

Abstract
The microbiome is essential for development, health and homeostasis throughout an animal’s life. Yet, the origins and transmission processes governing animal microbiomes remain elusive for non-human vertebrates, oviparous vertebrates in particular. Eggs may function as transgenerational carriers of the maternal microbiome, warranting characterisation of egg microbiome assembly. Here, we investigated maternal and environmental contributions to avian eggshell microbiota in wild passerine birds: woodlark Lullula arborea and skylark Alauda arvensis. Using 16S rRNA gene sequencing, we demonstrated in both lark species, at the population and within-nest levels, that bacterial communities of freshly laid eggs were distinct from the female cloacal microbiome. Instead, soil-borne bacteria appeared to thrive on freshly laid eggs, and eggshell microbiota composition strongly resembled maternal skin, body feather and nest material communities, sources in direct contact with laid eggs. Finally, phylogenetic structure analysis and microbial source tracking underscored species sorting from directly contacting sources rather than in vivo-transferred symbionts. The female-egg-nest system allowed an integrative assessment of avian egg microbiome assembly, revealing mixed modes of symbiont acquisition not previously documented for vertebrate eggs. Our findings illuminated egg microbiome origins, which suggested a limited potential of eggshells for transgenerational transmission, encouraging further investigation of eggshell microbiome functions in vertebrates.
Introduction
Host-microbe associations are universal to all animals [1] and increasingly recognised as a host trait in evolutionary biology [2]. An emerging paradigm concentrates on identification of the proximate mechanisms underlying natural microbiome variation and prediction of its fitness consequences when under selection [2, 3]. Understanding the origin, maintenance and transience of microbial symbionts across generations of animal hosts is crucial to predict potential host fitness consequences of microbiome variation [4, 5]. Although natural animal-microbiota investigations are mounting [6, 7, 8], enhanced coverage of animal lineages across diverse spatiotemporal scales, between and within host species, is essential to find general and differential features in the ecology and evolution of animal microbiomes.
A few studies already demonstrated that the establishment of a healthy microbiota can constitute a key aspect of vertebrate host fitness, eg, affecting later-life disease risk [9] and offspring growth rate [10]. Microbiome variation resulting from non-random transmission of symbionts from one generation to the next provides opportunities for selection through effects on host fitness [11]. Yet, non-random transgenerational transmission has only been documented in some animal clades and ranges from obligate vertical transmission in insects [12, 13], mixed vertical and horizontal acquisition in sponges [14], lizards [15] and humans [4], to highly selective horizontal symbiont acquisition in squids [16, 17]. This variety of transmission routes (reviewed in ref. [18]) calls for systematic investigations in a broad array of animal species—an effort that is crucial for identifying general patterns and developing general concepts of animal-microbiome dynamics [5, 19]. Since vertical transmission sensu stricto refers to maternal transmission through the germ line [12], it is increasingly considered more broadly by including environmental maternal effects, hereafter referred to as ‘transgenerational transmission’ [4]. As transgenerational transmission is understudied in non-human vertebrate systems [4, 19] compared to humans (eg, [20]), its investigation is fundamental to assess its meaning in evolutionary biology.
Symbiont transmission routes may have evolved alongside the different vertebrate reproductive modes (ie, oviparity, ovoviviparity and viviparity). In humans, birth mode and early-life maternal effects are important factors driving newborn microbiome assembly [20, 21], which might prove universal to most or all viviparous vertebrates [15]. Offspring of oviparous vertebrates, however, hatch (mostly) ex vivo in the exterior environment (eg, oviparous fish and amphibians in water, and reptiles and birds in nests). The lack of maternal-offspring inoculation upon hatching from an egg, especially in the absence of parental care, raises the question of how oviparous vertebrates acquire their (symbiotic) microbiome after hatching.
Considering the microbiome as a host trait [22, 2] and benefitting from birds as benchmark models in evolutionary biology, we argue that studying associations between avian hosts and their microbial communities could aid in uncovering microbiome effects on vertebrate host fitness. Identifying transmission routes and avian microbiome assembly may be a first step towards this goal. As fecundity of oviparous vertebrates heavily relies on egg survival and subsequent survival of offspring, we hypothesise that eggs could have evolved to function as transgenerational carriers of an initial inoculum to hatching offspring, and/or potentially providing a protective shield to invaders causing egg infection [23]. At laying, eggs pass through the distal intestine and then through the cloaca (hereafter ‘cloacal gut’), which serves to expel both faeces and eggs. Yet, healthy eggs of wild birds are thought to be internally sterile, but trans-shell and internal egg infection negatively affect hatchability and offspring survival probability [24–27]. Conversely, egg incubation [28–30] and eggshell smearing with symbiont-containing secretion induce compositional changes that increase hatchability [31]. Additional to such maternal effects, associations between nests and egg microbiota have been identified [32, 33], though high-throughput sequencing data integrating the multi-faceted maternal, nest and eggshell microbiota assembly are lacking. We previously demonstrated significant inter-individual variation among adult females in our lark study system [34], providing potential for non-random transgenerational microbiota transmission. If eggs function as carriers of maternal microbiota, we hypothesised similarity among eggshell and maternal cloacal microbiotas.
Here we used 16S ribosomal RNA (rRNA) gene sequencing to identify sources of avian eggshell microbiota in woodlarks Lullula arborea and skylarks Alauda arvensis. We evaluated alpha diversity and beta diversity of eggs and potential source communities to map egg-source microbiome associations by testing effects of host species, (individual) nests, and resemblance within nests between the first and second laid eggs of each clutch. We assessed phylogenetic structure of eggshell communities to evaluate how selective eggshells are in bacterial community assembly, and used cross-sectional and within-nest analyses of phylogenetic beta diversity to evaluate similarities between eggshell and source communities. Finally, we applied predictive modelling to estimate the proportional contribution of each source to eggshell communities.
Methods
Ethics statement
This study was carried out obeying the Dutch Law on animal experimentation under licence DEC6619B/C of the Institutional Animal Care and Use Committee of the University of Groningen.
Study species and sample collection
We monitored ground-nesting woodlarks L. arborea and skylarks A. arvensis at Aekingerzand, National Park Drents-Friese Wold, the Netherlands (N 52°55′; E 6°18′) between March and July 2014. Both species start incubation after completion of a clutch (four to six eggs for woodlarks and three to four eggs for skylarks). Females contact their eggs while sitting on the nest for protection against rain, heat and nest predation, as well as during overnight resting. Without handling eggs, we marked each first laid egg with a small dot using a water-resistant marker at the blunt end to distinguish it from second eggs. We collected both eggs when the second egg had been laid (first eggs within 36 h and second eggs within 12 h post laying), minimising egg age and incubation effects [35]. We aseptically stored eggs individually in sterile plastic bags (Whirl-Pak®, Nasco, Fort Atkinson, WI, USA), and replaced them by ethanol-sterilised homemade self-hardening clay eggs to encourage clutch completion. Females (96%) continued egg laying after sampling. We captured females as soon as possible after egg collection (mean ± s.e.: 10.3 ± 4.3 days, n = 19). We collected a cloacal swab, a swab sample (moistened with sterile PBS) of exposed brood patch skin, approximately five brood patch-aligning feathers, approximately three grass stems from the centre of the nest cup and surface soil within a 50 cm radius from the nest entrance. We handled birds and samples using new, clean latex gloves while handling and sampling birds, nests and eggs (changing gloves between samples), which we sterilised using 70% ethanol as an extra precaution to prevent sample contamination. In total, we collected 38 woodlark (19 nests) and 38 skylark eggs (19 nests). High nest predation [36] reduced our capture success of females, resulting in fewer female and nest samples than collected eggs (for full details see Table 1). Samples were kept on ice in the field (<12 h) and stored at −20 °C.
Table 1.
Summary of collected eggshell and source community samples of woodlarks and skylarks
| Woodlark | Skylark | |||
|---|---|---|---|---|
| No. of samples | No. of nests (with eggsa) | No. of samples | No. of nests (with eggsa) | |
| Egg | 38 | 19 (19) | 38 | 19 (19) |
| Cloacal gut | 12 | 12 (10) | 15 | 15 (8) |
| Brood patch skin | 13 | 13 (10) | 11 | 11 (8) |
| Feather | 13 | 13 (10) | 11 | 11 (8) |
| Nest material | 14 | 14 (11) | 7 | 7 (7) |
| Surface soil | 14 | 14 (11) | 9 | 9 (7) |
aNumber between brackets denotes the number of nests per sample type category for which the first two eggs of a clutch were successfully collected
DNA extraction and 16S rRNA gene amplicon sequencing
We aseptically isolated eggshells from randomly picked frozen eggs following [29] and powdered eggshells using liquid nitrogen and autoclaved-sterilised mortar and pestle. We aseptically peeled all cotton from swabs and moved it to extraction tubes. We transferred all brood patch-lining feathers and all nest lining grasses into individual sterile 15-ml tubes, added 978 μl sodium phosphate buffer and 122 μl MT buffer (kit reagents), vortexed tubes 10 s using a Vortex-Genie2 (MoBio Laboratories Inc., Carlsbad, CA), sonicated tubes for 15 min, and then vortexed 10 min to detach bacterial cells from the sources. We transferred the resulting suspensions to extraction tubes. We used (mean ± s.e.m.) 0.3 ± 0.01 g soil per sample for DNA extraction, and completed extractions of all samples using the FastDNA™ SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s protocol with minor adjustments: cell lysis was achieved by three times 1 min bead-beating and DNA elution in 100 μl PCR-grade water. We quantified DNA concentrations using the Quant-it PicoGreen dsDNA kit (Molecular Probes, Invitrogen, Eugene, OR, USA), normalised the concentrations to 1 ng template DNA per 25 μl reaction (triplicates per sample) and amplified the V4/V5 region of the 16S rRNA gene using primers 515 F and 926 R with partial Illumina adaptors and thermal cycler protocol: 95 °C for 5 min, 35 cycles at 95 °C for 40 s, 56 °C for 45 s, 72 °C for 40 s and 10 min at 72 °C. The use of negative controls (no template) during the amplification ensured no contaminations in the PCR reagents. Eleven eggshell samples failed PCR trials, leaving 65 eggshell samples for further analysis. We sent purified (QIAquick gel extraction Kit, QIAGEN GmbH, Hilden, Germany) pooled triplicates to GenoToul (INRA, Toulouse, France) for library preparation and Illumina MiSeq sequencing using 2 × 250 bp paired-end v2 chemistry.
Sequence data processing
We processed raw 16S rRNA gene sequence data using QIIME 1.9.0 [37]. We truncated reverse primers from demultiplexed, quality filtered reads (Phred score ≥ 25, maximum bad run length = 3, no primer mismatches), and used open-reference OTU-picking (Operational Taxonomic Unit) (default QIIME settings using Greengenes reference set (v. 13.8 [38]) with de novo clustering of non-matching sequences; 0.01%) using 97% identity in UCLUST [39]. After removal of singletons, assigning taxonomy with UCLUST (Greengenes v. 13.8, 97%), aligning representative sequences using PyNast [40], and removing chimeric sequences identified by UCHIME [41], we generated a phylogenetic tree using FastTree [42]. We removed OTUs belonging to Archaea, chloroplasts and mitochondria and filtered OTUs representing <0.01% of the total abundance.
Statistical analyses
We analysed bacterial community structure and diversity using rarefied data (5000 reads per sample) using phyloseq (v. 1.14.0 [43]), picante [44], and vegan (v. 2.4-0 [45]), and non-rarefied data using DESeq2 (v. 1.10.1 [46]) and a SourceTracker script [47] for R statistical software (v. 3.2.3 [48]). We analysed variation in alpha diversity metrics and beta diversity distances with analysis of variance (ANOVA) and Tukey’s post hoc contrasts using multcomp [49] or Mann–Whitney U-tests. R scripts are made accessible as supplementary information.
We used ANOVA to compare alpha diversity of eggshell communities with cloacal gut, brood patch skin, body feathers, nest material and surface soil communities. To explore the effect of post-laying time (categorical: <12 h and 24–36 h) on eggshell communities, we tested for differential abundance of phylotypes between first and second eggs, corrected for host species, using negative binomial models and Wald tests in DESeq2 (critical false discovery rate-corrected (FDR) q-value of 0.1). We determined relative abundances of differentially abundant phylotypes between first and second eggs in each potential source type. We then explored the prevalence of these phylotypes in maternal and nest-associated communities using indicator values for each differentially abundant phylotype [50]. Indicator values represent the strength of associations between taxa and community (sample) types, where larger values indicate greater sample type specificity and fidelity. To test correspondence of estimated alpha diversity metrics between first and second eggs within nests, we estimated the repeatability r of phylotype richness and Shannon diversity using linear mixed models (LMMs) in rptR [51]. To determine whether eggshells form a selective habitat for bacterial settlement, we analysed the degree of non-random phylogenetic community structure using the nearest taxon index (NTI) based on null modelling of mean nearest taxon distances (MNTDs) with 999 iterations of taxon label randomisations of our phylogenetic tree [34, 52, 53].
We analysed taxonomic (Bray–Curtis) and phylogenetic similarity (weighted UniFrac) of eggshell bacterial community composition in three ways: first, we analysed community clustering at the population-level by partitioning variation by lark species, by first vs. second eggs and by nest using constrained distance-based redundancy analysis [54]. Then, we compared phylogenetic composition of eggshell communities with potential source types (cloacal gut, brood patch skin, body feathers, nest material and surface soil communities) using principal coordinate analysis (PCoA) and permutational multivariate analysis of variance [55] at different taxonomic depths: phylum, family and phylotype level, using agglomerated OTU tables using tax_glom in phyloseq. Then, within each nest, we calculated pairwise weighted UniFrac distances to assess the phylogenetic similarities between eggs and potential sources and tested for differential egg-source similarities with LMM, using Nest ID as a random factor to prevent pseudo-replication bias of eggs from the same nest using nlme [56]. We reported post hoc Tukey’s contrasts with FDR-corrected q-values [57]. Finally, we employed SourceTracker [47] in QIIME as a Bayesian approach to predict the origins of eggshell microbiota, where we designated all maternal and environmental communities as ‘source’ pool and eggshell communities as ‘sink’ pool, independently for woodlark and skylark. We used unrarefied data as input for the SourceTracker model, which predicted source contributions using default model parameters. We tested differential proportional contributions of potential source community types using a Kruskal–Wallis test and a Dunn’s test for multiple group comparisons [58]. We visualised the relative contributions of maternal sources (cloacal gut, brood patch skin and feathers), and the three dominant sources (nest material, brood patch skin and feathers), separately, illustrated with ternary plots using ggtern (v. 2.2.0 [59]).
Results
The constructed OTU table contained 1148 phylotypes and sample coverage ranged between 5225 and 111376 sequences per sample. Rarefaction curves showed that Shannon diversity had levelled at 5000 reads (Supplementary Fig. S1). We rarefied the OTU table to 5000 reads per sample.
Alpha diversity of eggshell bacterial communities
Eggshell communities of woodlarks and skylarks harboured on average 67% (t = 4.62, P < 0.01) and 50% (t = 4.25, P < 0.01) more phylotypes than cloacal gut communities, respectively, but the number of phylotypes found on eggs was not different from other sample types (Fig. 1a). Similarly, Shannon diversity of eggshell communities was 29% higher in eggshell communities of both woodlarks (t = 5.84, P < 0.001) and skylarks (t = 6.07, P < 0.001) compared to cloacal gut communities (Fig. 1b). Phylotype richness (F1,62 = 1.36, P = 0.25) and Shannon diversity (W = 441, P = 0.26) of eggshells communities differed neither between woodlarks and skylarks (Fig. 1a, b), nor between freshly laid eggs (‘second egg’) and eggs laid a day earlier (‘first egg’) (phylotype richness: woodlark, t = 0.54, P = 0.95; skylark, t = 0.37, P = 0.98; Shannon diversity: woodlark, W = 163, P = 0.34; skylark, W = 112, P = 0.56; Fig. 1a, b).
Fig. 1.

Alpha diversity of eggshells of wild woodlarks and skylarks. Phylotype richness a and Shannon diversity b of eggshell bacterial communities, stratified by lark species and laying sequence. Alpha diversity metrics are compared to data from maternal and environmental communities (faded boxes) [25]. Significance of comparisons among first and second eggs, and between eggshells and other communities are shown (**P < 0.01, ***P < 0.001, ns not significant, other non-significant pairwise differences are not highlighted)
Relative taxon abundances of eggshell bacterial communities
Eggshells of woodlarks and skylarks each harboured six unique phylotypes that were not found in the maternal or nest-associated sources associated with each host species (Supplementary Table S1). One of these phylotypes was identified in eggshells of both woodlarks and skylarks. However, all of these eggshell-specific phylotypes were low in abundance and infrequently present in eggshell communities (Supplementary Table S1). Further analysis of bacterial abundance in eggshell communities revealed that the abundance of 111 taxa differed between first and second laid eggs, of which 109 taxa were more abundant in first laid eggs, which were present in the nest a day longer than second laid eggs at the moment of sampling (Fig. 2a). Relative abundances of these 111 phylotypes in potential sources showed that they were variably abundant in maternal and environmental communities (Fig. 2a). Our efforts to uncover potential links between eggshell and source communities revealed that 58 (52%) of these phylotypes were designated significant indicator taxa for one or more potential source community types (FDR q < 0.1; Fig. 2b). These indicator taxa were in all cases most indicative for either maternal or environmental communities, and particularly often for soil communities (Fig. 2b). Specifically, bacterial taxa that significantly proliferated on eggshells shortly after laying (<36 h) were not specific to the eggshell niche, but rather indicators of communities associated with females or the nest environment. Only two taxa that were most indicative for cloacal microbiota of larks, belonging to Ralstonia and Caulobacteriaceae, respectively, increased on eggshells (Fig. 2b), whereas the remaining proliferating eggshell-associated taxa were top indicators for skin (n = 2), feather (n = 4), nest material (n = 2) and soil (n = 46). These results suggest that the bacterial taxa thriving on eggshells shortly after they have been laid originate from alternative sources to the maternal cloacal community.
Fig. 2.

Phylotype abundance between first and second laid eggs, in comparison to maternal and environmental communities. a Differential abundance of 111 phylotypes between first and second eggs and abundance of each phylotype in maternal and nest environmental communities (grey scale). First and second eggs of the laying sequence are exposed to nest environmental conditions 24–36 h, and <12 h prior to collection, respectively. Mean (circles) and whiskers (95% CI) denote log2-fold change of phylotype abundance between first and second eggs, coloured by bacterial Phylum. b A subset of phylotypes (n = 58) was designated as significant indicator species (FDR q < 0.1) for one or more community types that potentially contribute to eggshell microbiota assembly. Relative abundances in a are calculated on sources only (ie, excluding eggshells). In b, the average indicator value (colour intensity) and relative abundance (bubble size) were calculated using the cumulative abundance across all communities (ie, including eggshell communities) as the denominator. Phylum-coloured connectors link phylotype IDs in a to each indicator taxon in b, which are labelled by the lowest taxonomic information available
Phylogenetic community structure analysis of eggshell bacterial communities
Of the phylotypes found in source samples, 99.4% of taxa was identified in at least one eggshell sample. This high fraction raised the question whether eggshells either form a selective niche or their microbiome primarily assembles through random processes, eg, random dispersal or ecological drift. Our null modelling analysis of phylogenetic community structure revealed significant deviations of observed MNTD distributions from the permutated null distribution in each eggshell subgroup (Fig. 3a) with average NTIs >2 in 86% of the eggshell communities (Fig. 3b), indicating predominantly phylogenetically clustered eggshell microbiota.
Fig. 3.

Phylogenetic structure in eggshell microbial communities. a Mean nearest taxon distance (MNTD) centred around zero and b nearest taxon index (NTI) for first and second eggs of woodlarks and skylarks. a The area under the curve is determined by the number of pairwise taxon distances (999 for the null distribution), where peak height (density) is negatively related to peak width, together representing the variation of MNTD in a given eggshell subgroup
Within-nest and between-nest variation of community resemblance of eggshells
The lack of repeatability of phylotype richness (r ± s.e. = 0 ± 0.157) and Shannon diversity (r ± s.e. = 0 ± 0.148) of eggshell microbiota within nests indicated that alpha diversity of bacterial communities of two consecutive eggs, originating from the same mother, were statistically not more similar than compared to a randomly selected egg from the population. Analysis of taxonomic (Bray–Curtis) and phylogenetic (weighted UniFrac) community structure showed that host species or egg age did not structure eggshell microbiota (Fig. S1a, b; Table 2). Instead, nest identity explained 63% of phylogenetic and taxonomic clustering of eggshell communities based on weighted UniFrac (Table 2) and Bray–Curtis dissimilarities, respectively (Fig. S1c; Table 2). Notably, PCoA showed very high similarity (clustering) for some nests, but not others (Supplementary Fig. S2), indicating substantial variation in how similar eggs originating from the same nest are. Nonetheless, the significant effect of nest identity on PCoA clustering was at least partly maintained at higher taxonomic levels (Table 2).
Table 2.
Taxonomic (Bray–Curtis) and phylogenetic (weighted UniFrac) beta diversity of eggshell bacterial communities analysed across lark species, laying sequence and nests, at the bacterial phylotype, family and phylum levels using ANOVA on constrained ordination (capscale analysis)
| Bray–Curtis | Weighted UniFrac | |||||||
|---|---|---|---|---|---|---|---|---|
| df | F | R 2 | P | df | F | R 2 | P | |
| Phylotype | ||||||||
| Lark species | 1 | 1.34 | 0.02 | 0.131 | 1 | 0.88 | 0.01 | 0.5 |
| Egg 1 vs. egg 2 | 1 | 1.28 | 0.02 | 0.184 | 1 | 1.19 | 0.02 | 0.282 |
| Nest | 35 | 1.45 | 0.63 | <0.001 | 35 | 1.41 | 0.63 | 0.013 |
| Residual | 27 | 0.34 | 27 | 0.34 | ||||
| Family | ||||||||
| Lark species | 1 | 0.84 | 0.01 | 0.562 | 1 | 0.7 | 0.01 | 0.633 |
| Egg 1 vs. egg 2 | 1 | 1.43 | 0.02 | 0.158 | 1 | 1.16 | 0.01 | 0.287 |
| Nest | 35 | 1.48 | 0.64 | 0.004 | 35 | 1.56 | 0.65 | 0.006 |
| Residual | 27 | 0.33 | 27 | 0.32 | ||||
| Phylum | ||||||||
| Lark species | 1 | 0.71 | 0.01 | 0. 474 | 1 | 0.84 | 0.01 | 0.485 |
| Egg 1 vs. egg 2 | 1 | 2.02 | 0.03 | 0.121 | 1 | 1.54 | 0.02 | 0.19 |
| Nest | 35 | 1.05 | 0.55 | 0.417 | 35 | 1.42 | 0.63 | 0.041 |
| Residual | 27 | 0.41 | 27 | 0.34 | ||||
aP values <0.05 are represented in bold
Eggshell community composition in relation to potential sources
Our female-egg-nest triad system allowed comparisons between eggshell microbiota and maternal and nest-associated sources. Separate beta diversity analysis between eggs and potential sources showed that eggshell communities least resembled cloacal and soil communities but were taxonomically and phylogenetically most similar to nest material, and maternal skin and feather communities (Fig. 4a, b, Table 3). Within nests, phylogenetic distances (weighted UniFrac) between eggs and potential sources showed that average distances between eggshell communities and each potential source varied significantly (Fig. 4c; LMM, F5, 105 = 12.6, P < 0.0001). Within-nest patterns showing that eggshell communities were more similar to feather and nest material communities than to cloacal gut, brood patch skin and soil communities were mostly concordant with population-level patterns (Fig. 4c, Supplementary Table S2).
Fig. 4.
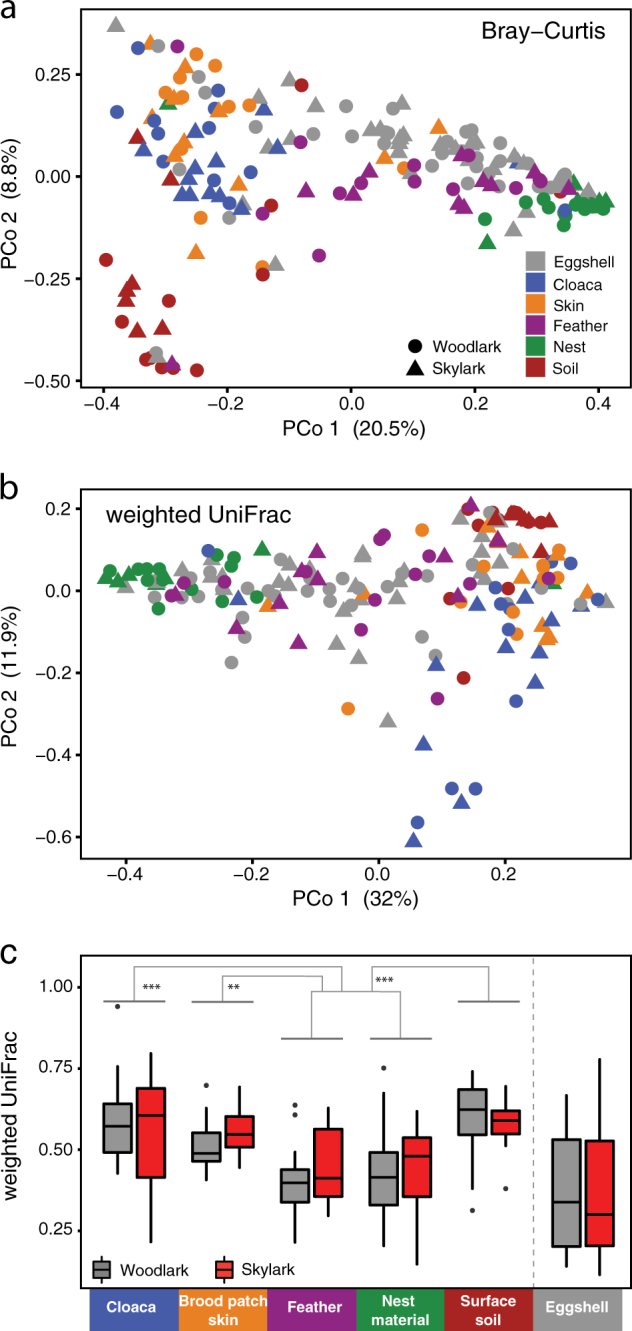
Beta diversity of eggshell, maternal and nest environmental communities. a PCoA of taxonomic community composition based on Bray–Curtis, and b phylogenetic community composition based on weighted UniFrac. c Within-nest pairwise weighted UniFrac distances between eggshells and other community types, and among two eggs in the same nest (two rightmost boxes). a, b Population-level PERMANOVA statistics are detailed in Table 3. c Eggshells communities were on average phylogenetically more similar to feathers and nest material than to cloacal gut, brood patch skin or surface soil communities (***FDR q < 0.001; **q < 0.01), full statistical details are in Supplementary Table S2
Table 3.
Population-level statistics of comparisons between eggshell communities and maternal and environmental source communities using PERMANOVA
| Bray–Curtis | Weighted UniFrac | ||||||
|---|---|---|---|---|---|---|---|
| Sample type | df | Pseudo-F | R 2 | P | Pseudo-F | R 2 | P |
| Cloacal gut | 1, 88 | 9.63 | 0.1 | <0.001 | 13.2 | 0.13 | <0.001 |
| Brood patch skin | 1, 85 | 7.87 | 0.08 | <0.001 | 11.2 | 0.12 | <0.001 |
| Feather | 1, 87 | 4.82 | 0.05 | <0.001 | 3.32 | 0.04 | <0.01 |
| Nest material | 1, 83 | 9.53 | 0.1 | <0.001 | 12.3 | 0.13 | <0.001 |
| Surface soil | 1, 82 | 13.7 | 0.14 | <0.001 | 16.6 | 0.17 | <0.001 |
P values <0.05 are represented in bold
Predictive source tracking of eggshell communities
Our population-level SourceTracker model predicted that both maternal and environmental sources contributed to eggshell community assembly (Fig. 5). The model estimated a minor role for cloacal gut communities in contributing to eggshell communities (Fig. 5a, b), corroborating patterns derived from beta diversity analyses. Regardless of the significant compositional dissimilarity between eggshell microbiota and the considered source communities (Table 3), SourceTracker identified skin, feathers and nest material communities as dominant sources to eggshell community assembly (Fig. 5, Table 4). Visualisation of predictions at the individual egg level revealed substantial variation among eggs with respect to the relative contributions of the three dominant sources (Fig. 5b, c). This variation among eggs complements population-level variation of beta diversity where eggshell communities spread widely in ordination space, indicating variable similarities to each (potential) source community (Fig. 4). The estimated proportional contribution of potential sources differed neither between woodlarks and skylarks (t < 0.70, df = 1, 305, P > 0.90; Fig. 5a), nor between first and second eggs of the laying sequence (t < 0.70, df = 1, 305, P > 0.90).
Fig. 5.

Predicted origins of bacterial communities of lark eggshells. a Mean (±95% CI) predicted contribution of bacterial source communities to eggshell communities for woodlarks and skylarks, separately presented for first and second laid eggs. b Predictions for individual eggs are plotted in three-axes ternary plots indicating the proportion of phylotypes originating from maternal sources (indicated by the triangle vertices) and c from each of the three dominant sources identified in a: nest material, brood patch skin and feathers. Each point represents a single eggshell community and its position represents the relative proportions of the potential source communities indicated at the triangle tips. a Dunn’s z statistics are detailed in Table 3
Table 4.
Pairwise contrasts of SourceTracker predicted proportional source contributions to eggshell microbiota using Dunn’s z statistic
| Cloacal gut | Brood patch skin | Feather | Nest material | |
|---|---|---|---|---|
| Brood patch skin | −6.48 (<0.001) | |||
| Feather | −6.32 (<0.001) | 0.16 (0.48) | ||
| Nest material | −7.70 (<0.001) | −1.22 (0.14) | −1.38 (0.12) | |
| Surface soil | 0.002 (0.50) | 6.49 (<0.001) | 6.32 (<0.001) | 7.71 (<0.001) |
Dunn’s z for pairwise contrasts (FDR q value; bold = q < 0.1)
Kruskal–Wallis χ2 (adjusted for ties) = 114.51, df = 4, P < 0.001
Discussion
Eggshell (bacterial) microbiomes of wild woodlarks L. arborea and skylarks A. arvensis were shaped through horizontal uptake from nest material-associated communities and transgenerational transmission from body feathers and brood patch skin of breeding females. The prominent differences in phylotype richness, Shannon diversity and phylogenetic beta diversity between eggshell and cloacal gut microbiota, and the negligible predicted contribution of maternal gut microbiomes to eggshell community assembly indicate that avian eggshells did not carry maternal gut communities shortly after laying. This suggests a limited potential for transgenerational gut symbiont transmission via the eggshell. Our results were congruent both at the population-level and within-nest level, and similar in both host species. Future studies in a wider range of bird species and habitats should prove the generality of these findings across the avian clade.
Our findings did not support the hypothesis that eggshells function as potent carriers of maternal gut symbionts, challenging the role of eggshells in transgenerational offspring inoculation. Higher alpha diversity of eggshell communities compared with maternal and nest environmental communities suggests that colonisation of eggshells continued after the eggs had been laid. Predicted contributions of nest material communities and proliferation of soil-borne bacteria corroborated these findings. Hence, we speculate that maternal gut bacteria that initially cover an eggshell in vivo are unable to survive and thrive ex vivo and create niche space for immigrants after experiencing inferior competitiveness in the novel (aerobic) niche. Our alpha diversity estimates of eggshell microbiomes contrast earlier egg microbiome studies, which concluded that egg communities were not richer than cloacal gut and were markedly less rich than nest material communities, respectively. Note that these patterns were based on low-resolution ARISA data [33, 60]. The lack of within-nest repeatability of alpha diversity metrics for first and second eggs could imply that colonisation occurred from multiple sources simultaneously, leading to composite communities from available sources, which could be expected when high metacommunity diversity and dispersal drove this early successional phase of community assembly [61]. Nevertheless, egg microbiota richness and diversity seemed not to be strictly pre-set by the maternal cloacal gut microbiome. One could wonder if amplification of relic DNA extracted from our samples may have caused the high variability in alpha diversity estimation [62]. However, if the drastic environmental change from in vivo to ex vivo impaired gut symbiont survival, and our DNA came from dead cells, we would expect higher similarities between cloacal and egg communities than we found, and hence we do not believe that this issue qualitatively confounded our results. Despite the limitations of DNA-based approaches [62], we believe that DNA-based surveys can provide informative insights into ecological interactions and processes in natural host-microbiome systems, and create avenues for further hypothesis-driven investigations. For our specific goal of assessing the transfer of microbes from parents to offspring, the next step would be to select specific taxa from our DNA-based community descriptions for tracking, eg, with in situ hybridisation techniques [63].
By evaluating taxon abundances as a proxy for live bacterial dynamics, we pursued a biologically informative understanding of bacterial taxon performance of first and second laid eggs. In two lark species, horizontally derived (ie, through direct contact) bacterial taxa appeared to outperform gut symbionts on freshly laid unincubated eggs under natural conditions, showing that eggshells form a particularly suitable niche for free-living bacteria. Changes in bacterial abundances between second laid eggs (<12 h post laying) and first laid eggs (24–36 h post laying), as a means of taxon-specific growth or thriving survivors, revealed that proliferating taxa on eggshells were not indicative for maternal cloacal microbiomes but for soil communities, and occasionally for nest material, feather and skin communities. The minimal success of gut symbionts on eggshells adds to the current understanding of egg microbiome dynamics: incubation induces an increase of bacterial abundance but a decrease of diversity of eggshell communities [29, 30, 35, 64] enhancing egg viability through reduction of the probability of trans-shell infection [25, 28]. Shawkey et al. [65] showed that mainly potential pathogenic bacteria were inhibited by incubation. Other studies also suggested incubation as a mechanism for preventing horizontally acquired (potentially pathogenic) microbes to thrive on eggshells and subsequently infect egg contents ([24, 60], but see refs. [66, 67]). Determining the identity and origin of bacterial taxa that thrive also during egg incubation up to the time of hatching, and subsequent monitoring of hatchling microbiome assembly will be essential and fertile ground to further explore the function of eggshells in transgenerational symbiont transmission.
Since phylogenetic community structure in egg microbiomes has not previously been described, we evaluated the phylogenetic structure of eggshell bacterial communities. Our data showed significant phylogenetic clustering of eggshell communities within two days after laying. This suggests bacterial species sorting (ie, some bacterial clades are more successful than others) through either environmental selection (niche-based selection) of the eggshell niche [53], or alternatively but not mutually exclusive, the outcome of competition among bacterial taxa. As eggshells harboured richer and more diverse communities than the average female cloaca, we hypothesised that multiple sources were involved in sourcing eggshell microbiomes, with some bacterial communities being more dominant than others. Compositional similarity among eggs and skin and feather communities also indicated that horizontal transmission contributed to shape the egg microbiome. As a technical note, the time lag between egg collection and sampling of females varied among nests, an aspect of our study that in theory may have reduced the resemblance between egg and cloacal gut communities. However, we do not expect major shifts in cloacal gut microbiota during the sampling timespan of this study [68, 69], but we acknowledge that testing this time lag effect warrants experimental investigation. Nevertheless, our results from population and within-nest level analyses were consistent and indicate substantially robust patterns.
Additionally, we sought to quantify the source contributions to eggshell microbiomes from the set of potential maternal and nest environmental bacterial communities sampled. Egg-source beta diversity comparisons and model-based source predictions for eggshell communities revealed that the phylogenetic composition of eggshell communities was not associated with cloacal gut microbiomes of breeding females. Cross-sectional (population-level) and within-nest analyses of phylogenetic composition between eggshells and the potential sources were congruent, except that eggshell and brood patch skin communities were not statistically more similar than eggshells with gut and soil communities in the within-nest analysis. Conversely, and most notably, the bare brood patch skin that females use to incubate eggs, the body feathers surrounding the bare skin, and the nest material adjoining the eggs were overall phylogenetically most similar to eggshell microbiomes. With respect to earlier egg-source investigations, it has been suggested that the cloacae of Eurasian hoopoes Upupa epops [33] and pied flycatchers Ficedula hypoleuca [70] might source their eggshell communities, although pairwise comparisons revealed mainly shared OTUs between hoopoe eggshells, beak and brood patch skin, whereas associations between uropygial secretion and eggshells were unexpectedly lacking [71]. Inferences from predicted contributions of potential sources underscored that eggshell microbiomes were largely derived through direct contact with sources, supported by beta-diversity patterns. As parental birds typically incubate their eggs, and with our findings indicating that skin, feathers and nest environment were dominant sources for eggshell microbiome assembly, we hypothesise that direct contact may be universally driving avian egg microbiome assembly. We refer to this idea as the ‘direct-contact inoculation hypothesis’. Because birds inhabit all biomes on Earth, we expect that egg microbiome compositions vary across large ecological scales, eg, seabirds vs. terrestrial birds [68, 72], and along climate gradients [67]. However, within each large-scale spatial setting, we expect that host-dependent ecological factors, such as nest type and nest materials [26, 73–75] may delimit the availability and composition of potential sources for eggshell microbiomes within a female-egg-nest triad context.
In light of symbiont transmission modes, one could argue that horizontal transmission appeared to be the main mode of eggshell community assembly, because bacterial inoculation of eggs seemed to occur through direct contact. However, because two out of three dominant sources were represented by maternal skin and feathers (~40–50% cumulative contribution), one could similarly justify dominance of (broad-sense) transgenerational transmission. Shifts in avian eggshell microbiota during the incubation phase have been reported [29] and could potentially lead to convergence of egg microbiota to more cloaca-like communities. Regardless, testing of our direct-contact inoculation hypothesis in more avian lineages may be a fruitful avenue, especially with experimental application of artificially assembled sources, and comparing eggshell microbiomes across different nest types and materials during the course of incubation.
Alternative symbiont acquisition by avian progeny expectedly occurs as proposed for lizards [76]: direct parent-offspring contact such as during food provisioning (only in altricial birds), horizontal transmission among individuals, ingestion of diet-associated microbes, passive environmental uptake and coprophagy. If these alternative routes constitute the dominant factors, contrary to vertical transmission during egg formation, it then implies (partial) build-up of avian microbiomes every generation anew, which would limit prospects for adaptive evolution of the microbiome through natural selection acting on microbiome variation [77]. Zooming out, egg(shell) microbiomes of other oviparous vertebrates are also only beginning to be elucidated, but community dynamics, forces shaping them and subsequent implications for development and fitness remain poorly resolved. Rooted in food production or public health frameworks, studies on fish and reptilian eggs identified egg microbiota effects on (mitigating) egg diseases [23, 78], offering insights in early-life microbiome acquisition [79]. Salmon egg surface communities associated more strongly with female gut microbiota than found here in birds [80], questioning generality of our direct-contact inoculation hypothesis to other oviparous vertebrate classes. While direct-contact inoculation of bird eggs, as we suggested here, needs experimental validation, our eggshell microbiome analysis in a natural context provides future avenues for developing and testing hypotheses regarding the functions of the egg microbiome and its consequential role in vertebrate biology.
Data availability
All 16S rRNA gene sequences have been deposited in the MG-RAST server under project numbers mgp21350 and mgp80058.
Electronic supplementary material
Phylogenetic tree of representative OTUs
Acknowledgements
We thank C. Burger, J. Dekker, T. Lamain, R. Voesten and S. Waasdorp for field assistance, F. Dijkstra, F. Dini-Andreote, M. van der Velde and M.A. Versteegh for bioinformatic and technical support, and Staatsbosbeheer Drents-Friese Wold for kindly allowing us to work in their area.
Funding
This work was funded by the Netherlands Organisation for Scientific Research with Vidi grant 864.10.012 to B.I.T. and by Dobberke Stichting grant UPS/BP/4405 to H.P.J.v.V.
Author contributions
H.P.J.v.V., J.F.S. and B.I.T. were involved in developing study concept and design. H.P.J.v.V. conducted sample collection and processing, data acquisition and analysis, and drafting of the manuscript. J.F.S. and B.I.T. contributed to developing conceptual ideas, critical revisions and improvement of the final manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0067-3) contains supplementary material, which is available to authorised users.
References
- 1.McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, et al. Animals in a bacterial world, a new imperative to the life sciences. Proc Natl Acad Sci U S A. 2013;110:3229–36. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hird SM. Evolutionary biology needs wild microbiomes. Front Microbiol. 2017;8:725. doi: 10.3389/fmicb.2017.00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohl KD. An introductory "how-to" guide for incorporating microbiome research into. Integrative and Comparative Biology. 2017;57:674–81. doi: 10.1093/icb/icx013. [DOI] [PubMed] [Google Scholar]
- 4.Funkhouser LJ, Bordenstein SR. Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 2013;11:e1001631. doi: 10.1371/journal.pbio.1001631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapira M. Gut microbiotas and host evolution: scaling up symbiosis. Trends Ecol Evol. 2016;31:539–49. doi: 10.1016/j.tree.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. 6:776-88. [DOI] [PMC free article] [PubMed]
- 7.Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ. The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol. 2014;23:1238–50. doi: 10.1111/mec.12510. [DOI] [PubMed] [Google Scholar]
- 8.Waite DW, Taylor MW. Exploring the avian gut microbiota: Current trends and future directions. Front Microbiol. 2015;6:673. doi: 10.3389/fmicb.2015.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knutie SA, Wilkinson CL, Kohl KD, Rohr JR. Early-life disruption of host microbiota decreases later-life resistance to infections. Nat Comm. 2017;8:86. doi: 10.1038/s41467-017-00119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacob S, Parthuisot N, Vallat A, Ramon-Portugal F, Helfenstein F, Heeb P. Microbiome affects egg carotenoid investment, nestling development and adult oxidative costs of reproduction in Great tits. Funct Ecol. 2015;29:1048–58. doi: 10.1111/1365-2435.12404. [DOI] [Google Scholar]
- 11.van Opstal EJ, Bordenstein SR. Rethinking heritability of the microbiome. Science. 2015;349:1172–3. doi: 10.1126/science.aab3958. [DOI] [PubMed] [Google Scholar]
- 12.Douglas AE. Mycetocyte symbiosis in insects. Biol Rev Camb Philos Soc. 1989;64:409–34. doi: 10.1111/j.1469-185X.1989.tb00682.x. [DOI] [PubMed] [Google Scholar]
- 13.Moran NA, Telang A. Bacteriocyte-associated symbionts of insects. Bioscience. 1998;48:295–305. doi: 10.2307/1313356. [DOI] [Google Scholar]
- 14.Reveillaud J, Maignien L, Eren AM, Huber JA, Apprill A, Sogin ML, et al. Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 2014;8:1198–209. doi: 10.1038/ismej.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohl KD, Brun A, Magallanes M, Brinkerhoff J, Laspiur A, Acosta JC, et al. Gut microbial ecology of lizards: insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol Ecol. 2017;26:1175–98. doi: 10.1111/mec.13921. [DOI] [PubMed] [Google Scholar]
- 16.McFall-Ngai MJ, Ruby EG. Symbiont Recognition and Subsequent Morphogenesis as Early Events in an Animal-Bacterial Mutualism. Science. 1991;254:1491–4. doi: 10.1126/science.1962208. [DOI] [PubMed] [Google Scholar]
- 17.Koch EJ, Miyashiro T, McFall-Ngai MJ, Ruby EG. Features governing symbiont persistence in the squid-vibrio association. Mol Ecol. 2014;23:1624–34. doi: 10.1111/mec.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bright M, Bulgheresi S. A complex journey: transmission of microbial symbionts. Nat Rev Microbiol. 2010;8:218–30. doi: 10.1038/nrmicro2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colston TJ, Jackson CR. Microbiome evolution along divergent branches of the vertebrate tree of life: what is known and what is unknown. Mol Ecol. 2016;25:3776–3800. doi: 10.1111/mec.13730. [DOI] [PubMed] [Google Scholar]
- 20.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Higraldo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the intestinal microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–5. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert SF. A holobiont birth narrative: the epigenetic transmission of the human microbiome. Front Genet. 2014;5:282. doi: 10.3389/fgene.2014.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohl KD. Diversity and function of the avian gut microbiota. J Comp Phys B. 2012;182:591–602. doi: 10.1007/s00360-012-0645-z. [DOI] [PubMed] [Google Scholar]
- 23.Sarmiento-Ramírez JM, van der Voort M, Raaijmakers JM, Diéguez-Uribeondo J. Unravelling the microbiome of eggs of the endangered sea turtle Eretmochelys imbricate identifies bacteria with activity against the emerging pathoges Fusarium falciforme. PLoS One. 2014;9:e95206. doi: 10.1371/journal.pone.0095206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cook MI, Beissinger SR, Toranzos GA, Rodriguez RA, Arendts WJ. Trans-shell infection of pathogenic micro-organisms reduces the shelf life of non-incubated bird’s eggs: a constraint on the onset of incubation? Proc R Soc Lond B Biol Sci. 2003;270:2233–40. doi: 10.1098/rspb.2003.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cook MI, Beissinger SR, Toranzos GA, Rodriguez RA, Arendts WJ. Microbial infection affects egg viability and incubation behavior in a tropical passerine. Behav Ecol. 2005;16:30–36. doi: 10.1093/beheco/arh131. [DOI] [Google Scholar]
- 26.Godard RD, Wilson CM, Frick JW, Siegel PB, Bowers BB. The effects of exposure and microbes on hatchability of eggs in open-cup and cavity nests. J Avian Biol. 2007;38:709–16. doi: 10.1111/j.0908-8857.2007.04052.x. [DOI] [Google Scholar]
- 27.Hansen CM, Meixell BW, Van Hemert C, Hare RF, Hueffer K. Microbial infections are associated with embryo mortality in Arctic-nesting geese. Appl Environ Microbiol. 2015;81:5583–92. doi: 10.1128/AEM.00706-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook MI, Beissinger SR, Toranzos GA, Arendts WJ. Incubation reduces microbial growth on eggshells and the opportunity for trans-shell infection. Ecol| Lett. 2005;8:532–7. doi: 10.1111/j.1461-0248.2005.00748.x. [DOI] [PubMed] [Google Scholar]
- 29.Grizard S, Dini-Andreote F, Tieleman BI, Salles JF. Dynamics of bacterial and fungal communities associated with eggshells during incubation. Ecol Evol. 2014;4:1140–57. doi: 10.1002/ece3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee WY, Kim M, Jablonski PG, Choe JC, Lee S. Effect of incubation on bacterial communities of eggshells in a temperate bird, the Eurasian magpie (Pica pica) PLoS One. 2014;9:e103959. doi: 10.1371/journal.pone.0103959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martín-Vivaldi M, Soler JJ, Peralta-Sánchez JM, Arco L, Martín-Platero AM, Martín-Bueno M, et al. Special structures of hoopoe eggshells enhance adhesion of symbiont-carrying uropygial secretion that increase hatching success. J Anim Ecol. 2014;83:1289–301. doi: 10.1111/1365-2656.12243. [DOI] [PubMed] [Google Scholar]
- 32.Goodenough AE, Stallwood B, Dandy S, Nicholson TE, Stubbs H, Coker DG. Like mother like nest: similarity in microbial communities of adult female pied flycatchers and their nests. J Ornithol. 2017;158:233–44. doi: 10.1007/s10336-016-1371-1. [DOI] [Google Scholar]
- 33.Martínez-García Á, Martín-Vivaldi M, Rodríguez-Ruano SM, Peralta-Sánchez JM, Valdivia E, et al. Nest bacterial environment affects microbiome of hoopoe eggshells, but not that of uropygial secretion. PLoS One. 2016;11:e0158158. doi: 10.1371/journal.pone.0158158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Veelen HPJ, Salles JF, Tieleman BI. Multi-level comparisons of cloacal, skin, feather and nest-associated microbiota suggest considerable influence of horizontal acquisition on the microbiota assembly of sympatric woodlarks and skylarks. Microbiome. 2017;5:156. doi: 10.1186/s40168-017-0371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grizard S, Versteegh MA, Ndithia HK, Salles JF, Tieleman BI. Shifts in bacterial communities of eggshells and antimicrobial activities in eggs during incubation in a ground nesting passerine. PLoS One. 2015;10:e0121716. doi: 10.1371/journal.pone.0121716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Praus L, Hegemann A, Tieleman I, Weidinger K. Predators and predation rates of Skylark Alauda arvensis and Woodlark Lullula arborea nests in a semi-natural area in the Netherlands. Ardea. 2014;102:87–94. doi: 10.5253/078.102.0112. [DOI] [Google Scholar]
- 37.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Buschman JD, Costello EK. QIIME allows analysis of high-throughput community sequence data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;28:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 40.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Anderson GL, Knight R. PyNast: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–7. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics. 2010;26:1463–4. doi: 10.1093/bioinformatics/btq166. [DOI] [PubMed] [Google Scholar]
- 45.Oksanen J, Blanchet GF, Friendly M, Kindt R, Legendre P, McGlinn D et al. (2016). vegan: Community ecology package. Ordination methods, diversity analysis and other functions for community and vegetation ecologists. Version 2.4.0. http://CRAN.R-project.org/package=vegan
- 46.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, et al. Bayesian community-wide culture-independent microbial source tracking. Nat Methods. 2011;8:761–3. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 49.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50:346–63. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 50.Dufrene M, Legendre P. Species assemblages and indicator species: the need for a flexible assymetrical approach. Ecol Monogr. 1997;67:345–66. [Google Scholar]
- 51.Nakagawa S, Schielzeth H. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol Evol. 2013;4:133–42. doi: 10.1111/j.2041-210x.2012.00261.x. [DOI] [Google Scholar]
- 52.Stegen JC, Lin X, Konopka AE, Fredrickson JK. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012;6:1653–64. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webb CO, Ackerly DD, McPeek MA, et al. Phylogenies and community ecology. Annu Rev Ecol Sys. 2002;33:475–505. doi: 10.1146/annurev.ecolsys.33.010802.150448. [DOI] [Google Scholar]
- 54.Legendre P, Anderson MJ. Distance-based redundancy analysis: multispecies responses in multifactorial ecological experiments. Ecology. 1999;84:511–25. [Google Scholar]
- 55.Anderson MJ. A new method for multivariate analysis of variance. Aust Ecol. 2001;26:32–46. [Google Scholar]
- 56.Pinheiro J, Bates D, DebRoy S, Sakar D, R Core Team. (2016). nlme: linear and nonlinear mixed-effects models. Version 3.1.128. http://CRAN.R-project.org/package=nlme
- 57.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical guide and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 58.Dinno A (2017). dunn.test: Dunn’s test of multiple comparisons using rank sums. Version 1.3.3. http://CRAN.R-project.org/package=dunn.test
- 59.Hamilton N (2016). ggtern: an extension to ‘ggplot2’, for the creation of ternary diagrams. Version 2.2.0. http://CRAN.R-project.org/package=ggtern
- 60.Brandl HB, van Dongen WFD, Darolová A, Krištovík J, Majtan J, Hoi H. Composition of bacterial assemblages in different components of reed warbler nests and a possible role of egg incubation in pathogen regulation. PLoS One. 2014;9:e114861. doi: 10.1371/journal.pone.0114861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci U S A. 2015;112:E1326–E1332. doi: 10.1073/pnas.1414261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carini P, Marsden PJ, Leff JW, Strickland MF, Fierer N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol. 2016;2:16242. doi: 10.1038/nmicrobiol.2016.242. [DOI] [PubMed] [Google Scholar]
- 63.Amann R, Fuchs BM. Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat Rev Microbiol. 2008;6:339–48. doi: 10.1038/nrmicro1888. [DOI] [PubMed] [Google Scholar]
- 64.Giraudeau M, Czirják GÁ, Duval C, Bretagnolle V, Gutierrez C, Guillon N, et al. An experimental test in mallards (Anas platyrhynchos) of the effect of incubation and maternal preen oil on eggshell microbial load. J Ornithol. 2014;155:671–7. doi: 10.1007/s10336-014-1050-z. [DOI] [Google Scholar]
- 65.Shawkey MD, Firestone MK, Brodie EL, Beissinger SR. Avian incubation inhibits growth and diversification of bacterial assemblages on eggs. PLoS One. 2009;4:e4522. doi: 10.1371/journal.pone.0004522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walls JG, Hepp GR, Eckhardt LG. Effects of incubation delay on viability and microbial growth of wood duck (Aix sponsa). eggs. Auk. 2011;128:633–70. doi: 10.1525/auk.2011.11124. [DOI] [Google Scholar]
- 67.Wang JM, Firestone MK, Beissinger SR. Microbial and environmental effects on avian egg viability: do tropical mechanisms act in a temperate environment. Ecology. 2011;92:1137–45. doi: 10.1890/10-0986.1. [DOI] [PubMed] [Google Scholar]
- 68.Hird SM, Sánchez C, Carstens BC, Brumfield RT. Comparative gut microbiota of 59 Neotropical bird species. Front Microbiol. 2015;6:1403. doi: 10.3389/fmicb.2015.01403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benskin CMH, Rhodes G, Pickup RW, Wilson Km Hartley IR. Diversity and temporal stability of bacterial communities in a model passerine bird, the zebra finch. Mol Ecol. 2010;19:5531–44. doi: 10.1111/j.1365-294X.2010.04892.x. [DOI] [PubMed] [Google Scholar]
- 70.Ruiz-de-Castañeda R, Vela AL, Lobato E, Briones V, Moreno J. Prevalence of potentially pathogenic culturable bacteria on eggshells and in the cloaca of female pied flycatchers in a temperate habitat in central Spain. J Field Ornith. 2011;82:215–24. doi: 10.1111/j.1557-9263.2011.00324.x. [DOI] [Google Scholar]
- 71.Martínez-García Á, Soler JJ, Rodríguez-Ruano SM, Martín-Bueno M, Martín-Platero AM, Juárez-García N, et al. Preening as a vehicle for key bacteria in hoopoes. Microb Ecol. 2015;70:1024–33. doi: 10.1007/s00248-015-0636-1. [DOI] [PubMed] [Google Scholar]
- 72.Dewar ML, Arnould JPY, Krause L, Dann P, Smith SC. Interspecific variations in the faecal microbiota of Procellariiform seabirds. FEMS Microb Ecol. 2014;89:47–55. doi: 10.1111/1574-6941.12332. [DOI] [PubMed] [Google Scholar]
- 73.Mennerat A, Mirleau P, Blondel J, Perret P, Lambrechts MM, Heeb P. Aromatic plants in nests of the blue tit Cyanistes caeruleus protect chicks from bacteria. Oecologia. 2009;161:849–55. doi: 10.1007/s00442-009-1418-6. [DOI] [PubMed] [Google Scholar]
- 74.Peralta-Sánchez JM, Martín-Vivaldi M, Martín-Platero AM, Martín-Bueno M, Oñate M, Ruiz-Rodríguez M, et al. Avian life history traits influence eggshell bacterial loads: a comparative analysis. Ibis. 2012;154:725–37. doi: 10.1111/j.1474-919X.2012.01256.x. [DOI] [Google Scholar]
- 75.Peralta-Sánchez JM, Møller AP, Martin-Platero AM, Soler JJ. Number and colour composition of nest lining feathers predict eggshell bacterial community in barn swallow nests: an experimental study. Funct Ecol. 2010;24:426–33. doi: 10.1111/j.1365-2435.2009.01669.x. [DOI] [Google Scholar]
- 76.Colston TJ. Gut microbiome transmission in lizards. Mol Ecol. 2017;26:972–4. doi: 10.1111/mec.13987. [DOI] [PubMed] [Google Scholar]
- 77.Byler KA, Carmi-Veal M, Fine M, Goulet TL. Multiple symbiont acquisition strategies as an adaptive mechanism in the coral Stylophora pistillata. PLoS ONE. 2013;8:e59596. doi: 10.1371/journal.pone.0059596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Y, De Bruijn I, Jack ALH, Drynan K, van den Berg AH, Thoen E, et al. Deciphering the microbial landscapes of fish eggs to mitigate emerging diseases. ISME J. 2014;8:2002–14. doi: 10.1038/ismej.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Llewellyn MS, Boutin S, Hoseinifar SH, Derome S. Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front Microbiol. 2014;5:3389. doi: 10.3389/fmicb.2014.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romero J, Navarrete P. 16 rDNA-based analysis of dominant bacterial populations associated with early life stages of coho salmon Oncorhynchus kisutch. Microb Ecol. 2006;38:1–26. doi: 10.1007/s00248-006-9037-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic tree of representative OTUs
Data Availability Statement
All 16S rRNA gene sequences have been deposited in the MG-RAST server under project numbers mgp21350 and mgp80058.