

Abstract
Group 3 tumors account for 28% of medulloblastomas and have the worst prognosis. FTY720, an immunosuppressant currently approved for treatment of multiple sclerosis, has shown antitumor effects in several human cancer cell lines. We hypothesized that treatment with FTY720 (fingolimod) would decrease tumorigenicity in medulloblastoma patient-derived xenografts (PDXs). Three Group 3 medulloblastoma PDXs (D341, D384 and D425) were utilized. Expression of PP2A and its endogenous inhibitors I2PP2A and CIP2A was detected by immunohistochemistry and immunoblotting. PP2A activation was measured via phosphatase activation kit. Cell viability, proliferation, migration and invasion assays were performed after treatment with FTY720. Cell cycle analysis was completed using flow cytometry. A flank model using D425 human medulloblastoma PDX cells was used to assess the in vivo effects of FTY720. FTY720 activated PP2A and led to decreased medulloblastoma PDX cell viability, proliferation, migration and invasion and G1 cell cycle arrest in all three PDXs. FTY720 treatment of mice bearing D425 medulloblastoma PDX tumors resulted in a significant decrease in tumor growth compared to vehicle treated animals. FTY720 decreased viability, proliferation, and motility in Group 3 medulloblastoma PDX cells and significantly decreased tumor growth in vivo. These results suggest that FTY720 should be investigated further as a potential therapeutic agent for medulloblastoma.
Introduction
Medulloblastoma is the most common malignant primary nervous system tumor in children1. Once considered a singular pathology, medulloblastoma is now more appropriately classified into four molecular subgroups, each having unique clinical and molecular characteristics: Wingless (WNT), Sonic Hedgehog (SHH), Group 3, and Group 42–4. Group 3 tumors account for approximately 25–30% of medulloblastomas and have the worst prognosis3,5,6. Despite current multimodal therapy with surgery, chemotherapy and radiation, infants and children with Group 3 tumors have a 5-year overall survival of 45 and 58%, respectively5. Furthermore, children who survive often suffer from long-term motor, sensory, endocrine, and neuropsychologic sequelae7,8. Novel therapeutic strategies to effectively treat this challenging malignancy are clearly needed.
FTY720 (2-Amino-2-[2-(4-octylphenyl)]-1,3-propanediol, fingolimod) is a synthetic sphingosine immunosuppressant that was approved by the United States Food and Drug Administration (FDA) for the treatment of multiple sclerosis9,10. Over the past decade, FTY720 has also been shown to have anti-tumor properties in several human malignancies11–13, including glioblastoma14–16. FTY720 produces these effects through multiple proposed mechanisms, including activation of the tumor suppressor protein phosphatase 2A (PP2A)17,18, down regulation of cyclin D119, inhibition of sphingosine kinase 1 (SphK1)20,21, and generation of reactive oxygen species (ROS)14,22,23. FTY720 is also thought to induce cell death through caspase-dependent apoptosis24 as well as necroptosis14,25 and autophagy14,25.
Because of the anti-tumor properties of FTY720 seen in other human malignancies, we hypothesized that it may also have anti-tumor effects on medulloblastoma. In this study, we demonstrated that FTY720 treatment led to decreased cell viability, migration and invasion, and caused cell cycle arrest and apoptosis in Group 3 medulloblastoma patient-derived xenografts (PDXs). FTY720 also significantly decreased tumor growth in vivo in mice bearing Group 3 medulloblastoma.
Results
FTY720 treatment increased PP2A activity
PP2A protein expression has been shown to be downregulated in medulloblastoma tumors and hypothesized to lead to upregulation of tumorigenic signaling pathways26. H&E staining confirmed that the PDXs represented medulloblastoma histologically (Fig. 1A, top panel). IHC demonstrated that PP2A and its endogenous inhibitors, PP2A inhibitor 2 (I2PP2A/SET) and cancerous inhibitor of PP2A (CIP2A), were present in all three medulloblastoma PDXs (Fig. 1A). Further, immunoblotting confirmed expression of these three proteins in whole cell lysates from the three human Group 3 medulloblastoma PDXs (Fig. 1B). Since activation of the tumor suppressor protein, PP2A, has been proposed as a mechanism by which FTY720 produces its anti-tumor effects17,18, the ability of FTY720 to activate PP2A was investigated. Following treatment of D341, D384, and D425 cells with FTY720 (5 µM) for 4 hours, the activity of PP2A was significantly increased over baseline relative to control in all three medulloblastoma PDX cell lines (Fig. 1C). It has been proposed that FTY720 targets the endogenous PP2A inhibitors, I2PP2A/SET and CIP2A, as a mechanism for PP2A activation27,28. Protein levels of CIP2A and I2PP2A/SET were measured following FTY720 treatment. CIP2A levels were diminished in the D341 and D384 MB PDXs (Fig. 1D, top left and middle panels), but was not changed in the D425 MB PDX (Fig. 1D, top right panel). I2PP2A levels were not altered (Fig. 1D, middle panels).
Figure 1.

FTY720 treatment increased protein phosphatase 2A activity. (A) H&E staining confirmed histology consistent with medulloblastoma in PDXs (top panels). Immunohistochemistry was performed to demonstrate I2PP2A, CIP2A and PP2A expression in formalin-fixed, paraffin-embedded human medulloblastoma PDXs. IgG negative controls reacted appropriately (second row panels, lower left insets). (B) Immunoblotting confirmed I2PP2A, CIP2A, and PP2A expression in whole cell lysates from these 3 medulloblastoma PDXs. (C) PP2A activity was measured in all 3 medulloblastoma PDXs. Treatment with FTY720 (5 µM) for 4 hours resulted in a significant increase in PP2A activity in all three PDX’s tested (*p ≤ 0.05). Experiments were repeated at least in triplicate and reported as mean ± SEM. (D) Protein levels of CIP2A and I2PP2A (SET) were measured following FTY720 treatment. CIP2A was diminished in the D341 and D384 MB PDXs (top left and middle panels), but not in the D425 MB PDX (top right panel). I2PP2A levels were not altered (middle panels).
FTY720 prevented progression through the cell cycle in medulloblastoma PDX cells
FTY720 is also known to affect cell cycle progression in other cancers19, so we hypothesized that medulloblastoma cells treated with FTY720 may fail to progress through the cell cycle. D425, D341 and D384 cells were treated with FTY720 (5 µM) for 24 hours and cell cycle analysis performed using PI staining. Representative histograms from all three PDX cell lines are presented in Fig. 2A. There was a significant increase in the percentage of cells in G1 phase in all three medulloblastoma PDX cell lines (Fig. 2B) indicating G1 cell cycle arrest. In addition, the percentage of cells in S phase in the D425 and D384 PDXs was significantly decreased over baseline controls, further indicating G1 cell cycle arrest (Fig. 2B). S phase in the D341 PDX was also decreased, but did not reach statistical significance. Data are presented in tabular form in Fig. 2C. These data indicate that FTY720 resulted in G1 cell cycle arrest in the medulloblastoma PDXs.
Figure 2.

FTY720 treatment of medulloblastoma PDXs led to cell cycle arrest. (A) Representative histograms for cell cycle analysis of D425, D341, and D384 human medulloblastoma PDX cells following treatment with FTY720 (5 µM for 24 hours). Cells were analyzed by flow cytometry following staining with propidium iodine. There was an increase in the percentage of cells in the G1 phase and a decrease in the percentage in S phase following FTY720 treatment. (B) Graphic representation of cell cycle analysis in D425, D341 and D384 medulloblastoma PDX cells treated with FTY720. There was a significant increase in the G1 phase in cells from all three PDXs (*p ≤ 0.05) and a significant decrease in S phase in the D425 and D384 cells (*p ≤ 0.05) after FTY720 treatment, indicating G1 cell cycle arrest. Experiments were repeated at least in triplicate and reported as mean ± SEM. (C) Cell cycle data presented in tabular form reporting mean ± standard error of the mean (SEM). Statistically significant changes are noted with bold type (p ≤ 0.05).
FTY720 treatment resulted in decreased viability and proliferation and led to apoptosis of medulloblastoma PDX cells
FTY720 has been shown to decrease cancer cell viability in a variety of tumor types11,13,24, so we examined whether FTY720 would decrease cell viability in medulloblastoma PDXs. Cells were treated with increasing concentrations of FTY720 for 24 hours and viability was measured. FTY720 significantly decreased viability in all 3 medulloblastoma PDX cell lines (Fig. 3A). The lethal dose 50% (LD50) for FTY720 was 7.5 µM in D425, 5.5 µM in D341, and 7.3 µM in D384 cells. Cell proliferation was also measured following treatment for 24 hours with increasing concentrations of FTY720. There was a significant decrease in proliferation seen in all 3 medulloblastoma PDXs following FTY720 treatment (Fig. 3B).
Figure 3.

FTY720 treatment resulted in decreased viability and proliferation of medulloblastoma PDXs. (A) Cell viability was measured using alamarBlue® assays. D425, D341 and D384 cells were treated with increasing concentrations of FTY720 for 24 hours. Viability was significantly decreased in the D341 beginning at a concentration of 4 µM (*p ≤ 0.01). D425 and D384 cells showed significant decreases in viability at concentrations of 6 µM (*p ≤ 0.01). (B) CellTiter 96® assays were used to measure proliferation. D425, D341 and D384 cells were treated with increasing concentrations of FTY720 for 24 hours. Proliferation was significantly decreased in the D341 and D384 cells at 4 µM (*p ≤ 0.01) and at 6 µM (*p ≤ 0.01) in the D425 cells. Experiments were repeated at least in triplicate and reported as mean ± SEM.
Since FTY720 treatment induced cancer cell apoptosis in other cancers16,22,23, we next determined whether the decrease in cell viability observed with FTY720 in medulloblastoma PDX cells may be secondary to apoptosis. Apoptosis was measured by immunoblotting for either loss of total PARP, or an increase in cleaved PARP or an increase in cleaved caspase 3, all of which are known to be indicative of apoptosis. In the D425 PDX, there was an increase in both cleaved PARP and cleaved caspase 3 with increasing concentrations of FTY720 (Fig. 4A, upper and lower panels). Cleaved PARP was also increased in the D341 cell line with FTY720 treatment (Fig. 4B). In the D384 cells, total PARP decreased (Fig. 4C, upper panels) and cleaved caspase 3 increased (Fig. 4C, lower panels) with increasing concentrations of FTY720. These data showed that FTY720 treatment resulted in apoptosis in the medulloblastoma PDX cells.
Figure 4.

FTY720 treatment resulted in apoptosis of medulloblastoma PDX cells. Western blotting for cleaved PARP, total PARP, and cleaved caspase 3 of whole cell lysates from medulloblastoma PDX cells treated with increasing concentrations of FTY720. (A) In D425 cells, FTY720 resulted in increased cleaved PARP and increased caspase 3 indicating apoptosis. (B) In D341 cells, increasing concentrations of FTY720 led to increased cleaved PARP, again indicating apoptosis. (C) D384 cells, treated with increasing concentrations of FTY720, showed decreased total PARP and increased cleaved caspase 3, both indicating apoptosis. Β-actin or GAPDH was utilized to demonstrate equal protein loading.
Migration and invasion of medulloblastoma PDX cells was decreased after treatment with FTY720
The ability to migrate and invade is a hallmark behavior of cancer cells. Therefore, we investigated whether FTY720 treatment decreased medulloblastoma cell migration and invasion. Since non-viable cells will not migrate or invade, cells were treated with concentrations of FTY720 (0 or 3 µM) well below their LD50 and migration and invasion was assessed after 24 hours. FTY720 resulted in a significant decrease in both migration (Fig. 5A,B) and invasion (Fig. 5C,D) in all 3 medulloblastoma PDXs.
Figure 5.

Migration and invasion of medulloblastoma PDX cells was decreased after treatment with FTY720. (A) Migration was determined using Transwell® inserts with 8 μM pores, coated on the bottom with laminin. Cells were treated with FTY720 (0, 3 µM) for 24 hours and allowed to migrate for 24 hours. Representative photographs of the inserts show decreased cell migration in all 3 PDXs with FTY720 treatment. (B) The number of cells migrating was quantitated and reported in graphic form, again showing over 50% decrease in migration in all 3 PDXs after treatment with FTY720. (C) Invasion was completed using Transwell® inserts with 8 μM pores, coated on the bottom with laminin and the inside of the inserts coated with Matrigel™. Cells were treated with FTY720 (0, 3 µM) for 24 hours and allowed to invade for 24 hours. Representative photographs of the inserts show decreased cell invasion in all 3 PDXs with FTY720 treatment. (D) The number of cells invading was quantitated and reported in graphic form, demonstrating significantly decreased invasion in all 3 PDXs after treatment with FTY720. Photographs are representative of three independent experiments showing similar results. Graphs are means of three experiments with data reported as mean ± SEM.
FTY720 decreased medulloblastoma tumor growth in vivo
For in vivo testing of FTY720 against medulloblastoma tumor growth, D425 human medulloblastoma PDX cells (2.5 × 106 in Matrigel™) were injected into the right flank of athymic nude mice (n = 15). The flank model was chosen as it allowed us to accurately measure changes in tumor size over time. Once tumors were palpable (100 mm3) animals were randomized to receive daily oral doses of vehicle (N = 7) or FTY720 (10 mg/kg/day) (N = 8) for 5 weeks. This dosage was chosen based on previous literature reports15,29,30. This time period was based upon prior experiments in our laboratory showing that 5 weeks was the usual maximal survival for animals with untreated tumors. At the end of 5 weeks of treatment, all surviving animals were euthanized. The animals treated with FTY720 showed a significant decrease in tumor volume when compared to vehicle treated animals (Fig. 6A). At 5 weeks, there were no surviving animals in the vehicle treated group and 3 in the FTY720 treated group. The individual tumor growth curves were also markedly different, with the vehicle-treated tumors doubling in size twice as fast as the FTY720 treated tumors (Supplemental Data Fig. 1). The median time for tumor doubling in the vehicle treated group was 4 days (range: 2–8 days) compared to 8 days (range: 5–13 days) in the FTY720 treated animals. FTY720 did not significantly affect the weight of the animals (Fig. 6B).
Figure 6.
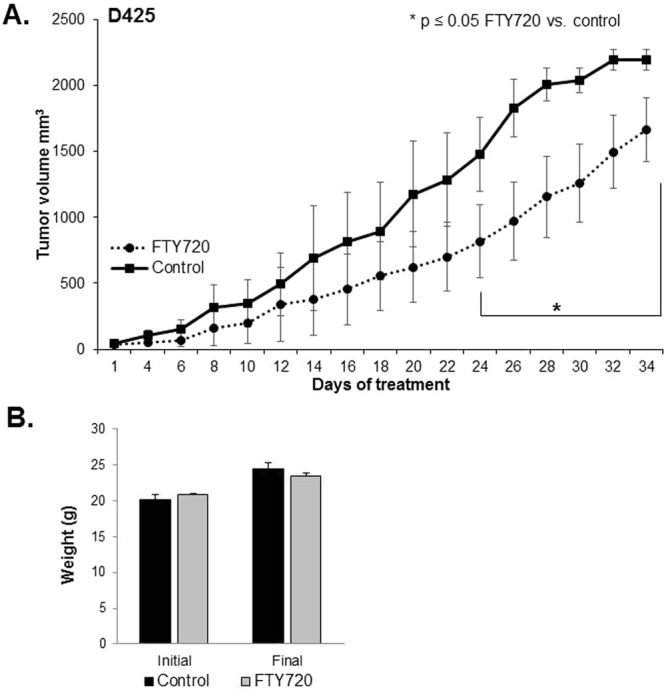
FTY720 decreased medulloblastoma tumor growth in vivo. (A) D425 cells (2.5 × 106 cells in 25% Matrigel™) were injected into the right flank of 6-week-old, female, athymic nude mice. When tumors reached an average of 100 mm3, mice were randomized to receive either vehicle (N = 7) or FTY720 (10 mg/kg/day) (N = 8) for 5 weeks. Animals treated with FTY720 (closed circles) had significantly smaller tumors than those treated with vehicle alone (closed squares). (B) Mice were weighed at the beginning of the experiment and at the time of euthanasia. There was no significant difference in animal weights between those treated with vehicle and those treated with FTY720.
Discussion
Despite aggressive therapy including surgery, multi-agent chemotherapy and craniospinal radiation, outcomes for children with Group 3 medulloblastoma remain poor, and survivors often suffer from debilitating life-long toxicities related to these interventions. There is a great need for new therapies to improve outcomes and reduce the toxicities from current treatments. Most current investigations are focused on discovery of new compounds, the repurposing of previously approved compounds, and the development of novel combinatorial therapies.
The use of immunosuppressant drugs to fight cancer is not a new concept and is one that has been implemented with a number of compounds31,32. FTY720, a metabolite from Isaria sinclairii, was developed as an immunosuppressant for use in transplantation, and is currently utilized in the treatment of multiple sclerosis. Recently, FTY720 was found to have an inhibitory effect upon human cancer cells. In 2001, Sonoda and colleagues reported that FTY720 induced apoptosis in human glioma cells33 and another group reported FTY720 treatment led to apoptosis of glioma stem cells16. Subsequent studies showed similar effects with decreased viability and apoptosis in other human tumor types including bladder cancer11, hepatoma34, pancreatic cancer24 and leukemia35. These promising findings prompted an investigation of the potential impact of FTY720 on medulloblastoma.
The mechanism of action of FTY720 in cancer cells is not completely defined. It has been hypothesized that FTY720 activates PP2A17,18 thereby leading to tumor cell death. FTY720-induced PP2A activation has been postulated to occur through direct effects or through targeting of I2PP2A/SET27 or CIP2A28, the endogenous inhibitors of PP2A. PP2A is a known tumor suppressor that is inactive or downregulated in many human tumors either through upregulation of its endogenous inhibitors, I2PP2A/SET36,37 or CIP2A38–40, or through mutations41 which limit the activity of PP2A. PP2A has been shown to be downregulated in medulloblastoma tumors leading to upregulation of tumorigenic signaling pathways26. In the current study, we demonstrated that PP2A and its two endogenous inhibitors I2PP2A/SET and CIP2A are expressed in medulloblastoma PDX cells. In addition, FTY720 treatment of human medulloblastoma PDXs resulted in activation of PP2A in the face of decreased overall PP2A protein expression, but changes in I2PP2A/SET or CIP2A expression were not consistently seen. These findings have been reported by other authors who have noted that CIP2A or I2PP2A/SET expression was not altered by FTY72042.
In this study, treatment of human medulloblastoma PDX cells with FTY720 arrested the cell cycle at G1. These effects were noted with low concentrations of FTY720 (5 µM). Other investigators have demonstrated similar findings. Permpongkosol et al. showed an increased percentage of DU145 prostate cancer cells in G1 and decreased percentage in S phase following FTY720, but at a much higher dose (20 µM)43. Increased percentages of cells in G1 were seen with FTY720 treatment of both gastric cancer cells44 and in acute myeloid leukemia cells45 in concentrations similar to those utilized in the current study (5 µM).
In addition to reducing cell viability, FTY720 has been shown to reduce tumor cell migration and invasion. Zhang and others showed that human glioma cells had a marked reduction in migration and invasion following FTY720 treatment15. Another study in human prostate cancer cells demonstrated a decrease in migration and invasion with FTY720 treatment46. Similarly, in the current study, FTY720 in doses well below the LD50 resulted in significant reductions in both migration and invasion in human medulloblastoma cells. Further, as postulated by previous investigators15, the AKT pathway may be involved in these findings as AKT phosphorylation was markedly decreased in D341 and D384 cells with increasing concentrations of FTY720 (Supplemental Fig. 2). FTY720-induced dephosphorylation of AKT has been noted in other tumor types including mesothelioma47, prostate cancer48 and breast cancer49.
Other investigators have proposed changes in ERK phosphorylation as a potential downstream target of FTY720. Rincon and colleagues showed decreased activation of ERK in BT-474 and MDA-MB-231 breast cancer cells following treatment with FTY72049. Cristóbal and colleagues reported similar results with PC-3 prostate cancer cells. When these cells were exposed to 10 µM FTY720, ERK phosphorylation was decreased48. In the current study, D384 medulloblastoma PDX cells showed decreased ERK phosphorylation without a change in total ERK expression following FTY720 treatment (Supplemental Data Fig. 2). On the contrary, phosphorylation of ERK was increased in the D341 PDX cells with increasing doses of FTY720. These data indicate that the effects of FTY720 on kinases may be cell line dependent, and along with the findings for AKT, will serve as a basis for further mechanistic studies of FTY720 effects in medulloblastoma.
Using an in vivo mouse xenograft model of D425 human medulloblastoma, we found that FTY720 treatment led to a significant decrease in tumor growth compared to untreated animals. A flank model was chosen to allow more careful determination of tumor growth over the time period of study and to adhere to the three R’s of animal use; reduce, replace, and refine. We believe that FTY720 would be applicable to the orthotopic location as it has been documented to pass the blood brain barrier and have effects upon intracranial glioblastoma16 as well as other intracranial pathologies50,51.
Our results provide evidence that FTY720 decreased medulloblastoma cell viability, migration, invasion, and in vivo tumor growth. These findings are novel as FTY720 has not yet been studied as a therapeutic tool for medulloblastoma. Furthermore, the use of human PDX medulloblastoma cell lines in these studies is also important as it provided a clinically relevant model for the preclinical study of this compound in medulloblastoma. These data suggest that FTY720 may be a novel therapeutic for treating medulloblastoma and further clinical evaluation is warranted.
Materials and Methods
Patient-derived medulloblastoma xenografts
Three Group 3 medulloblastoma xenografts52,53 established from pediatric patients were used for experiments: D341 Med (D341), D384 Med (D384), and D425 Med (D425). These tumors were generously provided by Darell D. Bigner, MD, PhD, Duke Medical Center29,30. The xenografts were maintained in athymic nude mice (Envigo, Pratville, AL). After tumors were harvested, the cells were dissociated using a Tumor Dissociation Kit (Miltenyi Biotec, San Diego, CA) per manufacturer’s protocol. All cell lines were maintained in neurobasal medium (Life Technologies, Carlsbad, CA) supplemented with B-27 supplement without Vitamin A (Life Technologies), N2 supplement (Life Technologies), amphotericin B (250 μg/mL), gentamicin (50 μg/mL), L-glutamine (2 mM), epidermal growth factor (10 ng/mL; Miltenyi Biotec) and fibroblast growth factor (10 ng/mL; Miltenyi Biotec). The cells were kept under standard conditions at 37 °C and 5% CO2. All three medulloblastoma PDXs were verified within the last 12 months using short tandem repeat analysis (Heflin Center for Genomic Sciences, UAB, Birmingham, AL).
Reagents and antibodies
FTY720 was purchased from Cayman Chemical (10006292, Cayman Chemical, Ann Arbor, MI). Primary antibodies used for Western blotting included the following: anti-I2PP2A (H-120) (sc-25564) from Santa Cruz Biotechnology (Santa Cruz, CA), anti-PP2A (ab32104) and anti-CIP2A (ab99518) from Abcam (Cambridge, MA), anti-total AKT (9272), anti-phospho-AKT (S473; 9271), p44/42 MAP Kinase [ERK1/2 (9102)], anti-phospho-p44/42 MAPK [phospho-ERK, T202/T204, (4377)], anti-total PARP (9542) and anti-cleaved Caspase-3 (9661) from Cell Signaling Technology (Danvers, MA), anti-β-actin from Sigma (A1978, Sigma Aldrich, St. Louis, MO), anti-cleaved PARP (MAB3565) and anti-GAPDH (MAB374, clone 6C5) from EMD Millipore (EMD Millipore, Billerica, MA).
Immunohistochemistry
Formalin-fixed paraffin-embedded xenograft tumor specimens were sectioned into 6 µm sections and baked at 70 °C for one hour on positive slides. Slides were deparaffinized, steamed, quenched with 3% hydrogen peroxide, and blocked with blocking buffer [bovine serum albumin (BSA), powdered milk, Triton X-100, phosphate buffered saline (PBS)] for 30 minutes at 4 °C. The primary antibodies anti-I2PP2A/SET (rabbit polyclonal, 1:400, sc-25564, Santa Cruz), anti-CIP2A (rabbit polyclonal, 1:200, ab99518, Abcam), and anti-PP2A (rabbit monoclonal, 1:200, ab32104, Abcam) were added and incubated for 1 hour in a humidity chamber at room temperature. After washing with PBS, the secondary antibody for rabbit (R.T.U. biotinylated universal antibody, Vector Laboratories, Burlingame, CA) was added for 1 hour at 22 °C. The staining reaction was developed with VECTASTAIN Elite ABC reagent (PK-7100, Vector Laboratories) and Metal Enhanced DAB Substrate (Thermo Fisher Scientific). Slides were counterstained with hematoxylin. Negative controls (rabbit IgG, 1 µg/mL, EMD Millipore) were included with each run. Routine hematoxylin and eosin staining was performed on each specimen.
PP2A activity assay
Cells (1 × 106 cells) were treated with FTY720 (5 µM) for 4 hours and then lysed using NP-40 lysis buffer. PP2A activity was measured using a PP2A Immunoprecipitation Phosphatase Assay Kit (17–313, EMD Millipore). This kit measures activity of the C subunit of PP2A (clone 106). Briefly, protein lysates were incubated with PP2A antibody at 4C with continuous rotation for 2 hours. Following the addition of assay buffers and malachite green solution, the plate was read at an absorbance of 650nm using a microplate reader (Epoch Microplate Spectrophotometer, BioTek Instruments, Winooski, VT). Phosphatase activity was determined using a standard curve. Experiments were repeated at least in triplicate and phosphatase activity was reported as mean fold change ± standard error of the mean (SEM) from the untreated sample for each cell line.
Immunoblotting
Briefly, cells were lysed on ice for 30 minutes in a buffer consisting of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton x-100, 1% sodium deoxcycholate, 0.1% SDS, phosphatase inhibitor (P5726, Sigma Aldrich), protease inhibitor (P8340, Sigma Aldrich), and phenylmethylsulfonyl fluoride (PMSF, P7626, Sigma Aldrich). The lysates were then centrifuged at 14 000 rpm for 30 minutes at 4 °C. Protein concentrations were determined using a Micro BCA™ Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL), separated by electrophoresis on SDS-PAGE gels, and transferred to Immobilon®-P polyvinylidene fluoride (PVDF) transfer membrane (EMB Millipore). Precision Plus Protein Kaleidoscope Standards (161-0375, Bio-Rad, Hercules, CA) were used for molecular weight markers to confirm expected size of target proteins. Antibodies were used in accordance with the manufacturers’ recommended protocol. Samples were visualized by enhanced chemiluminescence (ECL) using Luminata Classico and Luminata Crescendo Western horseradish peroxidase (HPR) substrates (EMD Millipore). Anti-β-actin was used an internal control to ensure equal protein loading between samples.
Cell cycle analysis
Cell cycle analysis was performed using propidium iodine (PI) staining and flow cytometric evaluation. Cells (1 × 106 cells) were plated and treated with FTY720 (5 µM). After 24 hours, a single cell suspension was achieved using Accutase® (Sigma). The cells were then washed with PBS and fixed in 1mL of ice-cold, 100% ethanol overnight. The cells underwent a second PBS wash and were stained for 1 hour with 200 μL of staining solution consisting of 20 μg/mL propidium iodide [(PI), Invitrogen, Carlsbad, CA], 0.1% Triton X (Active Motif, Carlsbad, CA) and RNAse A (0.1 mg/mL, Qiagen, Valencia, CA). The samples were analyzed with fluorescence-activated cell sorting (FACS) using a FACSCalibur™ Flow Cytometer (BD Biosciences, San Jose, CA). ModFit LT software (Verity Software House Inc., Topsham, ME) was used to analyze the data.
Cell viability and proliferation assays
An alamarBlue® assay (Thermo Fisher Scientific) was performed to assess cell viability following treatment with FTY720. Cells (1.5 × 103 cells) were plated and treated with increasing concentrations of FTY720. After 24 hours, 10 μL of alamarBlue® dye was added to each well. The plates were read at using a microplate reader (Epoch Microplate Spectrophotometer) to record the absorbance at 570 nm, using 600 nm as a reference wavelength. Experiments were completed in triplicate and viability reported as fold change ± SEM.
Proliferation was assessed using the CellTiter 96® Aqueous One Solution Cell Proliferation assay (Promega, Madison, WI). Medulloblastoma cells (5 × 103 cells) were plated and treated with FTY720 at increasing concentrations. After 24 hours, 10 μL CellTiter96® dye was added to each well and the absorbance was measured at 490 nm using a microplate reader (Epoch Microplate Spectrophotometer). Experiments were repeated in triplicate and proliferation reported as fold change ± SEM.
Cell migration and invasion assays
Cell migration and invasion assays were performed using 6.5 mm Transwell® inserts with 8 μM pore polycarbonate membrane (Corning Inc., Corning, NY) in 24-well culture plates. The bottoms of the inserts were coated with laminin (10 μg/mL). For invasion assays, the inside of the inserts were also coated with Matrigel™ (1 mg/mL, 50 µL; BD Biosciences) overnight at 37 °C and then washed with PBS. Cells were pretreated with FTY720 (0, 3 µM) for 24 hours and then 1.5 × 105 cells plated into the top of the insert. The insert was then placed into a well containing 300 μL of treated media containing 10% fetal bovine serum (FBS) as a chemo-attractant. After 24 hours, the cells on top of the inserts were removed using a cotton swab. The inserts were then fixed in 3% paraformaldehyde prior to staining with crystal violet. The imaging software SPOT Basic 5.2 (Diagnostic Instruments Inc., Sterling Heights, MI) was used to take pictures of the inserts at predetermined locations with a microscope at 100× and then the cells were quantified using ImageJ software (Ver 1.49, available online at http://imagej.nih.gov/ij)54. Experiments were repeated in triplicate and migration and invasion reported as fold change ± SEM.
Animal statement
Animal experiments were approved by the University of Alabama, Birmingham Institutional Animal Care and Use Committee (IACUC-09355) and were conducted within institutional, national, and NIH guidelines.
In vivo tumor growth
D425 cells (2.5 × 106 cells in 25% Matrigel™; Corning Inc.) were injected into the right flank of 6-week-old, female, athymic nude mice (Envigo, Pratville, AL). Once tumors were palpable (100 mm3), the mice were randomized to receive either 50 µL suspension vehicle (ORA-Plus®, Perrigo, Allegan, MI) or FTY720 10 mg/kg/day suspended in 50 µL ORA-Plus® once daily via oral gavage for 5 weeks. The FTY720 dosing was based on previous animal studies16,47,55. The flank tumors were measured twice weekly using calipers and tumor volume was calculated using the formula [(width2 × length/2] with length being the largest measurement. The mice remaining at the end of the treatment period were humanely euthanized.
Statistical analyses
Experiments were performed at a minimum of triplicate. Densitometry was performed utilizing Scion Image Program (http://www.nist.gov/lispix/imlab/prelim/dnld.html). Each band was normalized to background, then to their respective β-actin, and finally the phospho band was normalized to the total protein expression band. All bands were then normalized to 0 µM treatment group as previously reported56. Data reported as the mean ± standard error of the mean. Data between groups was compared using an analysis of variance or Student’s t test as appropriate. Statistical significance was defined as p < 0.05.
Data availability statement
No data sets were generated in the current studies.
Electronic supplementary material
Acknowledgements
This work was partially funded by the UAB Comprehensive Cancer Center Pilot Grant in Pediatric Cancer (5P30 CA013148-40; GK Friedman, E.A. Beierle) and in part by institutional grants from the National Cancer Institute including T32 CA091078 training grant in surgical oncology (E.F. Garner, L.L. Stafman), T32 CA183926 training grant in translational oncology (A.P. Williams) and flow cytometry core grants P30 AR048311 and P30 AI027767.
Author Contributions
E.F. Garner, A.P. Williams, L.L. Stafman, and J.M. Aye performed in vitro and in vivo experiments. E. Mroczek-Musulman reviewed pathology. J.E. Stewart and B.P. Moore passaged and maintained PDXs. G.K. Friedman and E.A. Beierle designed studies and E.A. Beierle prepared figures and wrote manuscript. All authors reviewed and edited the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-25263-5.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: clinical and biologic aspects. Neuro. Oncol. 1999;1:232–250. doi: 10.1215/15228517-1-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottardo NG, et al. Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol. 2014;127:189–201. doi: 10.1007/s00401-013-1213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Northcott PA, et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor MD, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kool M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho YJ, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011;29:1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massimino M, et al. Childhood medulloblastoma. Crit. Rev. Oncol. Hematol. 2016;105:35–51. doi: 10.1016/j.critrevonc.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Diller L, et al. Chronic disease in the Childhood Cancer Survivor Study cohort: a review of published findings. J. Clin. Oncol. 2009;27:2339–2355. doi: 10.1200/JCO.2008.21.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kappos L, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 10.Kappos L, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 11.Azuma H, et al. Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J. Urol. 2003;169:2372–2377. doi: 10.1097/01.ju.0000064938.32318.91. [DOI] [PubMed] [Google Scholar]
- 12.Azuma H, et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62:1410–1419. [PubMed] [Google Scholar]
- 13.Lu Z, et al. FTY720 inhibits proliferation and epithelial-mesenchymal transition in cholangiocarcinoma by inactivating STAT3 signaling. B.M.C. Cancer. 2014;14:783. doi: 10.1186/1471-2407-14-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Wang H, Ding K, Xu J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol. Lett. 2015;236:43–59. doi: 10.1016/j.toxlet.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Wang H, Zhu J, Ding K, Xu J. FTY720 reduces migration and invasion of human glioblastoma cell lines via inhibiting the PI3K/AKT/mTOR/p70S6K signaling pathway. Tumour Biol. 2014;35:10707–10714. doi: 10.1007/s13277-014-2386-y. [DOI] [PubMed] [Google Scholar]
- 16.Estrada-Bernal A, Palanichamy K, Chaudhury RA, Van Brocklyn JR. Induction of brain tumor stem cell apoptosis by FTY720: a potential therapeutic agent for glioblastoma. Neuro. Oncol. 2012;14:405–415. doi: 10.1093/neuonc/nos005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood. 2008;111:275–284. doi: 10.1182/blood-2006-10-053884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Huang Q, Lu Y, Li X, Huang S. Reactivating PP2A by FTY720 as a novel therapy for AML with C-KIT tyrosine kinase domain mutation. J. Cell Biochem. 2012;113:1314–1322. doi: 10.1002/jcb.24003. [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, et al. FTY720 shows promising in vitro and in vivo preclinical activity by downmodulating Cyclin D1 and phospho-Akt in mantle cell lymphoma. Clin Cancer Res. 2010;16:3182–3192. doi: 10.1158/1078-0432.CCR-09-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JW, et al. Sphingosine kinase 1 as a potential therapeutic target in epithelial ovarian cancer. Int. J. Cancer. 2015;137:221–229. doi: 10.1002/ijc.29362. [DOI] [PubMed] [Google Scholar]
- 21.Rosa R, et al. Sphingosine kinase 1 overexpression contributes to cetuximab resistance in human colorectal cancer models. Clin. Cancer Res. 2013;19:138–147. doi: 10.1158/1078-0432.CCR-12-1050. [DOI] [PubMed] [Google Scholar]
- 22.Wallington-Beddoe CT, Hewson J, Badstock KF, Bendall LJ. FTY720 produces caspase-independent cell death of acute lymphoblastic leukemia cells. Autophagy. 2011;7:707–715. doi: 10.4161/auto.7.7.15154. [DOI] [PubMed] [Google Scholar]
- 23.Pereira FV, et al. FTY720 induces apoptosis in B16F10-NEX2 murine melanoma cells, limits metastatic development in vivo, and modulates the immune system. Clinics (Sao Paulo) 2013;68:1018–1027. doi: 10.6061/clinics/2013(07)21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, et al. FTY720, a synthetic compound from Isaria sinclairii, inhibits proliferation and induces apoptosis in pancreatic cancer cells. Cancer Lett. 2007;254:288–297. doi: 10.1016/j.canlet.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 25.Zhang N, et al. FTY720 induces necrotic cell death and autophagy in ovarian cancer cells: a protective role of autophagy. Autophagy. 2010;6:1157–1167. doi: 10.4161/auto.6.8.13614. [DOI] [PubMed] [Google Scholar]
- 26.Włodarski P, Grajkowska W, Łojek M, Rainko K, Jóźwiak J. Activation of Akt and Erk pathways in medulloblastoma. Folia Neuropathol. 2006;44:214–220. [PubMed] [Google Scholar]
- 27.Saddoughi SA, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013;5:105–121. doi: 10.1002/emmm.201201283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, et al. FTY720-induced enhancement of autophagy protects cells form FTY720 cytotoxicity in colorectal cancer. Oncol. Rep. 2016;35:2833–2842. doi: 10.3892/or.2016.4668. [DOI] [PubMed] [Google Scholar]
- 29.He XM, et al. Phenotypic analysis of four human medulloblastoma cell lines and transplantable xenografts. J. Neuropathol. Exp. Neurol. 1989;48:48–68. doi: 10.1097/00005072-198901000-00005. [DOI] [PubMed] [Google Scholar]
- 30.He XM, et al. Differentiation characteristics of newly established medulloblastoma cell lines (D384 Med, D425 Med, and D458 Med) and their transplantable xenografts. Lab Invest. 1991;64:833–843. [PubMed] [Google Scholar]
- 31.Runowicz CD, Fields AL, Goldberg GL. Promising new therapies in the treatment of advanced ovarian cancer. Cancer. 1995;76:2028–2033. doi: 10.1002/1097-0142(19951115)76:10+<2028::AID-CNCR2820761320>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 32.Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit. Rev. Oncol. Hematol. 2005;56:47–60. doi: 10.1016/j.critrevonc.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Sonoda Y, et al. FTY720, a novel immunosuppressive agent, induces apoptosis in human glioma cells. Biochem. Biophys. Res. Commun. 2001;281:282–288. doi: 10.1006/bbrc.2001.4352. [DOI] [PubMed] [Google Scholar]
- 34.Lee TK, et al. FTY720 induces apoptosis of human hepatoma cell lines through PI3-K-mediated Akt dephosphorylation. Carcinogenesis. 2004;25:2397–2405. doi: 10.1093/carcin/bgh250. [DOI] [PubMed] [Google Scholar]
- 35.Neviani P, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J. Clin. Invest. 2007;117:2408–2421. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang SW, Xu S, Chen H, Liu J, Duan P. Oncogenic role of SET/I2PP2A for gynecologic cancers. Curr. Drug Targets. 2017;18:1152–1157. doi: 10.2174/1389450118666170328114506. [DOI] [PubMed] [Google Scholar]
- 37.Sobral LM, Coletta RD, Alberici LC, Curti C, Leopoldino AM. SET/I2PP2A overexpression induces phenotypic, molecular, and metabolic alterations in an oral keratinocyte cell line. F.E.B.S. J. 2017;286:2774–2785. doi: 10.1111/febs.14148. [DOI] [PubMed] [Google Scholar]
- 38.Dong QZ, et al. CIP2A is overexpressed in non-small cell lung cancer and correlates with poor prognosis. Ann. Surg. Oncol. 2011;18:857–865. doi: 10.1245/s10434-010-1313-8. [DOI] [PubMed] [Google Scholar]
- 39.Huang LP, Adelson ME, Mordechai E, Trama JP. CIP2A expression is elevated in cervical cancer. Cancer Biomark. 2010;8:309–317. doi: 10.3233/CBM-2011-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junttila MR, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 41.Colella S, et al. Reduced expression of the Aalpha subunit of protein phosphatase 2A in human gliomas in the absence of mutations in the Aalpha and Abeta subunit genes. Int. J. Cancer. 2001;93:798–804. doi: 10.1002/ijc.1423. [DOI] [PubMed] [Google Scholar]
- 42.Kim MO, et al. Antihelminthic drug niclosamide inhibits CIP2A and deactivates tumor suppressor protein phosphatase 2A in non-small cell lung cancer cells. Biochem. Pharmacol. 2017;144:78–89. doi: 10.1016/j.bcp.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Permpongkosol S, et al. Anticarcinogenic effect of FTY720 in human prostate carcinoma DU145 cells: modulation of mitogenic signaling, FAK, cell-cycle entry and apoptosis. Int. J. Cancer. 2002;98:167–172. doi: 10.1002/ijc.10178. [DOI] [PubMed] [Google Scholar]
- 44.Zheng T, et al. PTEN- and p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric cancer cells and nude mice. J. Cell Biochem. 2010;111:218–228. doi: 10.1002/jcb.22691. [DOI] [PubMed] [Google Scholar]
- 45.Chen L, et al. FTY720 induces apoptosis of M2 subtype acute myeloid leukemia cells by targeting sphingolipid metabolism and increasing endogenous ceramide levels. PLoS One. 2014;9:e103033. doi: 10.1371/journal.pone.0103033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou C, et al. FTY720, a fungus metabolite, inhibits invasion ability of androgen-independent prostate cancer cells through inactivation of RhoA-GTPase. Cancer Lett. 2006;233:36–47. doi: 10.1016/j.canlet.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 47.Szymiczek A, et al. FTY720 inhibits mesothelioma growth in vitro and in a syngeneic mouse model. J. Transl. Med. 2017;15:58. doi: 10.1186/s12967-017-1158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cristóbal I, et al. Activation of the tumor suppressor PP2A emerges as a potential therapeutic strategy for treating prostate cancer. Mar. Drugs. 2015;13:3276–3286. doi: 10.3390/md13063276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rincón R, et al. PP2A inhibition determines poor outcome and doxorubicin resistance in early breast cancer and its activation shows promising therapeutic effects. Oncotarget. 2015;6:4299–4314. doi: 10.18632/oncotarget.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rolland WB, et al. Fingolimod confers neuroprotection through activation of Rac1 after experimental germinal matrix hemorrhage in rat pups. J. Neurochem. 2017;140:776–786. doi: 10.1111/jnc.13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li, W., Xu, H., & Testai, F. D. Mechanism of action and dlinical potential of fingolimod for the treatment of stroke. Front.Neurol. 7, 139. [DOI] [PMC free article] [PubMed]
- 52.Snuderl M, et al. Targeting placental grwoth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152:1065–1076. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedman GK, et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro. Oncol. 2016;18:227–235. doi: 10.1093/neuonc/nov123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HS, et al. Sphingosine kinase 1 is a reliable prognostic factor and a novel therapeutic target for uterine cervical cancer. Oncotarget. 2015;6:26746–26756. doi: 10.18632/oncotarget.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillory LA, Stewart JE, Megison ML, Waters AM, Beierle EA. Focal adhesion kinase and p53 synergistically decrease neuroblastoma cell survival. J Surg Res. 2015;196:339–349. doi: 10.1016/j.jss.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data sets were generated in the current studies.