. 5C). USP22 lost its function in sg_USP13 cells during HSV-1 infection. Knockdown of USP22 promoted IFN-β expression and, in turn, restricted HSV-1 replication in sg_NT cells but not in sg_USP13 cells, indicating the cooperation of USP22 and USP13 in response to viral infection (Fig. 5, D and E). To identify which domain of USP22 is responsible for STING deubiquitination, we generated two deletion mutants containing the ZnF-UBP domain (ZNF) or the USP catalytic domain (UCH) (Fig. 5F). Similar to WT USP22, both USP22-C185A and USP22-UCH facilitated the interaction between STING and USP13, but USP22-ZNF failed to do so (Fig. 5G). Consistently, USP22-ZNF could not reduce STING K27-linked ubiquitination, whereas USP22-C185A and USP22-UCH could still restrain the K27-linked ubiquitination of STING, compared with WT USP22 (Fig. 5H). These results indicate that USP22 might recruit USP13 to remove ubiquitination of STING, and the UCH domain of USP22 was necessary.
Fig. 5. USP22 removes K27-linked ubiquitination on STING through cooperation with USP13.
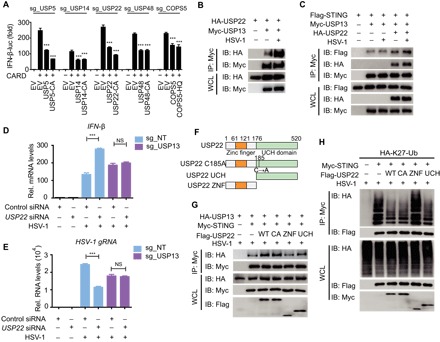
(A) Luciferase activity in indicated sg_DUB 293T cells transfected with an IFN-β luciferase reporter, together with vectors for RIG-I (2CARD), along with an empty vector or with expression vectors for the indicated WT DUB or catalytically inactive mutants. (B) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Myc-USP13 and HA-USP22 with or without HSV-1 infection with the indicated antibodies. (C) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-STING, Myc-USP13, and HA-USP22 with or without HSV-1 infection with the indicated antibodies. (D and E) sg_NT or sg_USP13 A549 cells were transfected with control siRNA or USP22 siRNA and then left uninfected of infected with HSV-1 for 18 hours. IFN-β mRNA levels (D) and HSV-1 (glycoprotein C mRNA) gRNA levels (E) were determined by qRT-PCR. Rel., relative. (F) Constructs of WT USP22, USP22 enzymatically inactive mutant (USP22-C185A), USP22 containing only the ZNF domain (USP22-ZNF), and USP22 containing only the UCH domain (USP22-UCH). (G) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Myc-STING, HA-USP13, and Flag-USP22-WT, USP22-C185A, USP22-ZNF, or USP22-UCH followed by HSV-1 infection with the indicated antibodies. (H) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-STING and HA-K27-Ub together with Myc-USP22, USP22-C185A, USP22-ZNF, or USP22-UCH followed by HSV-1 infection with the indicated antibodies. Data are representative of three independent experiments [mean and SEM in (A), (D), and (E)]. NS, nonsignificant (P > 0.05); ***P < 0.001.
We next investigated whether USP5 regulated IFN antiviral response by targeting RIG-I for degradation. We found that knockdown of USP5 led to higher protein levels of endogenous RIG-I after infection with VSV (fig. S7A). A slower degradation rate of the protein levels of RIG-I in USP5 knockdown cells was observed in the presence of the protein synthesis inhibitor cycloheximide (CHX) (Fig. 6A). To verify whether the function of USP5 depends on RIG-I, we generated RIG KO 293T cells and RIG-I-KO cells reconstituted with ectopically expressed RIG-I (RIG-I restored cells) as previously described (34) and checked the function of USP5 in RIG-I-KO 293T cells and RIG-I restored cells. Our data revealed that knockdown of USP5 enhanced IFN-β reporter activation and mRNA expression in WT and RIG-I restored cells but not in RIG-I-KO cells (fig. S7B and Fig. 6B). Consistently, knockdown of USP5 inhibited VSV replication in WT and RIG-I restored cells, whereas it failed to do so in RIG-I-KO cells (Fig. 6C and fig. S7, C and D). These results indicate that USP5 enhances VSV replication by mediating the degradation of RIG-I. We next found that USP5 could interact with STUB1 (STIP1 homology and U-box containing protein 1), one of the known E3 ligases of RIG-I (35), and SeV infection facilitated the interaction between USP5 and STUB1 (Fig. 6D). Moreover, USP5 promoted the interaction between RIG-I and STUB1 (Fig. 6E). To validate the role of STUB1 in the mechanism used by USP5, we also generated STUB1-deficient cells (sg_STUB1) (fig. S7E), and sg_STUB1-3# cells were used in the succeeding experiments. In sg_STUB1 293T cells, USP5 failed to enhance the K48-linked ubiquitination of RIG-I (fig. S7F) and subsequently abolished its function in promoting the degradation of RIG-I (Fig. 6F). Consistently, knockdown of USP5 failed to promote IFN-β reporter activation and mRNA expression (fig. S7G and Fig. 6G) as well as inhibit VSV replication (Fig. 6H and fig. S7, H and I) in sg_STUB1 cells. Together, these data suggest that USP5 inhibits IFN-β expression and supports VSV replication by recruiting STUB1 to degrade RIG-I.
Fig. 6. USP5 inhibits type I IFN signaling and enhances viral replication through the promotion of STUB1-mediated RIG-I degradation.
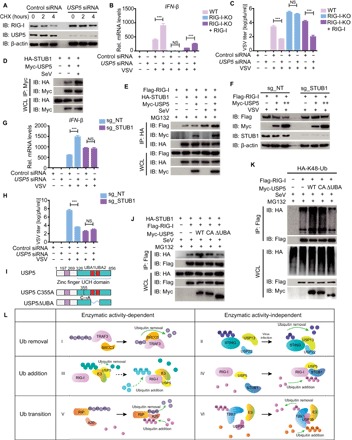
(A) Immunoblot analysis of extracts of A549 cells transfected with control siRNA or USP5 siRNA treated with CHX (100 μg/ml) for the indicated time after infection with VSV for 12 hours. (B) WT, RIG-I-KO 293T, or RIG-I-KO cells reconstituted with RIG-I were transfected with control siRNA or USP5 siRNA and then infected with VSV-eGFP for 18 hours. The mRNA level of IFN-β was determined by qRT-PCR. (C) Plaque titration of VSV-eGFP in the supernatant of the indicated cells transfected with control siRNA or USP5 siRNA followed by VSV-eGFP infection for 24 hours. pfu, plaque-forming units. (D) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Myc-USP5 and HA-STUB1 with or without SeV infection with the indicated antibodies. (E) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-RIG-I, Myc-USP5, and HA-STUB1 treated with MG132 and infected with SeV with the indicated antibodies. (F) Immunoassay of extracts of sg_NT or sg_STUB1 293T cells transfected with Flag-RIG-I and Myc-USP5 followed by VSV infection for 9 hours. (G) qRT-PCR analysis of IFN-β mRNA levels in sg_NT or sg_STUB1 293T cells transfected with control siRNA or USP5 siRNA followed by VSV-eGFP infection for 18 hours. (H) Plaque titration of VSV-eGFP in the supernatant of the indicated cells transfected with control siRNA or USP5 siRNA followed by VSV-eGFP infection for 24 hours. (I) Constructs of WT USP5, USP5 enzymatically inactive mutant (USP5-C335A), and USP5 without the UBA domain (USP5-ΔUBA). (J) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-RIG-I, HA-STUB1, and Myc-USP5-WT, USP5-C335A, or USP5-ΔUBA, followed by SeV infection and treatment with MG132 with the indicated antibodies. (K) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with Flag-RIG-I, HA-K48-Ub, and Myc-USP5-WT, USP5-C335A, or USP5-ΔUBA, followed by SeV infection and treatment with MG132 with the indicated antibodies. (L) The six working models of DUBs in modulating type I IFNs. Data are representative of three independent experiments [mean and SEM in (B), (C), (G), and (H)]. NS, nonsignificant (P > 0.05); ***P < 0.001.
To further explore the functions of USP5 in regulating RIG-I ubiquitination, we constructed a deletion of USP5 lacking ubiquitin-associated (UBA) domains (USP5-ΔUBA), which was considered as a ubiquitin and substrate binding motif (Fig. 6I). In contrast with WT USP5 and USP5-C355A, USP5-ΔUBA failed to promote the interaction between RIG-I and STUB1 as well as enhance the K48-linked ubiquitin chains of RIG-I (Fig. 6, J and K). This result indicates that although the enzyme activity of USP5 was not required to mediate RIG-I ubiquitination, its ability to bind to ubiquitin chains was critical to prompt this process.
In conclusion, the mechanisms of DUB function in the type I IFN pathway can be divided into six working models according to their mode of changing the ubiquitination status of their targets (Fig. 6L): (i) DUBs such as BRCC3 may cleave the ubiquitin chains on their target proteins, largely through their enzymatic activities; (ii) DUBs such as USP22 may reduce ubiquitination in a protease-independent manner; (iii) several DUBs may enhance ubiquitination through their DUB enzyme activities; (iv) DUBs such as USP5 may enhance ubiquitination on their targets independently of the enzyme activities; (v) DUBs such as A20, which has both DUB and E3 ligase activity, can mediate K63-K48 ubiquitination transition of RIP (receptor-interacting protein) by itself (36); and (vi) DUBs such as USP38 mediate ubiquitination transition with the help of other E3 ligases (27).
DISCUSSION
Here, we have reported a systematic screening study for the function of the DUB family in antiviral responses. Our data have revealed an unexpectedly large number of DUBs acting as novel regulators in controlling antiviral signaling. We also have confirmed the roles of CYLD, A20, USP3, USP19, USP21, OTUB1/2, MYSM1, etc. in dampening antiviral responses (19–21, 25, 26). Thus, the system reliability of our screening approach was validated by the consistency between our screening data and previous reports. In addition, the gene editing used by two different sgRNAs targeting different sites of each DUB exerted similar functions and led to the same conclusions, which can remarkably reduce systematic error and random error. Moreover, the consistent data from overexpression (Fig. 3 and fig. S3) and siRNA approaches (fig. S3) further confirmed the results from the sgRNA editing system.
On the basis of these three independent systems, we identified several previous unidentified DUBs, including USP5, USP14, USP22, USP48, USP52, COPS5, and BRCC3, as novel negative regulators in the type I IFN pathway. Nevertheless, apart from the negative regulation of type I IFN signaling, our study has further revealed that a small number of DUBs, including UCHL1, UCHL3, UCHL5, and POH1, could positively regulate type I IFN signaling. Many E3 ligases, especially the TRIM (tripartite motif-containing) family, function as positive regulators during antiviral responses (24). Because the DUB family could reverse the process of ubiquitin ligases, it is plausible that many DUBs function as negative regulators to form versatile negative feedback loops and restore immune homeostasis, thus eliminating the pathological effect of type I IFN in autoimmune disorders. On the other hand, several positive regulatory DUBs might reverse the degradation and maintain the protein stability of the key molecules in IFN signaling. In parallel, mRNA levels of an unprecedented large number of DUBs are found to be altered after viral infection or IFN stimulation, pointing to a broad aspect of feedback regulation that operates in type I IFN signaling. For example, the expression of USP18, USP26, and A20 was up-regulated after viral infection, thus forming negative feedback regulations. The mRNA level of UCHL3, UCHL5, and POH1 was also increased, which may result in possible positive feedback loops to amplify antiviral immune responses.
Recently, mounting evidence has revealed the increasing complexity and dynamics of the reversible ubiquitination. Ubiquitin can be linked to seven lysine (K) residues or to the N terminus of each other, leading to poly-ubiquitin chains that can encompass complex topologies (37). It has been well demonstrated that K63-linked poly-ubiquitin chains mediated signal complex assembly, which played crucial roles in type I IFN signaling (38). The K63-linked ubiquitination of RIG-I can be removed by USP3 (20), which resulted in the negative regulation of type I IFN signaling. K63-linked ubiquitin chains on TRAF3 and TRAF6 complexes can also be cleaved by MYSM1 (19), which dampen the innate immune response as well. A20 and CYLD can reduce the K63 ubiquitin chains on TBK1 to inactivate signal transduction (16, 17). Here, not only K63-linked ubiquitination of the key molecules but also other types of ubiquitin linkages can be reversibly regulated by DUBs. Because the fundamental role of DUBs is to cleave ubiquitin chains from substrates, the majority of reported DUBs regulate type I IFN signaling by removing the targets’ ubiquitination dependently of their DUB catalytic activity. The ubiquitination of some target proteins can be enhanced by DUBs. The functional patterns used by DUBs in altering different types of ubiquitination of their targets can be summarized into six working modes, which are dependent or independent of their enzyme activities (Fig. 6L). In detail, BRCC3 could remove the K63-linked ubiquitin chains on TRAF3 in a catalytic activity–dependent manner, and the enzyme active site mutant abolished its inhibition on IFN-β induction. Moreover, several DUBs such as USP22 might reduce the ubiquitin chains on their targets independently of their enzyme activity. Instead, USP22 recruits USP13 to cleave the ubiquitin chains. USP5 can promote K11-linked ubiquitination of RIG-I depending on its catalytic activity. In this case, we assume that DUBs may help to increase the stability of the E3 ligase or to reduce steric hindrance between the substrate and the E3 ligase by deubiquitination. On the other hand, several other DUBs can enhance the ubiquitin chains on their target proteins independently of their enzyme activities. For instance, USP5 can bridge STUB1 to RIG-I and promote the formation of the K48-linked ubiquitin chains on RIG-I, thus facilitating the degradation of RIG-I and inhibiting type I IFN signaling. During this process, the ubiquitin-binding domain of USP5 is critical to the enhancement of ubiquitination. Furthermore, several DUBs, such as USP38, promote the mediated K33/K48 ubiquitination transition (27). The cleavage of one type of ubiquitin chain and the occupation of another kind of ubiquitin chain may lead to the switching of the signal in the virus-triggered type I IFN pathway. Further studies are warranted to define the synergism and antagonism of ubiquitin ligases and DUBs in the extensive and complex ubiquitin system networks. The analysis from SpliceMiner (22) revealed that more than half of all DUBs had more than one splice variant (Fig. 1B). Several DUBs such as BRCC3 and OTUB1 have isoforms that lack the catalytic sites for protease activity. Thus, these variants might no longer regulate IFN signaling in response to virus infection (Fig. 4C). Several other DUBs, however, have incomplete interaction domain variants, such as UBA domains. For instance, the UBA domain of USP5 was critical to its function, whereas USP5 isoform 2, which misses amino acids in its UBA domain, might display impaired function compared to full-length USP5 during viral infection. Because most of the DUBs have more than one isoform, future research would ideally include a detailed study to evaluate the roles of these splice variants in the future.
Because it have been observed that DUBs have distinct expression patterns and protein abundance in different kinds of cells, we speculated that DUBs may exert diverse functions in different cell types. We performed VSV-eGFP infection screening and luciferase reporter activity analysis in DUB-deficient 293T cells; therefore, it is reasonable to hypothesize that some functional DUBs may have been missed because of their low expression level in 293T cells as well as the limited efficiency of specific sgRNAs. Here, we found that there was enhanced IFN activation in sg_USP7 cells, but it functioned as a negative regulator when we overexpressed USP7. USP7 is an NF-κB deubiquitinase and promotes NF-κB–mediated transcription (29). Previous studies have demonstrated that the HSV-1 protein ICP0 (infected cell protein 0) can mediate the translocation of USP7 from the nucleus to the cytoplasm, where USP7 binds to and deubiquitinates TRAF6 and NEMO (NF-κB essential modulator) (28). Thus, the function of USP7 can be transformed from a positive to a negative regulator of NF-κB signaling by altering subcellular localization. This may help to explain the contradiction in our study of USP7. Besides the different expression level and subcellular localization in different cells, one DUB could target different signaling proteins and function differently, which contributes to the complexity of DUB function and is worth further investigation. For example, our data showed that USP14 and USP18 negatively regulated the RLR-mediated type I IFN pathway at the TBK1 level, while promoting cGAS/STING signaling.
In summary, our study represents the first comprehensive revelation of a broad control of antiviral response by the family of DUBs. Novel regulatory DUBs are identified that regulate antiviral immunity at different levels along type I IFN signaling pathways through a diverse array of mechanisms. These findings thus have opened up a new avenue for follow-up studies to further uncover the complexity of ubiquitination regulation in immune responses. Given the importance of the stringent regulation of type I IFN signaling and the great potential of DUB functions, future studies hold great promise to exploit these novel mechanisms in immune regulation and to develop therapeutic interventions for tackling immune-related diseases.
MATERIALS AND METHODS
Cell culture and reagents
HEK293T and A549 cells were cultured in Dulbecco’s modified Eagle’s modified (DMEM) (Corning) with 10% fetal bovine serum (GenStar). Human PBMCs and THP-1 cells were maintained in RPMI 1640 medium (Gibco) with 10% fetal bovine serum in a 5% CO2 incubator at 37°C. HEK293T and A549 cells were from the cell bank of the Chinese Academy of Sciences. Blood from healthy donors was used for the isolation of PBMCs by Ficoll-Hypaque density-gradient centrifugation. The use of PBMCs was in compliance with institutional guidelines and approved protocols of Sun Yat-sen University. The cell lines were tested for mycoplasma contamination by the MycoAlert Mycoplasma Detection Kit. Recombinant human IFN-β was purchased from PeproTech. Horseradish peroxidase (HRP)–anti-Flag (M2) (A8592), anti–β-actin (A1978), and anti-USP14 (U8009) were purchased from Sigma-Aldrich. HRP–anti-hemagglutinin (HA) (12013819001) was purchased from Roche Applied Science. Anti–RIG-I (3743), anti-MAVS (3993), anti-STING (13647), anti-TBK1 (3504), anti-BRCC3 (18215S), anti-K48 linkage-specific polyubiquitin (4289), anti-K63 linkage-specific polyubiquitin (5621), and anti-CHIP (STUB1) (2080) were acquired from Cell Signaling Technology. Anti-IRF3 (sc-9082), anti-MAVS (sc-166583), anti-Ub (sc-8017), anti-USP5 (sc-390943), anti-USP13 (sc-366878), anti-USP48 (sc-100635), anti-USP52 (sc-517176), and anti-COPS5 (sc-13157) were from Santa Cruz Biotechnology. Anti-USP22 (55110-1-AP) was purchased from Proteintech. Anti-USP21 (ab38864) and anti-USP38 (ab72244) were purchased from Abcam. Anti-USP19 was purchased from ImmunoWay.
Plasmids and viruses
NF-κB, ISRE, and IFN-β promoter luciferase reporter plasmids and mammalian expression plasmids for HA- or Flag-tagged RIG-I (2CARD), RIG-I, STING, MAVS, TBK1, IKKi, IRF3, and IRF3 (5D) have been previously described (39). DUB expression plasmids were clones from cDNA, and mutants were constructed using standard molecular biology techniques. HSV-1 (KOS strain) was provided by G. Zhou (Guangzhou Medical University). VSV-eGFP and SeV have been previously described (25). Cells were infected at various multiplicities of infection (MOIs), as previously described (23).
Cell lines with stable Cas9 and sgRNA expression
After 16 hours, 293T and A549 cells were seeded, and medium was replaced with DMEM containing polybrene (Sigma-Aldrich) and lentiviruses expressing Cas9 and DUB-specific sgRNAs (table S3) for 18 hours. Cells were selected for puromycin resistance, and polyclonal pools of transduced cells were used for succeeding experiments.
sgRNA-mediated KO of DUBs in PBMCs
PBMC medium was replaced with RPMI 1640 containing polybrene and lentiviruses expressing Cas9 and DUB-specific sgRNAs. After centrifugation at 100g for 1 hour and incubation at 37°C for 24 hours, cells were selected for puromycin resistance (5 μg/ml) for 2 days. DUB-deficient PBMCs were seeded at the same amount (1 × 106) and left untreated or infected with SeV (MOI, 0.1) for 18 hours.
Luciferase reporter assays
293T cells were transfected with a mixture of luciferase reporter (firefly luciferase), pRL-TK (Renilla luciferase plasmid), and an indicated variety expression plasmid or an empty vector (pcDNA3.1) plasmid. Then, the cells were transfected with RIG-I (2CARD), MAVS, cGAS/STING, TBK1, or IRF3 (5D) or infected with SeV. Luciferase activity was measured at 24 hours after transfection or infection using a luminometer (Thermo Fisher Scientific) with a dual-luciferase reporter assay system according to the manufacturer’s instructions (Promega). Data represent relative firefly luciferase activity, normalized to Renilla luciferase activity.
Immunoprecipitation and immunoblotting
For immunoprecipitation, whole-cell lysates were prepared after transfection, followed by incubation overnight with the appropriate anti-Flag beads (Sigma-Aldrich) or anti-myc beads (Biotool). Beads were washed three to five times with low-salt lysis buffer. For ubiquitination assay, beads were washed three to five times with low-salt lysis buffer containing a low dose of SDS (0.5%) to reduce the effect of coimmunoprecipitated proteins, except for USP5 samples. Then, immunoprecipitates were eluted with 2× SDS loading buffer and resolved by SDS–polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad) and further incubated with appropriate antibodies. The LumiGlo Chemiluminescent Substrate System (Millipore) was used for protein detection.
RNA extraction and qRT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) and reverse-transcribed using oligo-dT primers and reverse transcriptase (Vazyme). qRT-PCR was performed using the SYBR Green qPCR Mix kit (GenStar), with the primers listed in table S3.
Measurement of cytokines
Human IFN-β in cell culture supernatants was detected with an enzyme-linked immunosorbent assay (ELISA) kit (SEA222Hu, Cloud-Clone Corp.) according to the manufacturer’s protocols. Human IL-6 in cell culture supernatants was detected with an ELISA kit (no. 555220, BD Biosciences), according to the manufacturer’s protocols.
Viral plaque titration
The VSV-containing supernatants were collected after viral infection for 24 hours. Vero cells were infected with VSV supernatants for 1 hour at room temperature as previously described (40). After washing with phosphate-buffered saline, the plate was overlaid with DMEM containing 1% low–melting point agarose and incubated at 37°C for 24 hours before crystal violet staining.
Statistical analysis
Data are represented as mean ± SEM when indicated, and two-tailed Student’s t test was used for statistical analysis. Differences between groups were considered significant when P < 0.05.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (31522018, 91629101, and 31601135), the National Key Basic Research Program of China (2015CB859800 and 2014CB910800), and the Training Program for Outstanding Young Teachers in Higher Education Institutions of Guangdong Province (YQ2015001), as well as by the CAMS Initiative for Innovative Medicine (CAMS-I2M, 2016-I2M-1-005), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2016ZX310194), the National Natural Science Foundation of China (81773058), the Ministry of Science and Technology Project Grant (No. 2014CB745203), and the Key Program for Innovative Drug Development of China (2015ZX09102023) to F.X.-F.Q. Author contributions: J.C., Q.L., Y.W., and Y.Q. designed and performed the experiments. J.H. and W.X. provided technical assistance. J.C., F.X.-F.Q., Q.L., and Y.W. wrote the manuscript. J.C. and F.X.-F.Q. supervised the entire project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaar2824/DC1
fig. S1. Knockdown efficiency of DUBs by sgRNAs.
fig. S2. Identification of functional DUBs in antiviral response by FACS analysis and luciferase assay.
fig. S3. Validation of identified DUB candidates in an overexpression system and by siRNA.
fig. S4. DUBs interact with the key molecules in type I IFN signaling pathways and the induction of the indicated genes in sg_NT A549 and PBMCs after viral infection.
fig. S5. Attenuated DUB expression in A549 cells and PBMCs affects virus-induced cytokine expression.
fig. S6. Total ubiquitination and different linkages of ubiquitination of the key molecules in the type I IFN pathway are regulated by DUBs.
fig. S7. USP5 inhibits type I IFN signaling and enhances viral replication through the promotion of STUB1-mediated RIG-I degradation.
table S1. Relative mRNA expression of DUBs in THP-1 monocytes or macrophages after VSV infection or IFN-β treatment for the indicated time points and the relative mRNA expression of DUBs in PBMCs after VSV infection, HSV-1 infection, or IFN-β treatment.
table S2. Summary of DUB expression patterns using different stimuli.
table S3. List of sgRNA sequences, qRT-PCR primer sequences, and siRNA sequences used in this study.
REFERENCES AND NOTES
- 1.Akira S., Uematsu S., Takeuchi O., Pathogen recognition and innate immunity. Cell 124, 783–801 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Collins S. E., Mossman K. L., Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev. 25, 525–531 (2014). [DOI] [PubMed] [Google Scholar]
- 3.O’Neill L. A., Bowie A. G., The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Yoneyama M., Fujita T., RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227, 54–65 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Chen G., Shaw M. H., Kim Y.-G., Nuñez G., NOD-like receptors: Role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4, 365–398 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Barber G. N., Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 23, 10–20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loo Y.-M., Gale M. Jr, Immune signaling by RIG-I-like receptors. Immunity 34, 680–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seth R. B., Sun L., Ea C. K., Chen Z. J., Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z., Yuan B., Bao M., Lu N., Kim T., Liu Y.-J., The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakobsen M. R., Bak R. O., Andersen A., Berg R. K., Jensen S. B., Jin T., Laustsen A., Hansen K., Østergaard L., Fitzgerald K. A., Xiao T. S., Mikkelsen J. G., Mogensen T. H., Paludan S. R., IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. U.S.A. 110, E4571–E4580 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L., Wu J., Du F., Chen X., Chen Z. J., Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S.-M., Maniatis T., IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Hu H., Sun S.-C., Ubiquitin signaling in immune responses. Cell Res. 26, 457–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nijman S. M., Luna-Vargas M. P., Velds A., Brummelkamp T. R., Dirac A. M., Sixma T. K., Bernards R., A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W., Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman C. S., O’Donnell M. A., Legarda-Addison D., Ng A., Cardenas W. B., Yount J. S., Moran T. M., Basler C. F., Komuro A., Horvath C. M., Xavier R., Ting A. T., The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parvatiyar K., Barber G. N., Harhaj E. W., TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 285, 14999–15009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ning S., Pagano J. S., The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 84, 6130–6138 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panda S., Nilsson J. A., Gekara N. O., Deubiquitinase MYSM1 regulates innate immunity through inactivation of TRAF3 and TRAF6 complexes. Immunity 43, 647–659 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Cui J., Song Y., Li Y., Zhu Q., Tan P., Qin Y., Wang H. Y., Wang R.-F., USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 24, 400–416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan Y., Mao R., Yu Y., Liu S., Shi Z., Cheng J., Zhang H., An L., Zhao Y., Xu X., Chen Z., Kogiso M., Zhang D., Zhang H., Zhang P., Jung J. U., Li X., Xu G., Yang J., USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 211, 313–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahn A. B., Ryan M. C., Liu H., Zeeberg B. R., Jamison D. C., Weinstein J. N., SpliceMiner: A high-throughput database implementation of the NCBI Evidence Viewer for microarray splice variant analysis. BMC Bioinformatics 8, 75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui J., Zhu L., Xia X., Wang H. Y., Legras X., Hong J., Ji J., Shen P., Zheng S., Chen Z. J., Wang R. F., NLRC5 negatively regulates the NF-κB and type I interferon signaling pathways. Cell 141, 483–496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Versteeg G. A., Rajsbaum R., Sanchez-Aparicio M. T., Maestre A. M., Valdiviezo J., Shi M., Inn K. S., Fernandez-Sesma A., Jung J., Garcia-Sastre A., The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38, 384–398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin S., Tian S., Chen Y., Zhang C., Xie W., Xia X., Cui J., Wang R. F., USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 35, 866–880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S., Zheng H., Mao A.-P., Zhong B., Li Y., Liu Y., Gao Y., Ran Y., Tien P., Shu H.-B., Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 285, 4291–4297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin M., Zhao Z., Yang Z., Meng Q., Tan P., Xie W., Qin Y., Wang R. F., Cui J., USP38 inhibits type I interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome. Mol. Cell 64, 267–281 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Daubeuf S., Singh D., Tan Y., Liu H., Federoff H. J., Bowers W. J., Tolba K., HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113, 3264–3275 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colleran A., Collins P. E., O’Carroll C., Ahmed A., Mao X., McManus B., Kiely P. A., Burstein E., Carmody R. J., Deubiquitination of NF-κB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 618–623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M., Meng Q., Qin Y., Liang P., Tan P., He L., Zhou Y., Chen Y., Huang J., Wang R. F. Y., Cui J., TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64, 105–119 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Zhang M., Zhang M.-X., Zhang Q., Zhu G.-F., Yuan L., Zhang D.-E., Zhu Q., Yao J., Shu H.-B., Zhong B., USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 26, 1302–1319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper E. M., Cutcliffe C., Kristiansen T. Z., Pandey A., Pickart C. M., Cohen R. E., K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 28, 621–631 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun H., Zhang Q., Jing Y.-Y., Zhang M., Wang H.-Y., Cai Z., Liuyu T., Zhang Z.-D., Xiong T.-C., Wu Y., Zhu Q.-Y., Yao J., Shu H.-B., Lin D., Zhong B., USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 8, 15534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xian H., Xie W., Yang S., Liu Q., Xia X., Jin S., Sun T., Cui J., Stratified ubiquitination of RIG-I creates robust immune response and induces selective gene expression. Sci. Adv. 3, e1701764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao K., Zhang Q., Li X., Zhao D., Liu Y., Shen Q., Yang M., Wang C., Li N., Cao X., Cytoplasmic STAT4 promotes antiviral type I IFN production by blocking CHIP-mediated degradation of RIG-I. J. Immunol. 196, 1209–1217 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Wertz I. E., O’Rourke K. M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D. L., Ma A., Koonin E. V., Dixit V. M., De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Swatek K. N., Komander D., Ubiquitin modifications. Cell Res. 26, 399–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heaton S. M., Borg N. A., Dixit V. M., Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 213, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin Y., Liu Q., Tian S., Xie W., Cui J., Wang R.-F., TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type I interferon by recruiting GSK3β to TBK1. Cell Res. 26, 613–628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xing J., Weng L., Yuan B., Wang Z., Jia L., Jin R., Lu H., Li X. C., Liu Y.-J., Zhang Z., Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 17, 1373–1380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaar2824/DC1
fig. S1. Knockdown efficiency of DUBs by sgRNAs.
fig. S2. Identification of functional DUBs in antiviral response by FACS analysis and luciferase assay.
fig. S3. Validation of identified DUB candidates in an overexpression system and by siRNA.
fig. S4. DUBs interact with the key molecules in type I IFN signaling pathways and the induction of the indicated genes in sg_NT A549 and PBMCs after viral infection.
fig. S5. Attenuated DUB expression in A549 cells and PBMCs affects virus-induced cytokine expression.
fig. S6. Total ubiquitination and different linkages of ubiquitination of the key molecules in the type I IFN pathway are regulated by DUBs.
fig. S7. USP5 inhibits type I IFN signaling and enhances viral replication through the promotion of STUB1-mediated RIG-I degradation.
table S1. Relative mRNA expression of DUBs in THP-1 monocytes or macrophages after VSV infection or IFN-β treatment for the indicated time points and the relative mRNA expression of DUBs in PBMCs after VSV infection, HSV-1 infection, or IFN-β treatment.
table S2. Summary of DUB expression patterns using different stimuli.
table S3. List of sgRNA sequences, qRT-PCR primer sequences, and siRNA sequences used in this study.