

Abstract
The processes mediating the repair of DNA double strand breaks (DSBs) are critical determinants of radiosensitivity and provide a source of potential targets for tumor radiosensitization. Among the events required for efficient DSB repair are a variety of post-translational histone modifications including methylation. Because trimethylation of histone H3 on lysine 27 (H3K27me3) has been associated with chromatin condensation, which can influence DSB repair, we determined the effects of radiation on H3K27me3 levels in tumor and normal cell lines. Irradiation of tumor cells resulted in a rapid loss of H3K27me3, which was prevented by the siRNA-mediated knockdown of the H3K27 demethylase UTX. Knockdown of UTX also enhanced the radiosensitivity of each tumor cell line. Treatment of tumor cells with the H3K27 demethylase inhibitor GSKJ4 immediately before irradiation prevented the radiation-induced decrease in H3K27me3 and enhanced radiosensitivity. As determined by neutral comet analysis and γH2AX expression, this GSKJ4 treatment protocol inhibited the repair of radiation-induced DSBs. Consistent with in vitro results, treatment of mice bearing leg tumor xenografts with GSKJ4 significantly enhance radiation-induce tumor growth delay. In contrast to results generated from tumor cell lines, radiation had no effect on H3K27me3 levels in normal fibroblast cell lines and GSKJ4 did not enhance their radiosensitivity. These data suggest that H3K27me3 demethylation contributes to DSB repair in tumor cells and that UTX, the demethylase responsible, provides a target for selective tumor cell radiosensitization.
Keywords: H3K27me3, histone demethylase, UTX, radiosensitization, GSKJ4
Introduction
The lesion primarily responsible for cell death after exposure to ionizing radiation is the DNA double strand break (DSB). The processes that mediate the response to and repair of DSBs are thus critical determinants of radiosensitivity and provide a source of potential targets for tumor radiosensitization. Among the events required for efficient DSB repair are a variety of post-translational histone modifications, which are involved in the relaxation of chromatin structure as detected locally around a DSB (1,2) and on a global level (3). In general, such changes in chromatin structure in response to radiation play a role in the sensing of DSBs (4) and facilitating the access of repair factors to sites of DNA damage (4). One of the most well defined post-translational histone modifications affecting radioresponse is phosphorylation, with the phosphorylation of H2AX established to play a critical role in activating the radiation-induced DNA damage response and the recruitment of repair proteins to DSBs (5). Acetylation and ubiquitination are also histone modifications that have been reported to participate in the repair of DSBs (6–8). Of note, inhibitors of histone deacetylation have been shown to enhance tumor cell radiosensitivity in a variety of preclinical models and have undergone clinical evaluation in combination with radiotherapy (9–11).
Histone methylation has also been implicated in the regulation of cellular radioresponse. Dimethylation of histone H3 on lysine 79 (H3K79me2) and H4 on lysine 20 (H4K20me2) have been linked to the recruitment of 53BP1 to DSBs (12,13), neither of those histone modifications, however, was induced by DNA damage (14). Ayrapetov et al, showed that H3K9me3 was increased locally around DSBs (15), which in the context of previous studies suggested that H3K9me3 is critical for the activation of Tip60-mediated repair of radiation-induced DSBs (2,15). At the global level, radiation was reported to increase the amount of H3K36me2, which was shown to enhance the recruitment of components of non-homologous end joining (NHEJ) repair (16,17). Although not as well defined, H3K27me3, which participates in local chromatin condensation and transcriptional silencing, has also been suggested to influence DSB repair (18–20). Support for H3K27me3 as a determinant of cellular radiosensitivity is derived primarily from studies of EZH2, the methyltransferase responsible for generating and maintaining H3K27me3 (21). EZH2 is recruited to radiation-induced DSBs (19,22) and its knockdown has been reported to enhance the radiosensitivity of U2OS cells (19). However, whereas pharmacological inhibition of EZH2 enhanced the radiosensitivity of atypical rhabdoid teratoid tumor cells (23), it had no effect on the radiosensitivity of a series of glioma cell lines (24).
Whether H3K27me3 plays a role in radioresponse thus remains unclear. Although recruitment of EZH2 to DSBs has been detected after laser micro-irradiation, conflicting data has been reported as to the concomitant enrichment of H3K27me3 at those sites (19,25). Moreover, based on immunoblot analysis, global levels of H3K27me3 were unaffected in U2OS cells (19) after irradiation yet they were decreased in a human fibroblast cell line (26). To better understand the role of H3K27me3 in radioresponse, we initially defined the effects of radiation on H3K27me3 levels in three human tumor cell lines initiated from a variety of solid tumor histologies. As shown here, after irradiation there was a rapid loss of H3K27me3, which could be attributed to the histone demethylase UTX. Knockdown of UTX or its inhibition with GSKJ4 prevented the radiation-induced decrease in H3K27me3 and enhanced tumor cell radiosensitivity. Finally, consistent with in vitro results, treatment of mice bearing leg tumor xenografts with GSKJ4 prevented the radiation-induced decrease in H3K27me3 and significantly enhanced radiation-induce tumor growth delay. These data suggest that loss of H3K27me3 contributes to cell survival after irradiation and that preventing this decrease via inhibition of UTX provides a strategy for tumor radiosensitization.
Materials and Methods
Cell lines and treatments
MDA-MB-231 (breast adenocarcinoma), A549 (lung adenocarcinoma), MRC5 (normal lung fibroblast) and MRC9 (normal lung fibroblast) were obtained from American Type Culture Collection (ATCC). U251 (glioblastoma) was obtained from the Division of Cancer Treatment and Diagnosis Tumor Repository (DCTD), National Cancer Institute (NCI). DCTD and ATCC employ short tandem repeat DNA fingerprinting, karyotyping, and cytochrome C oxidase to authenticate cell lines. All cells were cultured less than 3 months after resuscitation. MDA-MB-231 and U251 were grown in Dulbecco’s Modified Eagle’s Medium) supplemented with 10% FBS (Invitrogen). A549 was grown in Roswell Park Memorial Institute 1640 Medium and MRC5 and MRC9 were grown in Minimum Essential Medium, both supplemented with 10% FBS (Invitrogen). Cell cultures were maintained in an atmosphere of 5% CO2/95% air at 37°C. GSKJ4 (Selleck Chemicals) was dissolved in dimethyl sulfoxide. Cells were irradiated using a 320kV X-ray source (Precision XRay Inc.) at a dose rate of 2.3 Gy/min.
siRNA transfection
A pool of 4 siRNA duplexes (SMARTpool) targeted to UTX, JMJD3 and a nontargeted siRNA pool were purchased from Dharmacon Inc. Transfection with the respective siRNA pool was performed when cell cultures were 60% confluent using Dharmafect 4 transfection reagent (Dharmacon) per manufacturer’s protocol. All experiments were carried out 48h post-transfection.
Clonogenic Survival Assay
To evaluate radiosensitivity cells were plated at clonal density in six-well plates and irradiated 6h (U251, A549) to 16h (MDA-MB-231, MRC5, MRC9) later. 10 to 14d after seeding, plates were stained with 0.5% crystal violet, the number of colonies determined, and the surviving fractions were calculated. Radiation survival curves were generated after normalizing for the cytotoxicity induced by GSKJ4 or siRNA. Data presented are the mean ± SEM from at least 3 independent experiments.
Immunoblot analysis of whole cell lysates
Cells were lysed as described (27) and total protein was quantified using BCA protein assay (Thermo Scientific). Proteins were separated by SDS-PAGE; transferred to nitrocellulose (Bio-Rad) and probed with anti-UTX (KDM6A, Cell Signaling Technology), anti-JMJD3 (KDM6B, Abcam) and β-Actin (Cell Signaling Technology). Bands were visualized with IRDye secondary antibodies (LI-COR) and quantified with Image Studio (LI-COR).
Chromatin fractionation
Chromatin fractionation and immunoblot analysis of chromatin-associated proteins was performed as described by Sun et al (2). Briefly, cells were lysed in 10mM Tris-HCl (pH 7.5), 10mM KCl, 1.5mM MgCl2, 0.34M Sucrose, 10% Glycerol, 0.2% Triton X-100; supplemented with 1× phosphatase inhibitor cocktails II and III (Sigma-Aldrich), and 1× HALT protease inhibitor cocktail (Thermo Scientific) for 10 minutes on ice. Nuclei were pelleted and washed once in 10mM HEPES pH 7.9, 10mM NaCl, 3mM MgCl2, 1mM CaCl2 supplemented with 1× phosphatase inhibitor cocktails II and III (Sigma-Aldrich), and 1× HALT protease inhibitor cocktail (Thermo Scientific) and digested with 25KU Benzonase for 5min at 37°C. The reaction was stopped with 20mM EDTA and insoluble chromatin was separated from soluble chromatin by centrifugation. Supernatant (S1) was saved on ice and the insoluble chromatin pellet was resuspended in 10mM HEPES pH 7.9, 10mM KCl, 0.5M NaCl, 1mM EDTA, 0.1mM EGTA, 0.1% IGEPAL CA630, supplemented with 1× phosphatase inhibitor cocktails II and III (Sigma-Aldrich), 1× HALT protease inhibitor cocktail (Thermo Scientific), and sonicated at 25% amplitude for 2×5sec on ice. The sonicated chromatin samples were cleared by centrifugation and the supernatant (S2) was transferred to supernatant S1. Protein in the combined supernatant was quantified using BCA protein assay (Thermo Scientific) and separated by SDS-PAGE using 4–20% gradient gel (Bio-Rad), followed by protein transfer to a nitrocellulose membrane. For tumor cell lines 2μg of chromatin protein were subjected to immunoblot analysis; for normal cell lines 10μg of chromatin protein were analyzed. Membranes were probed with anti-H3K27me3 (Cell Signaling) and anti-H3 (Abcam) antibodies, which were visualized with IRDye secondary antibodies (LI-COR) and quantified with Image Studio (LI-COR).
Neutral Comet assay
Neutral comet assay was performed using a commercially available kit according to the recommendations from the manufacturer (Trevigen) with slight modifications. Briefly, monolayers were irradiated (10Gy) and returned to the incubator. At specified times, single-cell suspensions were generated, washed with PBS, mixed with low melting agarose (1:10), and transferred to the provided slides. Cells were lysed at 4°C for 1 h on wet ice, subjected to electrophoresis for 20 min at room temperature and fixed with 70% EtOH. DNA was stained with SYBR Green. Digital fluorescent images were analyzed with TriTek CometScore®as described (28). Data are expressed as % damage remaining, in which the Olive tail moment from cultures irradiated on ice and collected immediately after irradiation was set to 100% damage, with the remaining times post-irradiation normalized accordingly. All time points were corrected before normalization for DNA damage detected from GSKJ4 or vehicle treatment alone by subtracting the Olive tail moment of non-irradiated vehicle or GSKJ4 treated samples. Data represent the mean ± SEM of 3 independent experiments.
Flow cytometric analysis of γH2AX expression
Flow cytometry of γH2AX was performed as described by Karnak et al (29). Briefly, cells were trypsinized, washed with ice-cold PBS, and fixed at a concentration of 1×106 cells/mL in ice-cold 70% ethanol. Samples were incubated directly to Alexa488 conjugated mouse anti-γH2AX-specific antibody (Cell Signaling) overnight at 4°C. For quantification of γH2AX positivity, a gate was arbitrarily set on the control, untreated sample to define a region of positive staining for γH2AX of approximately 5%. This gate was then overlaid on the drug/radiation-treated samples. Cell cycle distribution and total DNA content was determined by adding Hoechst 33258 (1:50,000). Data were acquired on a LSR Fortessa flow cytometer (Becton Dickinson) with DIVA software and analyzed with FlowJo® (Tree Star).
Tumor Growth Delay
Eight to ten-week-old female athymic nude mice (NCr nu/nu; NCI Animal Production Program, Frederick, MD) were used in these studies. Animals were caged in groups of 5 or less and fed animal chow and water ad libitum. A single cell suspension U251 cells (1 × 106) was injected subcutaneously into the right hind leg. When tumors grew to a mean volume of approximately or 220mm3 mice were randomized into four groups: vehicle treated controls (10:90 DMSO:20% Captisol), drug treated (GSKJ4 100mg/kg/d), radiation (2Gy/day), or drug/radiation combination. The treatment protocol is described in detail in the results. Radiation was delivered locally using a Pantak X Ray source with animals restrained in a custom designed lead jig. To obtain tumor growth curves, perpendicular diameter measurements of each tumor were measured 3 times per week with a digital caliper and volumes were calculated using a formula (L x W2) / 2. Data are expressed as mean ± SEM tumor volume. Each experimental group contained between 7-10 mice. All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for Care and Use of Animals and approved by the Institutional Animal Care and Use Committee (IACUC).
Results
The effects of radiation on histone H3K27me3 levels were initially defined in three human tumor cell lines: U251 glioblastoma; MDA-MB-231 breast carcinoma and A549 lung carcinoma. For these studies, chromatin associated protein was isolated and 2μg per sample was subjected to immunoblot analysis of histone H3K27me3 with histone H3 used as a loading control. An initial experiment showed that at 1h after irradiation of U251 a small decrease in H3K27me3 was detectable after 2Gy, which decreased further as a function of dose out to 10Gy (Figure S1A). Thus, to evaluate the H3K27me3 levels after irradiation in more detail, each cell line was exposed to 10Gy and collected at times out to 6h. As shown in Figure 1, compared to control cells H3K27me3 levels were significantly reduced in each of the tumor cell lines after irradiation. Although there was some variability among cell lines regarding the time course, the maximum decrease was detected at 0.5 to 1h after exposure with H3K27m3 remaining significantly below unirradiated levels for at least 6h. These results indicate that, at least in these 3 tumor cell lines, after irradiation there is a rapid reduction in H3K27me3.
Figure 1.
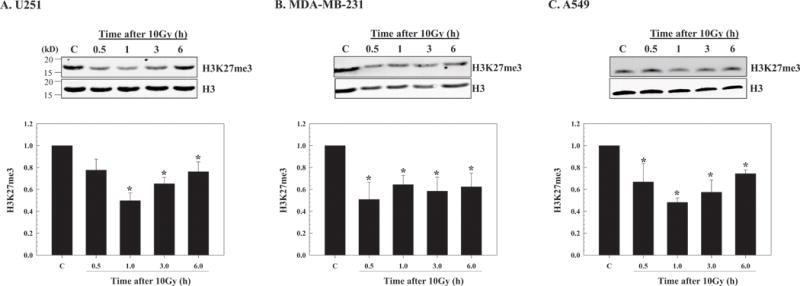
Effect of radiation on H3K27me3 levels in tumor cell lines. A) U251, B) MDA-MB-231 and C) A549 cells were irradiated (10Gy) and chromatin protein isolated at the indicated time points. Top panels show representative immunoblots for H3K27me3 along with the corresponding H3 levels from each cell line. Bottom panels show H3K27me3 levels as a function of time after irradiation normalized to the unirradiated controls (C). H3 was used as a loading control; the data expressed as the mean ± SEM of 3-4 independent experiments. *p≤0.04 according to Student’s t test.
The rapid reduction in this chromatin mark after irradiation suggested the participation of a H3K27me3 demethylase, of which two have been identified UTX and JMJD3 (30). As shown in figure 2A, UTX was readily detectable in the 3 tumor cell lines, whereas to detect JMJD3 it was necessary to use three times as much protein for immunoblot analysis (20μg versus 60μg, respectively). The relatively low expression level of JMJD3 in the tumor cell lines suggested that of the H3K27 demethylases UTX may mediate the radiation-induced decrease in H3K27me3. Of note, in normal lung fibroblasts (MRC5 and MRC9) levels of UTX and H3K27me3 were less than those detected in the tumor cell lines. To test this hypothesis, siRNA was used to knockdown UTX in U251, MDA-MB-231 and A549 cells. At 48h after siRNA transfection UTX expression was reduced by 90% in each of the cell lines as compared to cells receiving the non-targeted siRNA (Figure 2B). Whereas irradiation of cells transfected with the non-targeted siRNA decreased H3K27me3 level in each of the tumor lines (Figure 2C) knockdown of UTX prevented the radiation-induced decrease in H3K27me3 suggesting that UTX mediates the radiation-induced decrease in H3K27me3. This knockdown strategy was then used to determine whether UTX plays a role in tumor cell radiosensitivity (Figure 2D). As compared to cells receiving non-targeted siRNA, knockdown of UTX increased the radiosensitivity of each of the tumor cell lines with dose enhancement factors (DEFs) at a surviving fraction of 0.1 ranging from 1.2 to 1.3. Knockdown of UTX alone reduced survival to 0.38±0.10, 0.76±0.21 and 0.39±0.07 (mean ± SEM) in U251, MDA-MB-231 and A549 cells, respectively, as compared to cells receiving non-targeted siRNA. The data presented in figure 2 thus suggest that the UTX mediated decrease in H3K27me3 contributes to the survival of tumor cells after irradiation.
Figure 2.
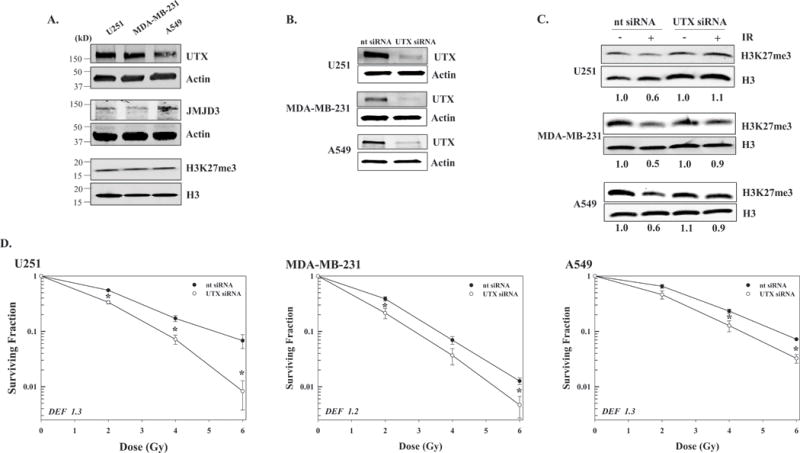
Effect of UTX knockdown H3K27me3 levels and tumor cell radiosensitivity. A) Expression of H3K27me3 demethylases (UTX and JMJD3) and H3K27me3 in the 3 tumor cell lines and two normal lung fibroblast cell lines (MRC5 and MRC9). Protein from whole cell lysates (20μg for UTX and 60μg for JMJD3), and chromatin protein (2μg) were subjected to immunoblot analysis with actin and H3 used as loading controls, respectively. B) Tumor cells were transfected with nontargeted (nt) siRNA or siRNA specific for UTX (UTX siRNA) and 48h post-transfection whole cell lysates was subjected to immunoblot analysis with actin used as a loading control. C) Transfected tumor cells were irradiated with 10Gy (IR) and collected at 1h later for immunoblot analysis of H3K27me3 with H3 used as a loading control. Representative immunoblots of 2 independent experiments are shown; ratios of H3K27me3 in irradiated versus unirradiated are shown below each blot. D) 48h after transfection U251, MDA-MB-231, and A549 cells were plated, irradiated after attachment and subjected to clonogenic survival analysis. Colony-forming efficiency was determined 10 to 14 days later and survival curves were generated after normalizing for cell killing from siRNA alone. DEFs were calculated at a surviving fraction of 0.1. Values shown represent the mean ± SEM for 3 to 4 independent experiments. *p≤0.04 according to Student’s t test.
These results also suggested that UTX provides a target for tumor cell radiosensitization. To investigate this possibility, we used the small molecule H3K27 demethylase inhibitor GSKJ4, the structure and specificity of which has been previously described (31–34). For each cell line, whereas radiation exposure reduced H3K27me3 levels at 1h, when GSKJ4 (4μM) was added to culture media immediately before irradiation H3K27me3 levels were essentially the same as in control (Figure 3A) indicating that GSKJ4 prevents the radiation-induced decrease in H3K27me3. The effects of GSKJ4 on cellular radiosensitivity were then evaluated using clonogenic survival analysis (Figure 3B). For this study, cells were plated at clonogenic densities and GSKJ4 added immediately before irradiation; 24h later cultures were rinsed and fresh, drug media was added. Treatment with GSKJ4 enhanced the radiosensitivity of each of the three tumor cell lines (Figures 3B) with (DEFs) at a surviving fraction of 0.1 ranging from 1.2-1.4. GSKJ4 treatment alone resulted in a minor amount of tumor cell killing, which was accounted for in the analysis of the combination protocol, with surviving fractions (mean ± SEM) of 0.62±0.08, 0.64±0.02 and 0.63±0.2 for U251, MD-MBA-231 and A549 cells, respectively. These results indicate that GSKJ4 enhances tumor cell radiosensitivity and suggest that the mechanism involves the prevention of the radiation-induced loss of H3K27me3.
Figure 3.
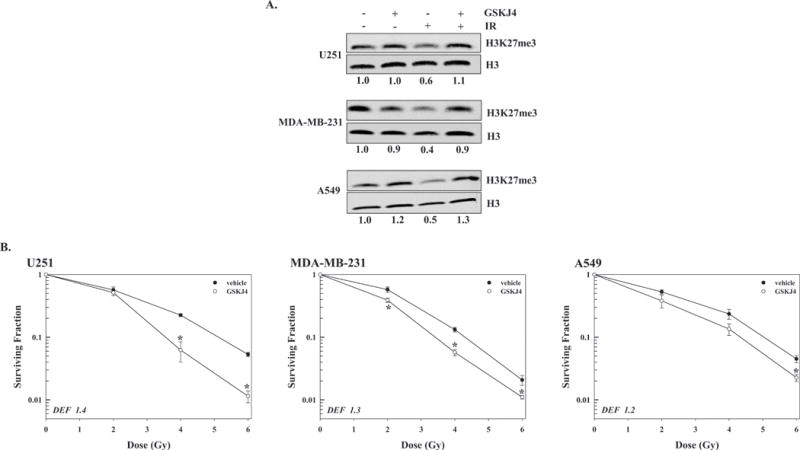
Influence of GSKJ4 on H3K27me3 levels in and radiosensitivity of the 3 tumor cell lines. A) GSKJ4 (4μM) was added to culture media immediately before irradiation (10Gy), 1h later chromatin protein was collected and subjected to immunoblot analysis for H3K27me3 with H3 used as a loading control. Representative immunoblots from each cell line are presented with the ratios comparing H3K27me3 levels in treated versus untreated cells shown below each blot. B) For clonogenic survival analysis GSKJ4 (4μM) was added to culture media immediately prior to irradiation, 24h later cultures were rinsed and fed fresh, drug-free medium. Colony-forming efficiency was determined 10 to 14 days later and survival curves were generated after normalizing for cell killing from drug alone. DEFs were calculated at a surviving fraction of 0.1. Values shown represent the mean ± SEM for 3 to 4 independent experiments. *p≤0.04 according to Student’s t test.
Because GSKJ4 was added immediately before irradiation, redistribution of cells into a radiosensitive phase of the cell cycle is unlikely to account for the observed radiosensitization. Moreover, GSKJ4 exposure alone ((4μM, 16h) had no effect on cell cycle phase distribution (Figure S1B). To gain insight into the mechanism through which GSKJ4 enhances tumor cell radiosensitivity, DSB repair was evaluated in U251 and MDA-MB-231 cells using the neutral comet and γH2AX expression assays (Figure 4). For the neutral comet assay GSKJ4 (4μM) was added to culture media immediately before irradiation (10Gy) and cells collected for analysis at times out to 24h (Figure 4A). For both cell lines, GSKJ4 treatment significantly slowed the repair of radiation-induced DSBs, which was detectable by 1h. The DSBs remaining at 24h reflect residual radiation-induced damage, which was increased in GSKJ4 treated cells, and is consistent with an increase in radiation-induced cell death. As an additional measure of DSB repair, the expression of phosphorylated histone H2AX (γH2AX), a surrogate marker for radiation-induced DSBs, was evaluated using flow cytometry (35). U251 and MDA-MB-231 cells were treated with GSKJ4 (4μM) immediately before irradiation (6Gy) and collected 16h later. In both cell lines (Figure 4B), GSKJ4 alone had no significant effect, whereas radiation alone increased the percentage of cells expressing γH2AX at 16h as compared to vehicle treated cells. For both cell lines, the combination of GSKJ4 and radiation increased the percentage of cells expressing γH2AX as compared to radiation alone suggesting that GSKJ4 inhibits DSB repair. Thus, data generated from the neutral comet and γH2AX assays suggest that the inhibition of H3K27m3 demethylation mediated by GSKJ4 inhibits the repair of radiation-induced DSBs, which could then account for the observed radiosensitization.
Figure 4.
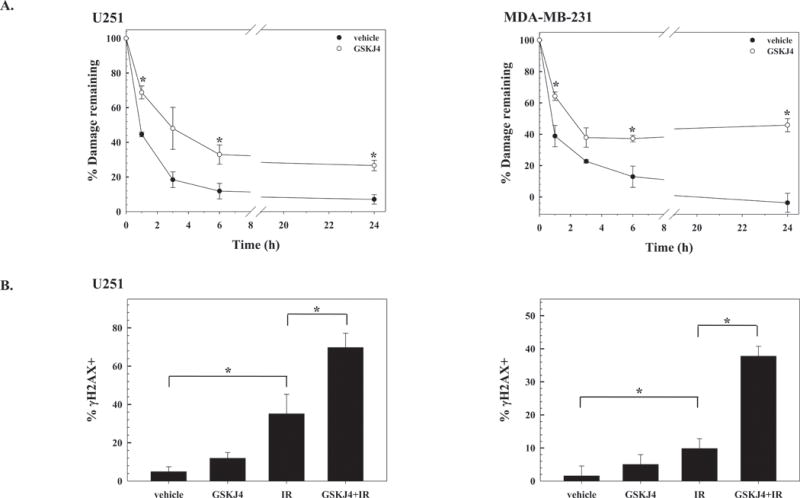
Influence of GSKJ4 on the repair of radiation-induced DNA damage. A) Neutral comet assay. U251 and MDA-MB-231 cells were treated with vehicle (DMSO) or GSKJ4 (4μM) immediately before irradiation (10Gy) and analyzed at times out to 24h. Data are expressed as percent damage remaining with the Olive tail moment immediately after radiation corresponding to 100% damage. Values shown represent the mean ± SEM for 3 to 4 independent experiments. B) γH2AX expression. U251 or MDA-MB-231 cells were treated with vehicle (DMSO) or GSKJ4 (4μM) immediately before irradiation (6Gy) and collected for analysis 16h later. Values shown represent the mean ± SEM for 3 independent experiments. *p≤0.04 according to Student’s t test.
The effects of radiation on H3K27me3 levels were also determined in the normal human fibroblast cell lines MRC5 and MRC9. As in the analysis of the tumor cell lines, MRC5 and MRC9 were exposed to 10Gy and chromatin protein collected and analyzed by immunoblot for H3K27me3 at times out to 6h. In contrast to the tumor cell lines, H3K27me3 levels in MRC5 and MRC9 cells were unchanged after irradiation (Figure 5A). Of note, histone H3K27me3 levels were considerably lower in the normal fibroblast cell lines compared to tumor cell lines (Figure 2A), which necessitated the higher chromatin protein (10μg) for immunoblot analysis. Consistent with the absence of a radiation-induced decrease in H3K27me3 and that treatment of the fibroblasts with GSKJ4 did not enhance their radiosensitivity (Figure 5B). Specifically, the demethylase inhibitor had no effect on the radiosensitivity of MRC9 and slightly reduced the radiosensitivity of MRC5 cells. GSKJ4 treatment alone resulted in surviving fractions (mean ± SEM) of 0.31±0.01 for MRC5 and 0.27±0.04 for MRC9. These data suggest that the radiation induced decrease of H3K27me3 methylation may provide a target for selective tumor cell radiosensitization.
Figure 5.
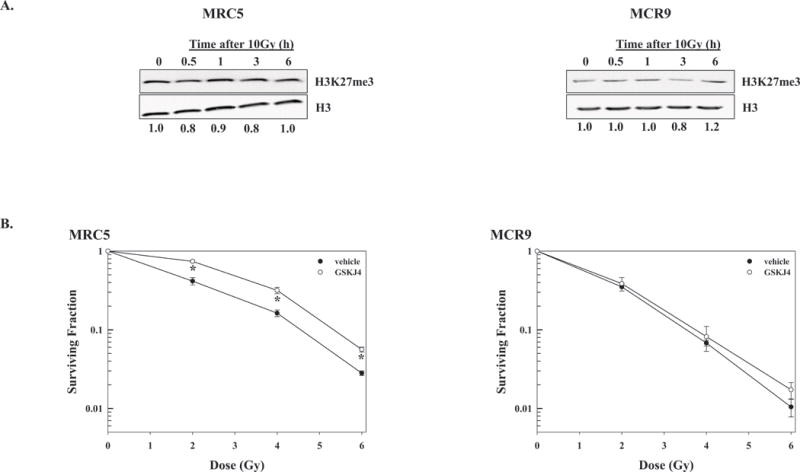
H3K27me3 levels in normal cells after irradiation and effects of GSKJ4 on normal cell radiosensitivity. A) H3K27me3 levels in MRC5 and MRC9 normal fibroblast cell lines after irradiation. Chromatin protein was collected at the designated times after exposure to 10Gy; 10μg was subjected to immunoblot analysis for H3K27me3 and H3 (loading control). Representative immunoblots from each cell line are shown; H3K27me3 ratios in irradiated versus unirradiated cells are shown below each blot. B) Clonogenic survival analysis of MRC5 and MRC9. GSKJ4 (4μM) was added immediately prior to irradiation and 24h later cultures were rinsed and fed fresh, drug-free medium. Colony-forming efficiency was determined 14 days later and survival curves were generated after normalizing for cell killing from drug alone. Values shown represent the mean ± SEM for 3 independent experiments. *p≤0.01 according to Student’s t test.
Finally, we determined whether the GSKJ4-mediated radiosensitization detected in vitro could be extended to an in vivo tumor xenograft model (Figure 6). Initially, the effect of radiation on H3K27me3 levels in tumors was determined as was whether this effect could be inhibited by GSKJ4. For this experiment, U251 were subcutaneously implanted into the right leg of nude mice and when the tumors reached (∼300 mm3), randomized into four groups: vehicle, GSKJ4 (100mg/kg), radiation (10Gy) and GSKJ4 plus radiation. Specifically, mice were treated with GSKJ4 1h before irradiation and tumors collected 3h after irradiation. As illustrated in Figure 6A and quantified in Figure 6B, GSKJ4 had no effect on the level of H3K27me3, whereas this chromatin mark was significantly reduced after irradiation. Consistent with in vitro results, administration of GSKJ4 prior to irradiation prevented the radiation-induced decrease of H3K27me3. Based on these findings, a protocol was designed to test the antitumor effectiveness of the GSKJ4/radiation combination. Mice bearing subcutaneous U251 leg tumors (∼220 mm3) were randomized into four groups: vehicle, GSKJ4, radiation, and GSKJ4 plus radiation. GSKJ4 was delivered twice a day (50mg/kg per intraperitoneal injection) at 1h before and 7h after local irradiation (2Gy) for three consecutive days. Two final doses of 50mg/kg GSKJ4 were given 16h and 24h after the last radiation dose. The growth rates of U251 tumors corresponding to each treatment are shown in Figure 6C. GSKJ4 treatment alone had no significant effect on tumor growth rate, although tumors in this group appeared to grow slightly faster than in vehicle treated mice. Radiation alone resulted in a tumor growth delay (the time in days for tumors in treated mice to grow from 220 to 1000 mm3 minus the time in days for tumors to reach the same size in vehicle-treated mice) of 5 days. The tumor growth delay in mice treated with the combination of GSKJ4 and radiation was 15 days, corresponding to a DEF of 3. Thus, as for tumor cells grown in vitro, these data indicate that GSKJ4 enhanced the tumor cell radiosensitivity in vivo.
Figure 6.
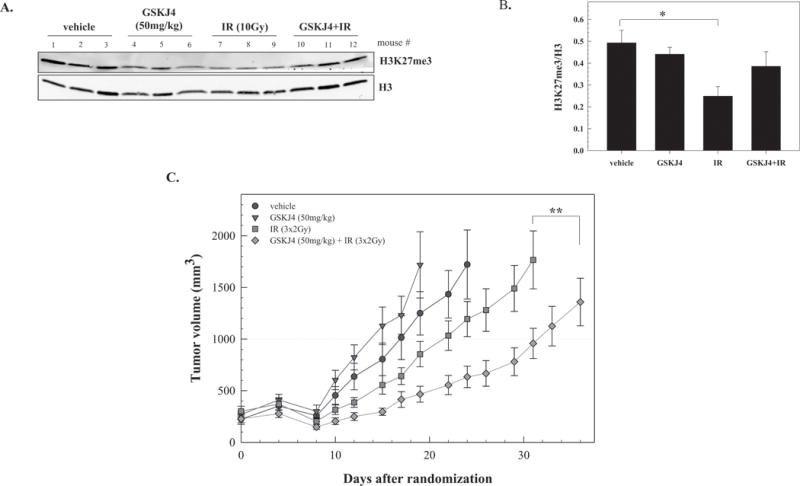
The effects of GSKJ4 on radiation-induced tumor growth delay. A) H3K27me3 levels in U251 leg tumor xenografts after treatment with GSKJ4 (50mg/kg), radiation (IR, 10Gy) or the combination. B) H3K27me3 versus H3 ratios calculated from immunoblot (shown in A) for vehicle, GSKJ4, radiation or combination (GSKJ4+IR) treated mice. Values represent the mean ± SEM of the 3 mice per group, *p≤0.03 according to Student’s t test. C) Mice bearing U251 leg tumor xenografts were treated with GSKJ4 (50mg/kg) 1h before and 7h after each tumor irradiation (2Gy) for 3 consecutive days; GSKJ4 only was also delivered on the 4th day (twice with each dose separated by 8h). Each group contained 7-10 mice. Values represent the mean tumor volumes ± SEM. **p≤0.006 according to ANOVA.
Discussion
Chromatin remodeling via the induction of post-translational histone modifications has been established as a critical process mediating the repair of radiation-induced DSBs (1,4,6). Whereas a number of such modifications have been identified, the studies presented here focused on the trimethylation at H3K27. Previous reports have shown that H3K27me3 levels are increased locally around DSBs induced UV laser micro-irradiation (19) and the endonuclease I-sceI (22). However, whether this histone modification influences the repair of the DSBs induced by ionizing radiation and, of significance with respect to cancer treatment, tumor cell radiosensitivity remained to be defined. In the study presented here, while H3K27me3 levels locally around DSBs were not addressed, X-irradiation of human tumor cell lines initiated from a variety of solid tumor types resulted in a rapid reduction in chromatin H3K27me3. Given that this histone modification has been causally associated with condensed chromatin (36,37) and thus putatively acts as an impediment to DSB repair (38), the loss of H3K27me3 would be expected to facilitate repair and contribute to cell survival after irradiation. Under these circumstances, targeting the molecule mediating the reduction in H3K27me3 may provide a strategy for enhancing tumor cell radiosensitivity.
The rapid reduction of H3K27me3 after irradiation suggested that rather than the inhibition of a methyltransferase (i.e., EZH2), the effect was mediated by one of the H3K27 demethylases. Along these lines, UTX was expressed at considerably higher levels in the 3 tumor cell lines than JMJD3. This is consistent with UTX being ubiquitously expressed and its suggested role as a “housekeeping” demethylase (39). Whereas the expression of JMJD3 has been reported to increase in response to a variety of stimuli, including by 6h after irradiation (39), the reduction in H3K27me3 reported here was detectable within 30 minutes. This rapid decrease is consistent with the action of UTX, which is already expressed at a relatively high level under basal conditions. Accordingly, knockdown of UTX prevented the radiation-induced reduction in H3K27me3, which was accompanied be increased tumor cell radiosensitivity. UTX-mediated changes in H3K27me3 have been mostly clearly associated with modifications in gene expression. Indeed, previous studies have implicated UTX in the regulation of genes critical to the radiation-induced DNA damage response (39,40). The siRNA based experimental protocol used to generate the data presented in figures 1–2 required 48h between transfection and irradiation and thus allowed for the potential contribution of changes in gene expression to the increase in radiosensitivity. However, in addition to the siRNA based knockdown approach, we also used the H3K27 demethylase inhibitor GSKJ4. Of mechanistic significance, GSKJ4 was added immediately before irradiation resulting in the prevention of the reduction in H3K27me3 along with an inhibition of DSB repair detectable at early as 1h and radiosensitization, which argues against a causal role for changes in gene expression. Thus, the data presented suggest that the radiation-induced loss of H3K27me3 relaxes chromatin structure to facilitate DSB repair. In general, histone methylation status has been linked to NHEJ (16,17) and the neutral comet analysis presented here showing that GSKJ4 inhibits the DSB repair by 6h after irradiation is consistent with a role for NHEJ. However, whereas results indicate that UTX mediates the loss of H3K27me3 after irradiation, the specific mechanism responsible for the increased UTX activity remains to be determined.
In contrast to the three human tumor cell lines, radiation had no effect on H3K27me3 levels in two normal human fibroblast cell lines. Consistent with the low basal protein expression of UTX and the lack of H3K27me3 reduction in response to radiation, GSKJ4 had no effect on the radiosensitivity of MRC5 and MRC9. These results suggest that the radiation-induced reduction in H3K27me3 and its role in DSB repair may be tumor cell specific. However, it was previously reported that after irradiation H3K27me3 levels were unchanged in the osteosarcoma cell line U2OS (19) and decreased in a human fibroblast cell line (26), although only a single time point post-radiation was evaluated in both studies. Thus, whereas the data presented here indicate a consistent response among 3 tumor cell lines, the molecular circumstances that allow for a UTX-mediated reduction in H3K27me3 after irradiation and the potential for this histone demethylase to provide a target for radiosensitization remains to be more completely defined.
GSKJ4 has been reported to inhibit the in vivo growth of diffuse intrinsic pontine glioma via JMJD3 inhibition (41) and T-cell acute lymphoblastic leukemia via UTX inhibition (42). In contrast, we found that GSKJ4 had no effect on the growth of U251 xenografts, which may be the result of the differences in the length of GSKJ4 administration, 5 days vs. 2-3 weeks in the previous studies (41,42). However, GSKJ4 enhanced the radiation-induced growth delay of U251 leg tumor xenografts, which in vitro data suggests is due to the inhibition of UTX. It is possible that GSKJ4, in addition to the H3K27 demethylases, may also inhibits other histone demethylases (31). Nevertheless, the data presented here showing that the GSKJ4-mediated inhibition of the H3K27me3 demethylation enhances the radiosensitivity of tumor but not normal cells supports further evaluation of this epigenetic approach to enhancing the effectiveness of radiotherapy.
Supplementary Material
Acknowledgments
Financial Information: Division of Basic Sciences, Intramural Program, National Cancer Institute (Z1ABC011372) to Philip J. Tofilon
Footnotes
Conflict of Interest: none
References
- 1.Gursoy-Yuzugullu O, House N, Price BD. Patching Broken DNA: Nucleosome Dynamics and the Repair of DNA Breaks. J Mol Biol. 2016;428(9 Pt B):1846–60. doi: 10.1016/j.jmb.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11(11):1376–82. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunt CR, Ramnarain D, Horikoshi N, Iyengar P, Pandita RK, Shay JW, et al. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiat Res. 2013;179(4):383–92. doi: 10.1667/RR3308.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152(6):1344–54. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146(5):905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar R, Horikoshi N, Singh M, Gupta A, Misra HS, Albuquerque K, et al. Chromatin modifications and the DNA damage response to ionizing radiation. Front Oncol. 2012;2:214. doi: 10.3389/fonc.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419(6905):411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 8.Cao J, Yan Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol. 2012;2:26. doi: 10.3389/fonc.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camphausen K, Cerna D, Scott T, Sproull M, Burgan WE, Cerra MA, et al. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer. 2005;114(3):380–6. doi: 10.1002/ijc.20774. [DOI] [PubMed] [Google Scholar]
- 10.Chinnaiyan P, Cerna D, Burgan WE, Beam K, Williams ES, Camphausen K, et al. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res. 2008;14(17):5410–5. doi: 10.1158/1078-0432.CCR-08-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krauze AV, Myrehaug SD, Chang MG, Holdford DJ, Smith S, Shih J, et al. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int J Radiat Oncol Biol Phys. 2015;92(5):986–92. doi: 10.1016/j.ijrobp.2015.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432(7015):406–11. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 13.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A. 2014;111(25):9169–74. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fnu S, Williamson EA, De Haro LP, Brenneman M, Wray J, Shaheen M, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci U S A. 2011;108(2):540–5. doi: 10.1073/pnas.1013571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu R, et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol. 2015;17(9):1158–68. doi: 10.1038/ncb3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Campbell S, Ismail IH, Young LC, Poirier GG, Hendzel MJ. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle. 2013;12(16):2675–83. doi: 10.4161/cc.25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Q, Oh JE, Chen W, Kim R, Kim RH, Shin KH, et al. Radioprotective effects of Bmi-1 involve epigenetic silencing of oxidase genes and enhanced DNA repair in normal human keratinocytes. J Invest Dermatol. 2011;131(6):1216–25. doi: 10.1038/jid.2011.11. [DOI] [PubMed] [Google Scholar]
- 21.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4(8):e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alimova I, Birks DK, Harris PS, Knipstein JA, Venkataraman S, Marquez VE, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15(2):149–60. doi: 10.1093/neuonc/nos285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gursoy-Yuzugullu O, Carman C, Serafim RB, Myronakis M, Valente V, Price BD. Epigenetic therapy with inhibitors of histone methylation suppresses DNA damage signaling and increases glioma cell radiosensitivity. Oncotarget. 2017;8(15):24518–32. doi: 10.18632/oncotarget.15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107(43):18475–80. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Hart RP, Mallimo EM, Swerdel MR, Kusnecov AW, Herrup K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat Neurosci. 2013;16(12):1745–53. doi: 10.1038/nn.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rath BH, Wahba A, Camphausen K, Tofilon PJ. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015;4(11):1705–16. doi: 10.1002/cam4.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol. 2004;26(3):249–61. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- 29.Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20(19):5085–96. doi: 10.1158/1078-0432.CCR-14-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A. 2007;104(47):18439–44. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinemann B, Nielsen JM, Hudlebusch HR, Lees MJ, Larsen DV, Boesen T, et al. Inhibition of demethylases by GSK-J1/J4. Nature. 2014;514(7520):E1–2. doi: 10.1038/nature13688. [DOI] [PubMed] [Google Scholar]
- 32.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488(7411):404–8. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakaki H, Okada M, Kuramoto K, Takeda H, Watarai H, Suzuki S, et al. GSKJ4, A Selective Jumonji H3K27 Demethylase Inhibitor, Effectively Targets Ovarian Cancer Stem Cells. Anticancer Res. 2015;35(12):6607–14. [PubMed] [Google Scholar]
- 34.Watarai H, Okada M, Kuramoto K, Takeda H, Sakaki H, Suzuki S, et al. Impact of H3K27 Demethylase Inhibitor GSKJ4 on NSCLC Cells Alone and in Combination with Metformin. Anticancer Res. 2016;36(11):6083–92. doi: 10.21873/anticanres.11198. [DOI] [PubMed] [Google Scholar]
- 35.Cuneo KC, Morgan MA, Davis MA, Parcels LA, Parcels J, Karnak D, et al. Wee1 Kinase Inhibitor AZD1775 Radiosensitizes Hepatocellular Carcinoma Regardless of TP53 Mutational Status Through Induction of Replication Stress. Int J Radiat Oncol Biol Phys. 2016;95(2):782–90. doi: 10.1016/j.ijrobp.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartova E, Krejci J, Harnicarova A, Galiova G, Kozubek S. Histone modifications and nuclear architecture: a review. J Histochem Cytochem. 2008;56(8):711–21. doi: 10.1369/jhc.2008.951251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Q, Wani AA. Histone modifications: crucial elements for damage response and chromatin restoration. J Cell Physiol. 2010;223(2):283–8. doi: 10.1002/jcp.22060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams K, Christensen J, Rappsilber J, Nielsen AL, Johansen JV, Helin K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS One. 2014;9(5):e96545. doi: 10.1371/journal.pone.0096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Hong Z, Ma W, Ma D, Qian Y, Xie W, et al. Drosophila UTX coordinates with p53 to regulate ku80 expression in response to DNA damage. PLoS One. 2013;8(11):e78652. doi: 10.1371/journal.pone.0078652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20(12):1394–6. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benyoucef A, Palii CG, Wang C, Porter CJ, Chu A, Dai F, et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016;30(5):508–21. doi: 10.1101/gad.276790.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.