

Abstract
Objective
To investigate the effects of genetic risk of Alzheimer disease (AD) dementia in the context of β-amyloid (Aβ) accumulation.
Methods
We analyzed data from 702 participants (221 clinically normal, 367 with mild cognitive impairment, and 114 with AD dementia) with genetic data and florbetapir PET available. A subset of 669 participants additionally had longitudinal MRI scans to assess hippocampal volume. Polygenic risk scores (PRSs) were estimated with summary statistics from previous large-scale genome-wide association studies of AD dementia. We examined relationships between APOE ε4 status and PRS with longitudinal Aβ and cognitive and hippocampal volume measurements.
Results
APOE ε4 was strongly related to baseline Aβ, whereas only weak associations between PRS and baseline Aβ were present. APOE ε4 was additionally related to greater memory decline and hippocampal atrophy in Aβ+ participants. When APOE ε4 was controlled for, PRS was related to cognitive decline in Aβ+ participants. Finally, PRSs were associated with hippocampal atrophy in Aβ− participants and weakly associated with baseline hippocampal volume in Aβ+ participants.
Conclusions
Genetic risk factors of AD dementia demonstrate effects related to Aβ, as well as synergistic interactions with Aβ. The specific effect of faster cognitive decline in Aβ+ individuals with higher genetic risk may explain the large degree of heterogeneity in cognitive trajectories among Aβ+ individuals. Consideration of genetic variants in conjunction with baseline Aβ may improve enrichment strategies for clinical trials targeting Aβ+ individuals most at risk for imminent cognitive decline.
Common genetic variants explain a large proportion of the heritability of sporadic Alzheimer disease (AD) dementia.1,2 However, the mechanisms underlying these genetic risk factors are unclear, and it is likely that multiple pathways are involved. Previous work has identified limited associations between genetic risk and β-amyloid (Aβ) accumulation3,4 and has suggested mechanistic pathways not directly linked to abnormal Aβ accumulation such as inflammatory pathways.5–7 Thus, genetic risk variants associated with AD dementia may not solely confer risk by promoting Aβ aggregation but may make an individual more likely to decline when faced with Aβ accumulation.
A synergistic effect between genetic risk and Aβ is consistent with a framework in which Aβ may be necessary but insufficient for clinical symptoms of AD dementia8 and may offer an explanation for the presence of clinically normal (CN) older individuals who have high levels of Aβ.9 Along these lines, it is known that although Aβ+ CN individuals show group-level reductions in cognitive performance over time,3,10–12 many Aβ+ individuals remain cognitively normal even after extended follow-up.13 Heterogeneity in cognitive decline has also been reported in symptomatic patients with AD dementia, which may be related to differences in genetic risk factors. For instance, although the APOE ε4 allele is consistently associated with abnormal accumulation of Aβ,14 Aβ+ individuals without dementia who are also APOE ε4 carriers show faster rates of cognitive decline than Aβ+ individuals who are APOE ε4 noncarriers.15,16 A similar effect modification has been shown for the BDNF gene such that Aβ+ individuals with the BDNF Met allele show faster cognitive decline than Aβ+ individuals without the Met allele.17 Given that these a priori genetic risk factors have been shown to influence cognitive decline among Aβ+ individuals, it is possible that other established genetic risk factors have a similar influence.
The overall goal of the present study was to investigate whether APOE ε4 and polygenic risk of AD dementia influence Aβ, cognition, and hippocampal volume measurements and to determine whether their associations with cognition and hippocampal volume are modified by Aβ status.
Methods
Alzheimer's Disease Neuroimaging Initiative
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI; adni.loni.usc.edu) as of March 25, 2017. This study used 702 ADNI-GO/2 participants (221 CN, 367 with mild cognitive impairment [MCI], and 114 with AD dementia) of European ancestry who had florbetapir PET images, longitudinal neuropsychological assessment, and genome-wide single nucleotide polymorphisms (SNPs). Of the 702 participants, 669 additionally had longitudinal brain MRI scans that passed quality control. Given that florbetapir PET was introduced midstudy, longitudinal neuropsychological and hippocampal volume measurements taken >1 year before the baseline PET scan were discarded from the analyses.
Standard protocol approvals, registrations, and patient consents
Institutional review boards approved study procedures across ADNI participating sites. Written informed consent for research was obtained from all participants.
Aβ imaging
Global standardized uptake value ratio florbetapir index values were averaged across 4 large bilateral regions (frontal, cingulate, parietal, and lateral temporal) and normalized by the whole cerebellum.18,19 A 1.11 cutoff of the baseline standardized uptake value ratio value was used to classify participants as Aβ+ and Aβ−.18,20
Neuropsychological assessments
Composite scores of memory21 and executive function22 were developed previously by using sophisticated neuropsychometric approaches (confirmatory factor analyses and item response theory) to account for different distributions and formats of individual scores, missing items, and different versions of neuropsychological tests.
Structural MRI
The 3T T1 structural brain MRI scans were processed with the FreeSurfer version 5.3 longitudinal pipeline and used to define bilateral hippocampal volume.23 We processed the MRI scans locally rather than using the imaging measurements from the ADNI website because this led to more longitudinal time points and the use of a more recent FreeSurfer version.
Processing of genetic data
Genome-wide SNP data from 793 ADNI-GO/2 participants were processed following standard pipelines.24 Non-European participants, population outliers, and closely related participants were removed. Genotype imputation was performed with the MaCH software.25 SNPs that had poor imputation quality, high missing rate (>2%), low minor allele frequency (<1%), or significant deviation from Hardy-Weinberg equilibrium (p < 1e−6) and participants who had a high missing genotype rate (>2%) were filtered out. Seven hundred sixteen participants with 7,694,586 autosomal SNPs remained after quality control. Of the 716 participants, 702 had florbetapir PET and longitudinal neuropsychological assessments.
Calculation of polygenic risk score
Polygenic risk score (PRS) was computed on the basis of the summary statistics from the stage 1 analysis of the International Genomics of Alzheimer's Project, the largest-to-date case-control genome-wide association studies (GWAS) (17,008 cases with AD and 37,154 CN controls) of AD dementia diagnosis,26 with the software package PRSice.27 Specifically, imputed genome-wide SNPs were pruned by use of p value informed clumping. PRS was computed as the sum of individual SNPs below a p value threshold, weighted by the log of the odds ratio estimated by the GWAS. PRS thus summarizes genetic effects among an ensemble of variants spanning the genome that may not individually achieve GWAS-level significance. A range of p value thresholds from p < 1e−7 to p < 1e−2 were used in this study.
Statistical analysis
The influences of APOE ε4 status and PRS on baseline florbetapir values and binary Aβ+/− groups were examined in the whole sample with linear regression and logistic regression, respectively, with adjustment for top 4 principal components of the genotype data, baseline diagnosis, baseline age, sex, and education. When assessing the effect of PRS, we included APOE ε4 status in the model.
The influences of APOE ε4 status and PRS on the baseline and longitudinal change in florbetapir values, neuropsychological composite scores, and hippocampal volume measurements were examined with longitudinal linear mixed-effects models (LMMs). All models included an intercept, time from baseline, top 4 genotype principal components, baseline diagnosis, baseline age, sex, education, and the interactions between baseline diagnosis, baseline age, sex, education, and time (i.e., baseline diagnosis × time, baseline age × time, etc) as fixed-effect covariates. Throughout the article, the calculation of PRS included the APOE ε4 allele, but when assessing the association between PRS and longitudinal change, we included APOE ε4 and its interaction with time in the model to assess the effect of PRS above and beyond APOE ε4 status. When analyzing hippocampal volume, we additionally adjusted for intracranial volume and its interaction with time as covariates. Random intercept and slopes were included in each LMM. All analyses were repeated in Aβ+ and Aβ− participants separately. All p values reported in this article are 2-sided uncorrected p values.
To compute a metric of variance explained, we extended the classic adjusted R2 statistic in multiple regression analysis to the context of LMM. Specifically, we assume that the unbiased estimate of the variance of the random slope in the full LMM is 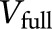 and the unbiased estimate of the variance of the random slope in a reduced model without genetic risk factors (APOE ε4 or PRS) is
and the unbiased estimate of the variance of the random slope in a reduced model without genetic risk factors (APOE ε4 or PRS) is 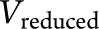 . The reduced model contained all covariates and their interactions with time. Then, variation in the rate of decline explained by each genetic risk factor is defined as
. The reduced model contained all covariates and their interactions with time. Then, variation in the rate of decline explained by each genetic risk factor is defined as 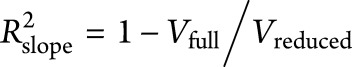 .
. 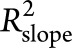 measures the proportion of interparticipant variation in the slope that can be explained by adding the genetic risk factor into the model.
measures the proportion of interparticipant variation in the slope that can be explained by adding the genetic risk factor into the model. 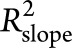 was calculated for APOE ε4 by adding APOE ε4 to the reduced model. PRS was then added to this model to estimate
was calculated for APOE ε4 by adding APOE ε4 to the reduced model. PRS was then added to this model to estimate 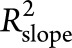 for PRS. Variance explained in baseline measurements by genetic factors can be defined similarly.
for PRS. Variance explained in baseline measurements by genetic factors can be defined similarly.
Data availability
All imaging, genetic, demographics, and neuropsychological composite scores used this article are publicly available and were downloaded from the ADNI website (adni.loni.usc.edu). Structural MRI data and GWAS data were downloaded and used to derive the measurements used here (hippocampus volume and PRS). The authors will provide a list of ADNI participant identifications on request for replication purposes.
Results
PRS, baseline Aβ, and clinical diagnosis
Table 1 summarizes the participant characteristics. In general, PRS was greater in the Aβ+ compared to the Aβ− group (2-sample t test p < 0.05 across diagnosis and PRS thresholds; figure 1). However, the associations between PRS and baseline Aβ were weak, regardless of whether Aβ is treated as a continuous variable or as a binary variable (table e-1, links.lww.com/WNL/A405). For instance, the most significant relationship between PRS and continuous Aβ explained only 0.75% of the Aβ variation (p = 0.013). As expected, APOE ε4 was strongly associated with elevated continuous Aβ at baseline, explaining 17.94% of the variance.
Table 1.
Participant demographics

Figure 1. PRSs of AD dementia by diagnosis and Aβ status.

Mean and standard error of the polygenic risk score (PRS) of Alzheimer disease (AD) dementia by diagnosis among (A) β-amyloid (Aβ)–positive and (B) Aβ-negative participants across PRS p value thresholds are shown. CN = clinically normal; MCI = mild cognitive impairment.
PRS was associated with clinical diagnosis, even in the Aβ+ group (PRS was higher in Aβ+ patients with MCI and Aβ+ patients with AD compared to Aβ+ controls; figure 1). This pattern was present for the majority of PRS thresholds (1-way analysis of variance p < 0.05). There were no significant diagnostic difference in PRS among the Aβ− group (figure 1). Diagnosis and its interaction with time were controlled for in all subsequent analyses to ensure that results related to cognitive decline were independent of diagnosis.
Genetic risk factors and Aβ accumulation
We next examined the associations between genetic risk factors and longitudinal accumulation of Aβ over time. Given the slow rates of Aβ accumulation, longitudinal Aβ was treated as a continuous variable and examined separately in the Aβ− and Aβ+ groups (defined at baseline). We found that Aβ− APOE ε4 carriers had faster Aβ accumulation over time than Aβ− noncarriers (p = 0.005). There was no effect of APOE ε4 on Aβ accumulation in the Aβ+ group. PRS had no influence on Aβ accumulation in either the Aβ+ or Aβ− group (figure 2 and tables e-2 and e-3, links.lww.com/WNL/A405).
Figure 2. Associations between genetic risk factors and Aβ, cognition, and hippocampal volume.

Variance explained by APOE ε4 and polygenic risk scores (PRSs) of Alzheimer disease dementia across longitudinal linear mixed effects models predicting (A and B) β-amyloid (Aβ), (C and D) memory, (E and F) executive function, and (G and H) hippocampal volume.
Genetic risk factors and cognition
Aβ+ APOE ε4 carriers had faster memory decline than noncarriers (p = 0.015). There was no effect of APOE ε4 on memory in the Aβ− group. APOE ε4 was weakly associated with executive function decline in the Aβ− group (p = 0.049) but not the Aβ+ group. Furthermore, controlling for APOE ε4, higher polygenic risk was strongly associated with higher rates of decline in memory and executive function in Aβ+ individuals across a range of PRS p value thresholds. These associations explained approximately 8% to 10% of the variation in the rate of cognitive decline across Aβ+ participants. PRSs were not associated with cognitive decline in Aβ− participants or baseline cognitive scores in either Aβ group (figures 2 and 3 and tables e-2 and e-3, links.lww.com/WNL/A405). All results remained similar when the analysis was restricted to participants without dementia (CN and MCI combined; table e-4 and figure e-1, links.lww.com/WNL/A404). The effects of PRS on cognitive decline among Aβ+ participants remained strong if we additionally controlled for baseline hippocampal volume and its interaction with time in the model (table e-5).
Figure 3. Illustration of the influences of genetic risk on cognitive decline and hippocampal atrophy in Aβ+ participants.

Model-based estimation of longitudinal changes in (A) memory, (B) executive function, and (C) hippocampal volume based on APOE ε4 status and the polygenic risk score (PRS) of Alzheimer disease dementia computed with p < 1e−5 as the p value threshold in β-amyloid–positive (Aβ+) participants. High PRS refers to the upper quartile (75%) of the PRS in the sample; low PRS refers to the lower quartile (25%) of the PRS in the sample. All fixed-effect covariates (principal components, baseline diagnosis, baseline age, sex, education, and their interactions with time) were set to the sample average.
Genetic risk factors and hippocampal volume
Among Aβ+ participants, both APOE ε4 status and polygenic risk were associated with baseline hippocampal volume. However, these effects on baseline hippocampus volume were small, accounting for <2% of the variance. Among Aβ−, PRS was strongly associated with longitudinal change in hippocampal volume but not hippocampal volume at baseline. PRS using the strictest threshold explained 8% of the variance in hippocampal atrophy in the Aβ− group (figures 2 and 3 and tables e-2 and e-3, links.lww.com/WNL/A405).
Discussion
In this study, we found that APOE ε4 and PRS of AD dementia show different patterns of associations with AD markers. Specifically, APOE ε4 was related to elevated baseline Aβ and the accumulation of Aβ over time in Aβ−, whereas PRS showed minimal associations with Aβ but was strongly associated with faster cognitive decline in Aβ+ participants. The influence of PRS on longitudinal cognitive trajectories was above and beyond the effect of APOE ε4 status, diagnosis, and hippocampal volume, highlighting the value of a priori genetic risk factors as a predictor of short-term decline among Aβ+ individuals. Furthermore, polygenic risk was not related to rates of cognitive decline in the Aβ− group, suggesting a synergistic effect between abnormal Aβ and genetic risk on cognitive decline. Finally, polygenic risk showed associations with hippocampal volume across both the Aβ+ and Aβ− groups, suggesting that genetic risk of AD may have an effect on non-Aβ pathways that influence brain structure in regions central to AD dementia.
We did not find strong evidence that an aggregate measure of genetic risk of AD dementia was directly associated with abnormal levels of amyloid, assessed either cross-sectionally or longitudinally. However, our analyses consistently showed an association between polygenic risk and cognitive decline that was specific to the Aβ+ group. This finding is in line with previous studies that have shown that Aβ and APOE ε4 interact to accelerate cognitive decline and hippocampal degeneration in CN individuals,16,17,28 highlighting synergistic interactions between genetic risk of AD and the presence of abnormal Aβ. Furthermore, we have previously observed that elevated PRS is associated with greater longitudinal cognitive decline in older individuals without dementia.29 However, other studies investigating PRS of AD dementia have failed to identify an association with cognitive decline30 or clinical progression.31 One source of this discrepancy may be that these previous investigations of polygenic risk did not examine decline in the context of AD biomarkers. Our results indicate that elevated PRS is associated with cognitive decline only among Aβ+ individuals, suggesting that genetic risk factors and Aβ act synergistically to influence cognitive decline. Recent work from Tan et al.32 and Desikan et al.33 also using the ADNI dataset has reported a similar pattern: cognitive decline was greatest in individuals with both abnormal CSF AD markers (amyloid and tau) and elevated genetic risk of AD as assessed with a polygenic hazard score. Although this study implemented a different methodological strategy to aggregate genetic risk across multiple genomic loci, the consistency across our study and theirs highlights a synergistic effect between genetic risk and AD pathology on cognitive trajectories. Given that genetic risk of AD has been implicated in non-Aβ pathways such as the immune system and cytoskeletal function,5–7 it is possible that the convergence of these non-Aβ pathways with late-life Aβ accumulation is an important predictor of subsequent decline.
Whereas polygenic risk was related to decline in both memory and executive function among Aβ+ individuals, APOE ε4 was specifically related to decline in memory and not executive function. This finding is consistent with analyses in patients with AD dementia that have suggested that APOE ε4 carriers tend to show memory impairment whereas patients with AD dementia who are APOE ε4 noncarriers show impairment in executive function.34 Likewise, examination of pathologically defined subtypes of AD dementia suggests that APOE ε4 is associated with a limbic predominant presentation among late-onset AD cases.35 The finding that polygenic risk influences both memory and executive function implies that polygenic risk of AD dementia may influence a more distributed set of brain regions compared to APOE ε4. Along these lines, we previously found that polygenic risk was associated with multiple cortical regions, including frontal, parietal, and temporal cortices,36 which may explain the broad effect of polygenic risk on cognitive decline. Given that calculation of polygenic risk aggregates information across many genomic loci and potentially many genetic pathways, it is unclear whether there are certain subgroups of genetic risk factors that relate to more specific brain networks and would demonstrate an effect on cognition that was domain specific. If so, a participant's composition of these genetic risk factors may be predictive of the specific cognitive domain that is most vulnerable in an individual and provide an explanation for the heterogeneity in clinical expression of AD dementia that exists across individuals. Future work that refines polygenic risk to encapsulate variability across different cognitive domains may offer insights into the known heterogeneity of the clinical expression of AD.37
In contrast to memory and executive function, we found that in Aβ+ participants, PRS did not strongly influence longitudinal hippocampal atrophy but was associated with baseline volume, while in the Aβ− group, PRS was strongly associated with the longitudinal change in hippocampal volume but not its baseline measurements. This finding highlights that although polygenic risk influences hippocampal volume regardless of Aβ status, the time course of this effect varies. It is possible that the effect of polygenic risk on hippocampal atrophy occurs before late-life Aβ accumulation and that once abnormal levels of Aβ have occurred, there are stronger drivers of subsequent atrophy compared to polygenic risk (such as tau-mediated neurodegeneration38). This framework is consistent with previous work that has shown a relationship between polygenic risk of AD dementia and hippocampal volume in young participants,29,39 well before the age at which abnormal Aβ is anticipated.40 Thus, the effect of polygenic risk on hippocampal volume in stages preceding Aβ accumulation (as supported by findings in both younger participants29,39 and older Aβ− participants in the current study) may ultimately make an individual more vulnerable to cognitive decline in late life when additionally faced with Aβ accumulation.
Our analyses have several limitations. First, the PRSs were calculated from summary statistics from cross-sectional case-control GWAS.26 Therefore, the estimated effect sizes of genetic variants may not reflect their strength of influences on longitudinal change in AD markers, and the constructed PRS may thus be suboptimal for investigating the genetic basis of cognitive decline and AD progression. Conducting large-scale GWAS of longitudinal neuropsychological assessments and neuroimaging biomarkers of AD may improve genetic prediction of longitudinal decline. Second, although we focused on hippocampus volume, other MRI-derived neuroimaging markers of AD progression may also be relevant, especially given the association we identified between PRS and executive function. Third, although we report that ≈10% of the variance in cognitive decline among Aβ+ individuals can be explained by genetic risk factors, the majority of variance remains unexplained. Thus, the consideration of additional factors such as the regional deposition of tau (via PET), as well as vascular, lifestyle, and other genetic risk factors that were not captured in the International Genomics of Alzheimer's Project GWAS analysis, will be necessary to optimize our ability to predict risk at the individual participant level. Lastly, replication of the findings in this study is needed in other large cohorts with biomarker data and prospective neuropsychological follow-up.
We observed that APOE ε4 status and higher polygenic risk of AD dementia were associated with higher rates of decline in neuropsychological assessments of memory and executive function in Aβ+ individuals. The effect of polygenic risk on cognitive decline was above and beyond APOE ε4 and specific to the Aβ+ group, suggesting a synergistic effect between abnormal Aβ and genetic risk. Polygenic risk was also associated with hippocampal volume, an effect that was not dependent on Aβ status. These findings suggest that although this aggregate measure of genetic risk is not strongly associated with amyloid, it may moderate an individual's risk of decline once abnormal levels of amyloid are present, ultimately improving efforts to identify individuals at risk for cognitive decline.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CN
clinically normal
- GWAS
genome-wide association studies
- LMM
linear mixed-effects model
- MCI
mild cognitive impairment
- PRS
polygenic risk score
- SNP
single nucleotide polymorphism
Author contributions
Conception and design of the study: T.G., and E.C.M. Statistical analysis: T.G. Drafting the manuscript: T.G., and E.C.M. Interpretation of data: T.G., M.R.S., J.W.S., R.A.S., and E.C.M. Revising manuscript: T.G., M.R.S., J.W.S., R.A.S., and E.C.M.
Study funding
This work was funded by NIH grants K99AG054573 (T.G.); R01NS083534, R01NS070963, K25EB013649, R21AG050122, and R41AG052246 (M.R.S.); K24MH094614 and R01MH101486 (J.W.S.); and K01AG051718 (E.C.M.). J.W.S. is a Tepper Family MGH Research Scholar and was also supported in part by a gift from the Demarest Lloyd, Jr. Foundation. This research used resources provided by the Center for Functional Neuroimaging Technologies and a P41 Biotechnology Resource Grant (P41EB015896) supported by NIH. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program (S10RR023043 and S10RR023401). Data collection and sharing for this project was funded by the ADNI (NIH grant U01 AG024904) and Department of Defense ADNI (Department of Defense award W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, by the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc; Biogen; Bristol-Myers Squibb Co; CereSpir, Inc; Cogstate; Eisai Inc; Elan Pharmaceuticals, Inc; Eli Lilly and Co; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co Inc; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corp; Pfizer Inc; Piramal Imaging; Servier; Takeda Pharmaceutical Co; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Disclosure
T. Ge received funding from K99AG054573. M. Sabuncu received funding from R01NS083534, R01NS070963, K25EB013649, R21AG050122, and R41AG052246. J. Smoller received funding from U01HG008685, R56MH110872, R01MH101486, R01MH106547, OT2OD024612, and K24MH094614. He is a Tepper Family MGH Research Scholar and was also supported in part by a gift from the Demarest Lloyd, Jr. Foundation. R. Sperling has served as a paid consultant for Abbvie, Biogen, Bracket, Genentech, Lundbeck, Roche, and Sanofi. She has served as a coinvestigator for Avid, Eli Lilly, and Janssen Alzheimer Immunotherapy clinical trials. She has spoken at symposia sponsored by Eli Lilly, Biogen, and Janssen. She receives research support from Janssen Pharmaceuticals and Eli Lilly and Co. R. Sperling received funding from P01AG036694, U01AG032438, U01AG024904, P50AG005134, U19AG010483, R01AG037497, R01AG034556, R01AG053798, R01AG027435, R01AG054029, K24AG035007, Fidelity Biosciences, Harvard NeuroDiscovery Center, and the Alzheimer's Association. E. Mormino has served as a paid consultant for Eli Lilly and Biogen. E. Mormino received funding from K01AG051718. Go to Neurology.org/N for full disclosures.
References
- 1.Escott-Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain 2015;138:3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ridge PG, Mukherjee S, Crane PK, Kauwe JS. Alzheimer's disease: analyzing the missing heritability. PLoS One 2013;8:e79771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mormino EC, Papp KV, Rentz DM, et al. Early and late change on the preclinical Alzheimer's cognitive composite in clinically normal older individuals with elevated amyloid-β. Alzheimer's Dement 2017;13:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beecham GW, Hamilton K, Naj AC, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet 2014;10:e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desikan RS, Schork AJ, Wang Y, et al. Polygenic overlap between C-reactive protein, plasma lipids, and Alzheimer disease. Circulation 2015;131:2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.International Genomics of Alzheimer's Disease Consortium. Convergent genetic and expression data implicate immunity in Alzheimer's disease. Alzheimer's Dement 2015;11:658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 2015;77:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and “wingmen.” Nat Neurosci 2015;18:800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer's disease: definition, natural history, and diagnostic criteria. Alzheimer's Dement 2016;12:292–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim YY, Maruff P, Pietrzak RH, et al. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer's disease. Brain 2014;137:221–231. [DOI] [PubMed] [Google Scholar]
- 11.Petersen RC, Wiste HJ, Weigand SD, et al. Association of elevated amyloid levels with cognition and biomarkers in cognitively normal people from the community. JAMA Neurol 2016;73:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snitz BE, Weissfeld LA, Lopez OL, et al. Cognitive trajectories associated with β-amyloid deposition in the oldest-old without dementia. Neurology 2013;80:1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roe CM, Fagan AM, Grant EA, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology 2013;80:1784–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 2015;313:1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mormino EC, Betensky RA, Hedden T, et al. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology 2014;82:1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim YY, Villemagne VL, Pietrzak RH, et al. APOE ε4 moderates amyloid-related memory decline in preclinical Alzheimer's disease. Neurobiol Aging 2015;36:1239–1244. [DOI] [PubMed] [Google Scholar]
- 17.Lim YY, Villemagne VL, Laws SM, et al. BDNF Val66Met, Aβ amyloid, and cognitive decline in preclinical Alzheimer's disease. Neurobiol Aging 2013;34:2457–2464. [DOI] [PubMed] [Google Scholar]
- 18.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated β-amyloid deposition in elderly subjects. Brain 2009;132:1310–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 2012;6:502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav 2012;6:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. NeuroImage 2012;61:1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case-control association studies. Nat Protoc 2010;5:1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2010;34:816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Euesden J, Lewis CM, O'Reilly PF. PRSice: polygenic risk score software. Bioinformatics 2014;31:1466–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim YY, Villemagne VL, Laws SM, et al. APOE and BDNF polymorphisms moderate amyloid β-related cognitive decline in preclinical Alzheimer's disease. Mol Psychiatry 2015;20:1322–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mormino EC, Sperling RA, Holmes AJ, et al. Polygenic risk of Alzheimer disease is associated with early-and late-life processes. Neurology 2016;87:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris SE, Davies G, Luciano M, et al. Polygenic risk for Alzheimer's disease is not associated with cognitive ability or cognitive aging in non-demented older people. J Alzheimer's Dis 2014;39:565–574. [DOI] [PubMed] [Google Scholar]
- 31.Lacour A, Espinosa A, Louwersheimer E, et al. Genome-wide significant risk factors for Alzheimer's disease: role in progression to dementia due to Alzheimer's disease among subjects with mild cognitive impairment. Mol Psychiatry 2017;22:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan CH, Fan CC, Mormino EC, et al. Polygenic hazard score: an enrichment marker for Alzheimer's associated amyloid and tau deposition. Acta Neuropathol 2017;24:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med 2017;14:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolk DA, Dickerson BC, Weiner M, et al. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional–executive network function in Alzheimer's disease. Proc Natl Acad Sci USA 2010;107:10256–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabuncu MR, Buckner RL, Smoller JW, Lee PH, Fischl B, Sperling RA. The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb Cortex 2012;22:2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lam B, Masellis M, Freedman M, Stuss DT, Black SE. Clinical, imaging, and pathological heterogeneity of the Alzheimer's disease syndrome. Alzheimer's Res Ther 2013;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 2007;8:663–672. [DOI] [PubMed] [Google Scholar]
- 39.Foley SF, Tansey KE, Caseras X, et al. Multimodal brain imaging reveals structural differences in Alzheimer's disease polygenic risk carriers: a study in healthy young adults. Biol Psychiatry 2017;81:154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:351–357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All imaging, genetic, demographics, and neuropsychological composite scores used this article are publicly available and were downloaded from the ADNI website (adni.loni.usc.edu). Structural MRI data and GWAS data were downloaded and used to derive the measurements used here (hippocampus volume and PRS). The authors will provide a list of ADNI participant identifications on request for replication purposes.