

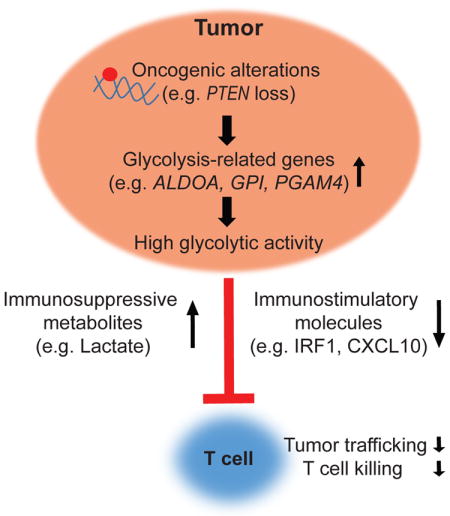
SUMMARY
Adoptive T cell therapy (ACT) produces durable responses in some cancer patients; however, most tumors are refractory to ACT and the molecular mechanisms underlying resistance are unclear. Using two independent approaches, we identified tumor glycolysis as a pathway associated with immune resistance in melanoma. Glycolysis-related genes were upregulated in melanoma and lung cancer patient samples poorly infiltrated by T cells. Overexpression of glycolysis-related molecules impaired T cell killing of tumor cells, whereas inhibition of glycolysis enhanced T cell-mediated antitumor immunity in vitro and in vivo. Moreover, glycolysis-related gene expression was higher in melanoma tissues from ACT-refractory patients, and tumor cells derived from these patients exhibited higher glycolytic activity. We identified reduced levels of IRF1 and CXCL10 immunostimulatory molecules in highly glycolytic melanoma cells. Our findings demonstrate that tumor glycolysis is associated with the efficacy of ACT and identify the glycolysis pathway as a candidate target for combinatorial therapeutic intervention.
eTOC Blurb
XXX et al identify tumor glycolysis to be associated with immune resistance to adoptive T cell therapy (ACT). In ACT-treated patients, increased tumor glycolytic activity is associated with lower therapeutic response, highlighting the therapeutic potential of combining glycolysis inhibition with ACT in cancer patients.
INTRODUCTION
Adoptive T cell therapy (ACT) is a form of personalized cancer therapy that involves the administration of ex vivo expanded autologous tumor-infiltrating lymphocytes (TILs) to cancer patients. Ideally, the intravenously injected TILs arrest in the tumor microvasculature, migrate into the tumor tissue, encounter tumor antigens and reject established tumors (Chen and Mellman, 2013). ACT has demonstrated efficacy in multiple cancer types, particularly in patients with melanoma, where it produces objective responses in approximately 50% of patients (Dudley et al., 2005; Rosenberg and Restifo, 2015). However, most patients are refractory to ACT and only 25% of patients achieve durable, complete tumor regression (Goff et al., 2016). Therefore, there is strong rationale to improve our understanding of the molecular mechanisms of tumor resistance to ACT in order to develop more effective combinatorial therapies.
Two rate-limiting steps of the cancer immunity cycle are the trafficking of T cells to tumor and their subsequent eradication of tumor cells (Chen and Mellman, 2013). An expanding body of evidence suggests that activation of oncogenic signaling pathways, either by activating mutations of oncogenes (e.g. CTNNB1) or loss of function of tumor suppressors (e.g. PTEN), may impair TIL recruitment and effector function (Frederick et al., 2013; Khalili et al., 2012; Liu et al., 2013; Peng et al., 2016; Spranger et al., 2015). For example, we recently demonstrated that loss of the phosphatase and tensin homolog (PTEN) impedes trafficking of effector T cells to tumors, reduces the sensitivity of melanoma cells to T cell-mediated killing, and correlates with inferior outcomes in patients treated with immune checkpoint blockade. Moreover, we found that targeting PTEN-regulated signaling pathway improves the efficacy of immunotherapy, which has led to an ongoing clinical trial (NCT03131908) (Peng et al., 2016). These results support the rationale to identify additional tumor intrinsic pathways that may promote resistance to the antitumor immune response, and could represent targets to potentiate the efficacy of T cell-based therapies.
Results from recent studies emphasize that tumor metabolism plays a role in cancer development and therapeutic resistance (Altman et al., 2016; Nakazawa et al., 2016; Yang and Vousden, 2016). The activation of several oncogenic signaling pathways, including mTOR, BRAF, and c-Myc has been shown to increase cancer cell glycolysis and lead to lactate accumulation within the tumor microenvironment (Renner et al., 2017). The dysregulated glycolysis of tumor cells promotes tumor growth and invasiveness, and is positively correlated with the incidence of metastases in cancer patients (Walenta et al., 2000). The glycolytic switch of cancer cells also limits the development of an effective antitumor immune response (Chang et al., 2015; Pilon-Thomas et al., 2016; Singer et al., 2011). However, the impact of tumor glycolysis on T cell-mediated antitumor immunity has not been well characterized.
To address this knowledge gap, we used two unbiased and independent approaches to identify alternative pathways that may mediate immune resistance. These studies identified glycolysis as a candidate pathway of resistance, which prompted us to examine the levels of glycolysis-related genes in melanoma and non-small cell lung cancer (NSCLC). We correlated patterns of glycolysis-related genes to T cell trafficking to tumors and to the sensitivity of tumors to T cell-mediated killing. Upregulated tumor glycolytic activity was observed in melanoma patients who failed to respond to ACT. Our work highlights the critical role of tumor intrinsic glycolysis in modulating T cell-mediated antitumor activity and lays the foundation for the development of glycolysis inhibitors to improve the effectiveness of ACT for cancer treatment.
RESULTS
Identification of tumor glycolysis as a candidate mediator of immune resistance
Recently, we discovered that loss of PTEN enables melanomas to evade immune surveillance by limiting T cell trafficking into tumors and preventing T cell-mediated killing of tumor cells (Peng et al., 2016). We leveraged this finding and employed two independent and unbiased approaches to identify additional tumor pathways that may modulate T cell-mediated tumor killing. First, we used the PTEN loss melanoma model to identify potential immunosuppressive mechanisms shared by PTEN loss and other dysregulated pathways in melanoma. We performed Ingenuity Pathway Analysis (IPA) on gene expression profiles of melanoma cells with and without silenced PTEN. That analysis identified tumor glycolysis as a differentially regulated pathway with one of the highest confidence scores (Figure 1A). We next evaluated the energy metabolism of PTEN-silenced melanoma cells. We noted that PTEN-silenced melanoma cells had an increased extracellular acidification rate (ECAR), a measure of intrinsic glycolysis, compared with wild-type PTEN melanoma cells (Figure 1B, top panel). The mitochondrial oxygen consumption rate (OCR), an index of mitochondrial oxidative phosphorylation (OXPHOS), was similar in both cell types (Figure 1B, bottom panel). To confirm that tumor PTEN expression plays a regulatory role in tumor glycolysis, we performed an unbiased metabolic profiling of melanoma cells with and without silenced PTEN. As shown in Figure S1A, we found a defined clustering of these cell lines by principal component analysis based on their metabolite profiles. Indeed, levels of intermediate products of tumor glycolysis, including glucose-1 phosphate, 6-phospho-D-gluconate, pyruvate and lactate, were significantly higher in PTEN-deficient melanoma cells, as shown in Figure S1B. These findings indicate that enhanced tumor intrinsic glycolysis is a characteristic feature of immune resistant, PTEN-loss melanoma cells.
Figure 1. Glycolysis-related genes are identified as candidate genes that promote resistance to T cell-mediated antitumor activity.

A. List of pathways that are significantly modulated by silencing PTEN in melanoma cells. The mRNA expression profiles of PTEN-silenced and control cell lines were established from a melanoma cell line (A375) by microarray analysis. The two PTEN-silenced cell lines expressed shRNA hairpins that target different positions of the PTEN sequence (PTEN-silenced Tu-17 and Tu-60), and the corresponding control cell line (Control Tu) expressed a non-targeting shRNA hairpin. IPA was used to determine the pathways that are directly regulated by PTEN expression. B. Bioenergetic profiles of melanoma cells with or without silenced PTEN are shown. The Seahorse XF cell Mito stress test was used to define the bioenergetic profiles of PTEN-silenced and control cells. The changes in ECAR and OCR for the time indicated during a XF24 extracellular flux analyzer run are plotted. Oligomycin (1 uM) and FCCP (0.3 uM) were sequentially injected to the assay medium. The dash lines indicate the injection time points. *P<0.05. FCCP: carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone. C. A modified dropout screen used to identify genes that promote resistance to T cell-mediated tumor killing is shown. Patient-derived melanoma cells were transduced with a pooled shRNA library, followed by puromycin selection to eliminate non-transduced cells. After a 2-week cell culture to remove the genes that directly modulate cell survival, the cells were selected by autologous TILs with an effector to target (E:T) ratio of 1:1 that caused approximately 30% killing efficiency. The shRNA-targeted genes that promote resistance to T cell-induced tumor killing were underrepresented in the samples with TIL treatment. D. The identified candidate immune regulators involved in enzymatic steps of glycolysis are shown. LogFC: the base 2 log of the fold change of TIL-treated groups vs. control; FDR: false discovery rate. ALDOA and ALDOC: aldolase A and C; ENO3: enolase 3, GPI: glucose-6-phosphate isomerase; PGAM2: phosphoglycerate mutase 2.
Next, we employed a loss of function shRNA library screen in order to discover candidate metabolic molecules that may modulate the sensitivity of tumor cells to T cell-mediated killing. Briefly, we transduced patient-derived melanoma cells with a pooled library of barcoded shRNAs targeting genes involved in cellular metabolism. Our metabolomic library included 3,430 shRNA sequences that target 343 genes involved in multiple metabolic pathways. The shRNA-transduced melanoma cells were either cultured alone or exposed to their autologous TILs, and the intensities of gene-specific shRNA sequences in melanoma cells with or without TIL treatment were analyzed. This screen is based on the principle that the individual gene-specific shRNA sequences that sensitize tumor cells to T cell-mediated killing would be depleted in TIL-treated samples (Figure 1C). The consistency across two repeated experiments (R=0.42) provided high confidence in the identified hits (Figure S1C). Among the top ten underrepresented shRNA-targeted genes identified from these experiments, we observed two glycolysis-related genes, PGAM2 and ALDOC, whose false discovery rates were less than 5% (Figure S1D). When we filtered the hits derived from our metabolomic screen using a P value < 0.05 and focused on glycolysis-related genes, we discovered that ALDOA, ALDOC, ENO3, GPI, PGAM2 and PKM were underrepresented in melanoma cells exposed to TILs when compared with controls (Figure 1D). We also noted that multiple shRNAs silenced the same gene involved in glycolysis, including ALDOA and PKM, confirming that glycolysis-related molecules are associated with resistance to T cell killing of melanoma cells. Taken together, two unbiased and independent approaches identified tumor glycolysis-related genes as candidate molecules that may promote resistance to T cell-mediated antitumor immune response.
Expression of glycolysis-related genes is negatively correlated with T cell infiltration in tumor samples from melanoma and NSCLC patients
T cell infiltration is one of the key rate-limiting steps in ACT (Chen and Mellman, 2013). To determine whether tumor glycolysis is associated with T cell recruitment to melanomas, we evaluated the relationship between the expression of glycolysis-related genes and T cell infiltration in cutaneous melanoma samples from TCGA (Cancer Genome Atlas, 2015). Tumor infiltration of lymphocytes was determined by pathologic review of cutaneous melanoma samples and reported as the lymphocyte score (L score), where a high L score indicates a high abundance of TILs (Cancer Genome Atlas, 2015). We found that the expression of the glycolysis-related genes ALDOA, ALDOC, ENO2, GAPDH, GPI and PFKM was significantly higher in samples that were poorly infiltrated by T cells (Figure 2A). When we categorized the melanoma TCGA samples by PTEN copy number (CN) and L score, we noted that the expression of ALDOA was still inversely correlated with the L score, regardless of PTEN copy number (Figure S2A). These findings suggest that the effects of tumor intrinsic glycolysis on T cell infiltration of tumors from melanoma patients may include, but are not limited to, PTEN loss.
Figure 2. Increased expression of glycolysis-related genes is associated with poor T cell infiltration in clinical samples of melanoma and NSCLC.

A. Boxplots correlating the lymphocyte infiltration of tumors (L score) and the mRNA expression levels of tumor glycolytic genes are shown in melanoma samples (TCGA). L score in cutaneous melanoma patients was evaluated by pathologists and reported as numerical value on a 0–6 scale, where a score >3 indicates high abundance of tumor-infiltrating T cells. The mRNA gene expression is plotted as transcripts per million. B. Boxplots correlating the mRNA expression of the glycolytic genes with the levels of T cell signature genes in NSCLC samples from PROSPECT are shown. C. Significant correlations between the mRNA expression of the indicated glycolysis-related genes and the IHC immune markers scored in NSCLC samples from PROSPECT dataset are shown as Pearson correlation coefficients. P<0.05 was used to determine statistical significance. The gray squares represent no statistically significant correlations. BPGM: bisphosphoglycerate Mutase; ENO2: enolase 2; FBP1, FBP2: fructose-bisphosphatase 1 and 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; LDHA, LDHB: lactate dehydrogenase A, B; PFKM: phosphofructokinase, muscle; PFKP: phosphofructokinase, platelet; PGAM1, PGAM4: phosphoglycerate mutase 1 and 4; PGK1: phosphoglycerate kinase; PGK2: phosphoglycerate kinase; SLC2A1: solute carrier family 2 member 1; GZB: granzyme B.
In addition to melanoma, immunotherapy has induced durable responses in patients with other types of cancer, including NSCLC (Liu and Giaccone, 2016). To determine whether the association between tumor glycolysis and tumor infiltration of T cells can be extended to other forms of cancer, we analyzed clinical samples from NSCLC patients. We applied a previously validated 13-gene signature of T cell infiltration (Spranger et al., 2015) to a dataset of surgically resected NSCLC samples from the PROSPECT cohort to characterize tumors as either enriched in T cells (T cell-inflamed) or poor in T cells (non T cell-inflamed). Evaluation of mRNA levels of glycolytic genes showed that ENO2, ENO3, GAPDH and PFKM were significantly higher in non T cell-inflamed samples compared with T cell-inflamed tumors (Figure 2B). We also clustered NSCLC samples from TCGA into T cell-inflamed and non T cell-inflamed groups, as shown in Figure S2B. Expression of ENO3 was again significantly higher in NSCLCs from TCGA that were poorly infiltrated by T cells compared with T cell-inflamed NSCLCs (Figure S2C).
To confirm these findings, we correlated the expression of tumor glycolysis-related genes with the immunohistochemical staining (IHC) scores of immune markers in PROSPECT tumor samples (Kadara et al., 2017; Parra et al., 2016). Expression of glycolysis-related genes, including ALDOA, ENO2, ENO3, GAPDH, GPI, LDHA, PFKM, PFKP, PGAM1, PGAM4 and SLC2A1 was negatively correlated with markers of cytotoxic T cells (CD3, CD8, GZB), T helper cells (CD4), memory T cells (CD45RO), macrophages (CD68), or natural killer (NK) cells (CD57) (Figure 2C). In contrast, expression levels of FBP1 and FBP2 were positively correlated with immune markers of T cell, macrophage or NK cell infiltration. The genes FBP1 and FBP2 encode two enzymes that catalyze the conversion of intermediate products that oppose lactic acid production. These findings indicate that increased tumor glycolytic activity is associated with poor tumor infiltration of T cells in melanoma and NSCLC patient samples.
Tumor glycolysis impairs T cell-mediated apoptosis of melanoma cells
Because T cell-mediated tumor killing is another rate-limiting step in ACT, we next examined the impact of tumor glycolysis on tumor resistance to T cell-mediated apoptosis. We ectopically expressed the melanoma antigen gp100 and the murine MHC Class I molecule H-2Db, which can be recognized by Pmel T cells derived from the TCR transgenic Pmel-1 mouse, in patient-derived melanoma cell lines. This system allows the evaluation of tumor cell sensitivity to T cell-mediated cytotoxicity without the confounding influences of variable expression of tumor antigens or MHC Class I molecules. The patient-derived melanoma cell lines displayed a diverse sensitivity to killing by tumor-reactive T cells (Figure S3A). Comparison of the cell lines with high versus low T cell-mediated killing revealed that ALDOA expression was significantly higher in the insensitive melanoma cell lines (Figure S3B). Indeed, we observed an inverse correlation between ALDOA levels in tumor cells and the percentage of melanoma cells killed by T cells (Figure 3A). These results suggest that highly glycolytic melanoma cells are more resistant to T cell-mediated apoptosis. To functionally validate these results, we first transduced a patient-derived melanoma cell line with a lentiviral vector encoding ALDOA. Bioenergetic studies of the cell metabolic activity revealed significantly higher ECAR levels in ALDOA-overexpressing cells compared with controls (Figure S4A), while no significant difference was noted in OCR (Figure S4B). Overexpression of ALDOA significantly increased the glucose uptake and lactate production of melanoma cells (Figure S4C and S4D, respectively), although the dependency of ALDOA-overexpressing cells on different nutrients, including pyruvate, glutamine and fatty acids as mitochondrial fuels, was comparable to controls (Figure S4E). These studies further confirmed that ALDOA-overexpressing cells display a glycolytic phenotype. Next, we evaluated whether increasing tumor intrinsic glycolytic activity by ALDOA overexpression could have an impact on T cell-mediated apoptosis of melanoma cells. We found that ALDOA overexpression significantly reduced the vulnerability of melanoma cells to autologous TIL-mediated apoptosis compared with controls, but had no effect on tumor cell apoptosis in the absence of T cells (Figure 3B). Furthermore, pretreatment of TILs with increasing concentrations of lactic acid, a terminal product of glycolysis, impaired the antitumor effector function of TILs in a dose-dependent manner (Figure 3C).
Figure 3. Increased tumor glycolytic activity impairs T cell-mediated apoptosis of melanoma cells.

A. The linear regression of mRNA expression of ALDOA in patient-derived melanoma cell lines correlated with the percentage of T cell-mediated tumor killing is shown. Patient-derived melanoma cell lines (n=12) overexpressing Gp100 and H-2Db, which can be recognized by Pmel T cells derived from the TCR transgenic Pmel-1 mouse, were generated by lentivirus-based genetic manipulations. Transduced melanoma cells were co-cultured with Pmel T cells at an effector to target ratio of 10:1 for 3 hours. The percentage of T cell-induced tumor killing was then calculated by quantifying the number of cleaved caspase-3+ cells with respect to the total number of tumor cells by flow cytometry. The mRNA expression of glycolysis-related genes in patient-derived melanoma cells was determined by microarray analysis. The R and P-value of the correlation between the ALDOA expression and the sensitivity to T cell killing in this set of patient-derived melanoma cell lines are listed. B. Overexpression of ALDOA in melanoma cells promotes resistance to T cell-mediated tumor cell apoptosis. Patient-derived melanoma cells (Mel2400) were transduced with lentiviral vectors encoding either ALDOA-RFP or GFP-RFP. The percentage of cleaved caspase-3+ transduced melanoma cells (based on the expression of the reporter gene RFP) was determined by flow cytometry. C. The impaired effector function of TILs by lactic acid is shown. TILs (TIL2559) derived from a melanoma patient were treated overnight with different concentrations of lactic acid. *P<0.05 (with versus without lactic acid treatment) D. LDHAi increases T cell-induced killing of melanoma cells. Patient-derived melanoma cells (Mel2792) were pre-treated with 1 uM of GSK2837808A, and followed by co-culture with autologous TILs (TIL2792) for 4 hours. E. Targeting glycolysis pathway enhances the antitumor activity of T cells. B16 melanoma cells were implanted subcutaneously in C57BL/6 mice. Tumor-bearing mice were treated with either GSK2837808A (6 mg/kg) orally and/or adoptively transferred Pmel T cells. Mice treated with the relevant solvent were used as control. Tumor sizes were monitored by measuring the perpendicular diameters of the tumors and are shown as mean tumor area ± SEM. All experiments were carried out in a blinded, randomized fashion (n=4–5 mice per group). F. Kaplan-Maier plots show survivals of tumor-bearing mice treated with vehicle, GSK2837808A, T cell therapy and combination of GSK2837808A and T cell therapy (n=4–5 mice per group). *P<0.05, **P<0.01, and ****P<0.0001.
To determine the impact of inhibiting glycolysis pathway on T cell-mediated tumor killing, we inhibited the activity of lactate dehydrogenase A (LDHA), which catalyzes the interconversion of pyruvate and lactate, in our preclinical melanoma models. Pretreatment of patient-derived melanoma cells with GSK2837808A, an LDHA-specific inhibitor (LDHAi), markedly increased the susceptibility of tumor cells to autologous TIL-mediated killing compared with cells that were treated with either LDHAi or TILs alone (Figure 3D). Furthermore, our in vivo studies demonstrated a significant decrease in the levels of lactate produced by tumors treated with LDHAi as compared with tumors from control mice (Figure S4F). To evaluate the role of tumor glycolysis in controlling resistance to T cell-mediated antitumor immunity, we determined the efficacy of LDHAi alone and in combination with adoptively transferred tumor-reactive T cells in a syngeneic melanoma model. We found that both LDHAi and T cell transfer were moderately effective as single agent treatments at reducing tumor growth when compared to control mice. However, the combination treatment with T cells and LDHAi produced the most profound reduction in tumor growth when compared with control and either monotherapy (Figure 3E). Moreover, the combination therapy was found to elicit the greatest increase in overall survival compared with any of the treatment modalities (Figure 3F). Collectively, these results provide functional evidence that increased tumor intrinsic glycolysis and its end products promote resistance of melanomas to T cell-induced killing and that targeting tumor glycolysis significantly enhances the response of melanomas to T cell-mediated antitumor activity.
Increased tumor intrinsic glycolytic activity is associated with poor response of melanoma patients to ACT
To determine the clinical relevance of our findings, we evaluated the expression levels of glycolysis-related genes in tumor tissue samples from melanoma patients who were treated with ACT. Patients were classified as responders or non-responders to ACT, and the expression of glycolytic genes was determined by RNA sequencing. That analysis revealed that expression levels of GPI and PGAM4 were significantly higher in tumor samples from non-responders when compared with responders (Figure 4A). Based on these results, we used the averaged expression of GPI and PGAM4 genes to represent the overall expression of glycolytic genes for further studies in melanoma patients. We found that the overall expression of glycolytic genes was significantly higher in patients who failed to respond to ACT (stable disease/progression of disease, n=12) compared with patients who experienced clinical objective responses (complete response/partial response, n=7; Figure 4B), suggesting that tumor glycolysis may predict response to ACT in melanoma patients.
Figure 4. Increased tumor glycolytic activity is correlated with resistance to ACT in melanoma patients.

A. The expression of glycolysis-related genes in FFPE samples (n=19) from melanoma patients treated with ACT was determined by RNA sequencing. Response to ACT was determined on the basis of Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1). The FC indicates the gene expression fold change between non-responders (N) and responders (R). Non-responders are patients with stable disease (SD) or progressive disease (PD) after treatment with ACT. Responders are patients with complete or partial response (CR or PR) to ACT. A significantly higher expression of GPI and PGAM4 in non-responders compared to responders is highlighted. B. The association between the overall glycolytic gene expression measured in FFPE tumor samples from melanoma patients and the radiographic response of their tumor to TIL therapy is shown. The overall expression of glycolytic genes was determined as averaged mRNA expression of GPI and PGAM4 genes. C. The different bioenergetic profiles of melanoma cell lines are plotted. The levels of ECAR and OCR in the Mito stress tests were evaluated in patient-derived melanoma cell lines and used to generate cell energy phenotype profiles by the XF cell Energy Phenotype Report Generator. The baseline activity (open symbols) and the metabolic activity in response to mitochondrial stressors (closed symbols) of each cell line are connected by dashed lines. D, E. Elevated tumor glycolytic activity in melanoma cells from non-responders to ACT. Patient-derived melanoma cell lines were used to perform the Seahorse XF glucose stress test as described in the manufacturer’s protocol. The ECAR and OCR of each cell line were recorded 10 minutes after glucose injection in tumor cells. Cell lines were stratified into two groups based on their corresponding response to ACT. The ECAR (D) and OCR (E) levels of both groups are shown. F. Increased expression of glycolysis-related genes in glycolytic melanoma cells. Patient-derived melanoma cell lines were stratified into high ECAR cells vs. low ECAR cells based on the ECAR level determined by glucose stress test. G. Association between the overall expression of glycolytic genes and radiographic tumor response in melanoma cells derived from ACT-treated patients is shown. The averaged gene expression of GPI and PGAM4 genes was used to represent the overall expression of glycolytic genes in melanoma cell lines.
Because tumor samples are comprised of tumor cells and several different populations of non-transformed cells, we wanted to exclude the possibility that our analysis was influenced by a confounding cell type and thus, we used melanoma cell lines derived from patients enrolled in our ACT clinical trial, and constructed cell-based assays to examine the impact of tumor glycolytic activity on the immune resistant phenotype of melanoma cells. Bioenergetic studies demonstrated that these cells displayed different energy profiles (Figure 4C). To better understand the observed differences in the metabolic phenotypes of these cell lines, we selected the cell lines from ACT-treated patients, who were not included in the cohort used for FFPE tumor tissue analysis (Figure 4A and 4B). We correlated ECAR and OCR with the response to ACT observed in patients from whom the cells were derived. ECAR levels in melanoma cells from patients who were refractory to ACT (n=8) were significantly higher compared to cell lines from patients who responded to ACT (n=6) (Figure 4D). There were no significant differences in OCR between the two groups (Figure 4E). The overall expression of glycolytic genes was also significantly higher in cells with high ECAR levels compared to cells with low ECAR levels (Figure 4F). Indeed, the overall expression of glycolytic genes was also significantly higher in cell lines from non-responders compared with responders (Figure 4G). Taken together, using two non-overlapping ACT-treated melanoma patient cohorts, we demonstrated that tumor intrinsic glycolytic activity is associated with resistance to ACT.
To identify potential mechanisms by which tumor intrinsic glycolysis contributes to resistance to ACT, we performed microarray and RPPA analyses on the cell lines with the highest and lowest quartile ECAR levels. These studies revealed that the molecules differentially expressed in high versus low ECAR melanoma cell lines showed similar patterns of expression at both mRNA and protein levels (Figure 5A). Interferon regulatory factor 1 (IRF1), which has previously been shown to be downregulated in patients whose tumors were refractory to immunotherapy, was among the differentially modulated molecules.
Figure 5. Phenotypic characterization of tumors with high glycolytic activity.

A. Differentially expressed molecules between patient-derived melanoma cell lines with high versus low glycolytic activity are shown. Cell lines with the highest and lowest quartile ECAR levels were selected to compare the expression profiles determined by microarray and RPPA analyses. The log2 FC expression of molecules between high ECAR and low ECAR cells was calculated. The log2 FC that was statistically significant in both microarray and RPPA analyses is plotted (n=4 per group). On the x-axis, the direction and length of each bar represents the log2FC of the indicated molecules in high vs. low ECAR cells at the protein level. The color of each bar encodes quantitative value of the log2FC of the indicated molecules on the y-axis in high vs. low ECAR cells at the mRNA level. B. Reduced CXCL10 production by high ECAR cells compared to low ECAR cells. C. A negative correlation between the mRNA level of CXCL10 and the expression of glycolysis-related genes in melanoma and NSCLC datasets is shown.
To further investigate the relationship between glycolysis and T cell infiltration in melanoma patients that were refractory to ACT, we determined the association between the mRNA levels of 13 glycolysis-related genes and of T cell markers (LCK and CD3E) in non-responders using melanoma tissue samples derived from patients treated with ACT. This analysis revealed a significant negative correlation between the expression of GPI, PFKM and LDHB and mRNA levels of T cell markers in ACT-refractory melanoma samples (Figure S5). These findings suggest that in melanoma patients refractory to ACT, a negative correlation exists between T cell infiltration and glycolysis. Because levels of cytokines/chemokines within the tumor microenvironment can dictate T cell trafficking into tumors (Speiser et al., 2016), we measured the expression of 75 different cytokines/chemokines produced by patient-derived melanoma cells with different glycolytic activities. We found that melanoma cells with low glycolytic activity produced significantly higher concentrations of CXCL10, a critical T cell-attracting chemokine, when compared to highly glycolytic tumor cells (Figure 5B). To validate these findings, we correlated the mRNA levels of CXCL10 with the expression of glycolysis-related genes in melanoma and NSCLC patient samples. We found negative correlations between CXCL10 and ALDOA, ENO2, GAPDH, GPI and PFKM expression in melanomas, and ENO2, ENO3, and PFKM in NSCLCs (Figure 5C). These results further support that highly glycolytic tumors can evade T cell-mediated antitumor immune responses by impeding efficient tumor trafficking of T cells.
DISCUSSION
Over the past two decades, there has been a growing interest in the aberrant metabolism of cancer cells. Indeed, metabolic rewiring was recently identified as one of the emerging hallmarks of cancer (Hanahan and Weinberg, 2011). Among different types of metabolic rewiring, accelerated aerobic glycolysis is the first recognized cancer metabolism reprogramming phenotype. By favoring aerobic glycolysis, cancer cells can obtain required macromolecules to produce new cells and maintain cellular redox during proliferation (Lunt and Vander Heiden, 2011). Much of the renewed interest in tumor glycolysis stems from recent studies demonstrating that oncogenic pathways promote tumor development by modulating tumor glycolysis. For example, the loss of liver kinase B1 (LKB1) expression, a tumor suppressor that couples bioenergetics to cell growth control through regulation of mTOR activity, promotes a metabolic profile that accelerates lung cancer cell growth and proliferation (Faubert et al., 2014). However, the biological events controlled by the glycolysis pathway, and in particular its immune regulatory role, are not well defined.
In this study, we identified an immunosuppressive role for tumor intrinsic glycolysis using two independent approaches, namely the PTEN approach and the shRNA library screen. We then used patient-derived tumor cell lines and confirmed that the upregulated expression of a glycolysis-related gene, ALDOA, promotes resistance of tumor cells to T cell-mediated killing. In addition, we observed that increased tumor glycolytic activity is associated with poor T cell tumor infiltration in melanoma and NSCLC clinical samples. This finding is consistent with recent work demonstrating that tumor infiltration of CD8+ T cells is inversely correlated with elevated expression of glycolysis-related genes in head and neck squamous cell carcinoma (Ottensmeier et al., 2016).
Using tumor samples from melanoma patients treated with ACT, we found that the expression of GPI and PGAM4 was significantly higher in tumor tissues from non-responders than in responding patients. Moreover, our analysis of independent patient-derived cell lines validated that melanoma cells from non-responders displayed increased glycolytic activity and increased overall expression of glycolytic genes. These results provide the first clinical evidence that a high level of tumor glycolysis limits the response of melanoma patients to ACT. Further investigations are needed to determine the correlation of tumor glycolysis and response to immune checkpoint inhibitors in patients with melanoma. To extend our findings to other cancer lineages, we examined clinical samples of NSCLC and also noted an association between increased expression of glycolysis-related genes and poor tumor infiltration of T cells, suggesting that similar correlative studies in NSCLC patients receiving immunotherapy may advance our knowledge regarding immune resistance across multiple cancer types.
The mechanisms whereby tumor glycolysis induces immune resistance are incompletely understood. It has been shown that highly glycolytic tumors may attenuate the effector function of TILs by depleting glucose and nutrition within the tumor microenvironment (Chang et al., 2015). However, our cell-based studies emphasize that tumor cells that overexpress glycolytic genes are refractory to T cell-mediated killing independent of the amount of glucose in the surrounding microenvironment, indicating that, in addition to glucose deprivation, tumor glycolysis may activate additional signaling networks that promote immune resistance. Metabolites produced by the glycolytic pathway, such as lactic acid, are also known to contribute to the development of an immunosuppressive tumor microenvironment. For example, recent studies demonstrated that increased lactic acid impairs T cell proliferation and cytokine production (Brand et al., 2016; Fischer et al., 2007). In addition, the administration of bicarbonate to neutralize the acidic tumor microenvironment has shown to be an effective strategy to improve the efficacy of T cell-based immunotherapy in preclinical models (Pilon-Thomas et al., 2016). Consistent with these results, we demonstrated that pretreatment with lactic acid impairs the tumor killing ability of TILs. We also found that highly glycolytic melanoma cells can produce sufficient levels of lactic acid to impair T cell effector functions in vitro and that inhibition of lactic acid production by an LDHA-specific inhibitor can enhance the antitumor activity of tumor-reactive T cells in vitro and in vivo. These results emphasize the importance of lactate in contributing to the immunosuppressive phenotype of highly glycolytic tumors.
A comparison of the transcriptomic and proteomic profiles of melanoma cells with high versus low glycolytic activity revealed that IRF1, a critical transcription factor in the IFN-γ signaling pathway, was downregulated in highly glycolytic tumors. Impaired tumor IFN-γ signaling pathway was recently identified as a mediator of resistance to cancer immunotherapy (Benci et al., 2016; Gao et al., 2016). Given that the expression pattern of IRF1 in tumors with different glycolytic activities is very consistent between two unbiased and independent approaches (microarray and RPPA), IRF1 could play a critical role in mediating immune resistance in glycolytic tumors. Further studies are needed to determine the role of the IFN-γ pathway in therapeutic resistance of highly glycolytic tumor cells. In addition, our data point to the downregulation of the T cell chemoattractant CXCL10 as a potential mechanism that may be responsible for the impaired localization of T cells to highly glycolytic tumors and one that warrants additional investigation. These findings suggest that the mechanisms whereby glycolysis impairs the antitumor immune response are likely multifactorial.
Several glycolysis inhibitors have been shown to prevent tumor development and progression (Galluzzi et al., 2013; Xie et al., 2014). Among those inhibitors, 2-deoxy-D-glucose (2DG) and TLN-232, a glucose analog and a pyruvate kinase (PKM2) inhibitor, respectively, have been tested in the clinical setting (Dwarakanath et al., 2009; Sborov et al., 2015). Unfortunately, neither agent provided a significant benefit in delaying tumor growth when administered as monotherapy. However, both agents were overall well tolerated in patients treated in the context of clinical trials, suggesting that targeting tumor glycolysis may represent a safe therapeutic strategy in combination with immunotherapy. In our studies, we demonstrated that inhibition of tumor glycolysis pathway using an LDHAi increased the sensitivity of patient-derived melanoma cells to T cell killing and improved the responses to ACT. These results support targeting tumor glycolysis as a therapeutic strategy to overcome immune resistance. However, it is known that T cells also rely on glycolysis to generate the required building blocks for their proliferation, activation, and some effector functions (Palmer et al., 2015). Therefore, additional studies will be necessary to explore the impact of glycolysis inhibitors on T cell function at various stages of tumor development in different types of cancer.
Limitations
As outlined above, our results demonstrate that highly glycolytic tumors have reduced sensitivity to T cell-mediated killing, and that tumor glycolysis may impair tumor trafficking and function of T cells. Each of these mechanisms may contribute to the development of tumor immune resistance. However, delineating the individual contribution or importance of these regulatory pathways to the development of an immune resistant phenotype remains challenging due to a lack of appropriate preclinical models. Moreover, the limited availability of patient samples has restricted the scope of our work to only the relationship between tumor glycolytic activity and clinical response of melanomas to ACT. Therefore, additional investigation is needed to determine whether these findings translate to other types of tumors and T cell-based immunotherapies, including immune checkpoint inhibitors. Finally, understanding the cellular and molecular mechanisms that dictate how tumor glycolysis regulates T cell-mediated killing of cancer cells and T cell infiltration of tumors is essential for the development of therapeutic strategies to overcome immune resistance. Nevertheless, our findings provide an innovative perspective on the immune regulatory role of tumor glycolysis in melanoma and NSCLC, and the rationale for the clinical development of glycolysis inhibitors in combination with T cell-based immunotherapy to overcome tumor immune resistance.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Recruitment of participants for clinical study
Men and women with metastatic melanoma were recruited for participation in a phase II study (NCT00338377) evaluating ACT using autologous TILs plus high-dose interleukin-2 (IL-2). Patient enrollment, TIL expansion and infusion, and high-dose IL-2 therapy were carried out under a protocol (2004-0069) approved by the Institutional Review Board (IRB) of the University of Texas MD Anderson Cancer Center. Research tumor tissues and cell lines derived from the research tissues were collected or established under the IRB-approved research protocol (LAB06-0755). The content of the informed consent forms was discussed with the subjects at the time of study entry and all subjects gave their verbal and written consent to participate.
Patient-derived cell lines
Patient-derived melanoma cell lines and their autologous TILs were established according to previously described protocols (Chacon et al., 2013). All melanoma cell lines were maintained in RPMI 1640 complete medium supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) and normocin (Invitrogen). TIL cell lines were maintained in RPMI 1640 with 10% human type AB serum (GEMINI, West Sacramento, CA), 3000U/ml IL-2 (Prometheus Laboratories, San Diego, CA) and penicillin-streptomycin.
METHOD DETAILS
In vitro pooled shRNA barcode library screen
The human lentiviral shRNA library used in this study comprises 3430 distinct hairpin constructs targeting 343 genes involved in multiple metabolic pathways. shRNAs were cloned with a unique molecular barcode and expressed under the control of a constitutive U6 promoter (Cellecta, Inc. Mountain View, CA). The shRNA library pool, along with the packaging plasmids psPAX2 and pMD2.G, were co-transfected into human embryonic kidney 293 (HEK293) cells to generate high titer viral supernatants. Patient-derived melanoma cells were transduced at a multiplicity of infection (MOI) of 0.25 to ensure expression of a single barcoded shRNA per cell. The screen was performed such that each shRNA was present in at least 1000 cells. Three days after transduction, GFP-expressing melanoma cells were sorted by flow cytometry and cultured for two weeks. Then, shRNA expressing tumor cells were either exposed to autologous TILs at an effector to target ratio (E:T) of 1:1 or maintained in growth medium for 24 hours. Genomic DNA was extracted and shRNA abundance determined through deep sequencing of triplicate samples using HiSeq 2000 (Illumina), as described previously (Bossi et al., 2016; Carugo et al., 2016). The frequency of shRNA was calculated in tumor samples with or without TIL treatment by dividing each hairpin count by the total number of aligned reads. For each shRNA hairpin, the log2 ratio was calculated as the base 2 log of the fold change of hairpin frequency between TIL-treated and untreated samples.
TCGA and PROSPECT datasets
Public TCGA (https://portal.gdc.cancer.gov/ and http://gdac.broadinstitute.org/) data repositories for skin cutaneous melanoma (SKCM) (Cancer Genome Atlas, 2015), lung adenocarcinomas (LUADs) (Cancer Genome Atlas Research, 2014) and squamous cell lung cancers (LUSQs) (Cancer Genome Atlas Research, 2012) were used as source of sample data. For the analysis of the SKCM and NSCLC TCGA sets, we used copy number (by SNP array), mRNA expression (by RNA sequencing), and tumor infiltration of lymphocytes (by gene signatures and pathological analyses).
The MD Anderson Cancer Center cohort obtained from the Profiling of Resistance patterns and Oncogenic Signaling Pathways in Evaluation of Cancers of the Thorax (PROSPECT) study (Cardnell et al., 2015; Skoulidis et al., 2015; Tang et al., 2013) was also used as source of lung cancer samples for this work. The PROSPECT cohort analyzed for this study included 209 surgically resected samples, 152 non-squamous and 57 squamous lung cancers.
Lentiviral transduction of tumor cells
The PTEN-silenced and control cell lines used are established from the melanoma cell line A375. Two PTEN-silenced cell lines expressing shRNA hairpins that target different positions of PTEN sequence (PTEN-silenced Tu-17 and Tu-60), and one corresponding control cell line expressing a non-targeting shRNA hairpin were generated, as previously described (Peng et al., 2016). To overexpress ALDOA in melanoma cells, the plasmid encoding full-length, sequence-verified human ALDOA cDNA (Ref sequence BC010660) was purchased from DNA SU plasmid repository (Tempe, AZ). The ALDOA cDNA was cloned into a modified pCDH-CMV-IRES-RFP (System Biosciences). The pCDH-CMV-IRES-RFP vector including GFP cDNA served as a control vector. High titer lentivirus was prepared in 293T cells using established protocols and standard viral packaging systems. Transduced tumor cells were sorted based on the expression of RFP to establish the ALDOA-overexpressing and control cell lines.
Targeted metabolomics analysis
Melanoma cells with or without silenced PTEN were cultured for three days. The day before collection, cells were plated in fresh medium. Cells were collected by centrifugation, washed three times and samples were then immediately lysed in methanol:water (80:20) at dry-ice temperature. The quantity of the metabolite fraction was adjusted to the corresponding protein concentration calculated upon processing a parallel sample. The samples were submitted to the Mass Spectrometry Facility at Harvard University where metabolite fractions were processed and analyzed by targeted LC-MS/MS via selected reaction monitoring (SRM), as previously described (Yuan et al., 2012).
Characterization of ALDOA-overexpressing melanoma cells
The glucose uptake of ALDOA-overexpressing cells was measured by a fluorescent probe, 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2-NBDG), as described previously (Zou et al., 2005). Briefly, Mel2400-ALDOA and Mel2400-RFP cells were seeded in 6-well plates and allowed to stabilize overnight. The next day, the cells were washed with PBS and subsequently incubated with glucose-free culture medium with 50 uM of 2-NBDG (Molecular Probes) at 37°C for 30 minutes. The 2-NBDG uptake reaction was stopped by washing with cold PBS and maintained in PBS at 4°C. Flow cytometry analysis was performed within 30 min after the completion of 2-NBDG incubation. Cells without 2-NBDG incubation were used to determine the baseline glucose uptake levels. The glucose uptake rate was determined by the changes of fluorescent intensity between cells after 2-NBDG incubation and baseline. To measure the lactate production, Mel2400-ALDOA and Mel2400-RFP cells were seeded in 6-well plates at 6 × 105 cells/well and cultured at 37 °C with 5% CO2 for 48 hours. The lactic acid concentration of conditioned medium was determined by the Accutrend Lactate Analyzer using BM lactate test strips (Roche).
Lactate treatment and caspase-3 cleavage assay
Patient-derived melanoma cells were co-cultured with autologous TILs at indicated effector to target ratios (E: T) at 37 °C for 3 hours. After incubation, the cell mixtures were permeabilized with Fix/Perm solution (BD Biosciences) for 20 min at RT and then stained with a PE or APC-conjugated anti-cleaved caspase-3 monoclonal antibody (BD Biosciences; RRID:AB_1727414), as previously described (He et al., 2005). Samples were analyzed by flow cytometry and tumor cells were gated based on the expression of either fluorescent report protein or pre-labeled DDAO Dye. The percentage of cleaved caspase-3+ cells in total tumor cells was calculated and used to determine the extent of T cell-induced tumor apoptosis. To evaluate the in vitro effects of lactic acid on T cell function, the number of T cells plated per well prior to overnight treatment with lactic acid was counted by two independent investigators in a blinded fashion. Lactic acid was administered at the shown concentration and the cells incubated overnight. The next day, T cells were washed and recounted prior to being co-cultured with melanoma cells.
Lactate level measurements in melanoma-bearing mice
Tumors were generated by implanting 5 × 105 B16 melanoma cells (RRID:CVCL_0159) into the subcutaneous space of C57BL/6Ncr mice. Seven days later, tumor-bearing mice were treated with GSK2837808A (6 mg/kg) suspended in vehicle (3% DMSO, 1% methylcellulose) by oral gavage daily for 5 days. After day 5 of treatment, animals were euthanized and tumor samples were harvested for mass spectrometry (MS)-based lactate measurements using a modified, previously described method (Chuang et al., 2009).
Adoptive T cell transfer and LDHA inhibitor in vivo treatment
The adoptive T cell transfer was performed as previously described (Peng et al., 2010). On day 0, we implanted 5 × 105 B16 melanoma cells into the subcutaneous space of C57BL/6 mice. On day 6, lymphopenia was induced by administering a nonmyeloablative dose (350 cGy) of radiation. On day 7, we adoptively transferred 1 × 106 transduced Pmel T cells into tumor-bearing mice and this was followed by intravenous injection of 0.5 × 106 peptide-pulsed DCs. IL-2 (5 × 105 I.U./mouse) was intraperitoneally administered twice daily for 3 days after T cell transfer. GSK2837808A (6 mg/kg) suspended in vehicle (3% DMSO, 1% methylcellulose) was administered by oral gavage daily beginning on day 7. The relevant solvent was administrated to control animals. Tumor sizes were monitored by measuring the perpendicular diameters of the tumors. Mice were euthanized when the length of tumors reached 1.5 cm. All experiments were carried out in a blinded, randomized fashion.
Seahorse Bioanalyzer
To characterize the cell energy phenotype and glycolytic activity of patient-derived melanoma cell lines, cells were counted blindly by two independent investigators and seeded at a density of 2 × 104 cells/well in XF96 plates and allowed to stabilize overnight. Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were next measured by the Seahorse XF96 analyzer as described in manufacturer’s instructions for the XF cell mito stress, the XF cell glucose stress and the XF cell mito fuel flex test kits. The results were analyzed using Wave software (Seahorse/Agilent).
Cytokine/Chemokine measurement
Patient-derived melanoma cells were seeded at a density of 1 × 105 cells/well in 96-well plates and incubated overnight. The levels of 75 different cytokines/chemokines in culture supernatants of patient-derived melanoma cells were determined by the multiplex human cytokine/chemokine panels with a Luminex-200 system according to the manufacturer’s instructions.
RNA and protein analysis
Total RNAs were isolated from either patient formalin-fixed paraffin embedded (FFPE) tumor tissue blocks or patient-derived tumor cell lines using the RNeasy FFPE or RNA mini isolation kits (QIAGEN), respectively, following the manufacturer’s recommendation. RNA quality was confirmed using the Agilent 2100 Bioanalyzer. The RNA expression levels in tumor tissues were determined by high-throughput RNA sequencing, as described previously (Tirosh et al., 2016). For each patient-derived tumor cell line, 40 ug of total RNA was used for the labeling reaction. The microarray-based gene expression profiling was performed following the previously described protocol (Ma et al., 2010). The raw data files used for the microarray-based gene expression analysis have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE101644.
Total proteins were isolated from patient-derived tumor cells by lysis buffer as previously described (Cheng et al., 2005). Protein concentration was determined by BCA reaction (Pierce Biotechnology, Inc., Rockford, IL) and adjusted to 1 ug/ul using the lysis buffer. 50 ug of total proteins from each sample were submitted to the Functional Proteomics RPPA Core Facility at MDACC for RPPA analysis.
QUANTIFICATION AND STATISTICAL ANALYSES
Summary statistics (e.g., mean, standard error) of the data were reported. Assessments of differences in continuous measurements between two groups were made using two-sample t-test after data transformation (typically logarithm, if necessary). Differences among several treatments were evaluated using analysis of variance (ANOVA) models with repeated measures. Two-way hierarchical clustering was performed on the expression values on 13-T cell signature genes in TCGA and PROSPECT to determine group’s membership (T cell-inflamed versus non T cell-inflamed). Two sample t-test was then used to compare the expression values between the two groups. We adjusted for multiple test comparisons by applying Beta-Uniform model (BUM) to resulting p-values and chose appropriate FDR cutoff to identify significant genes (Pounds and Morris, 2003). Spearman correlation was calculated pair-wisely between glycolysis factors and the immune marker immunohistochemical (IHC) scores in PROSPECT dataset, and between glycolysis gene expression and tumor apoptosis induced by T cells. The Kaplan-Meier method and log-rank test were used to compare survival between mice receiving different treatments. P values of less than 0.05 were considered significant (two-tail), the corresponding FDR q-value was 0.20. Graph generation and statistical analyses were performed using the Prism software program, Tableau 10.1 software program and R software programming language (version 3.2). The sample size for each experiment was chosen based on the study feasibility given its exploratory nature. Samples that did not meet proper experimental conditions were excluded from the analysis. All experiments were performed in a blinded manner.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-Active Caspase-3 | BD Bioscience | Cat#560626; RRID:AB_1727414 |
| Biological Samples | ||
| Human Melanoma Tissues | Obtained from patients enrolled in the clinical Trial (2004-0069, NCT00338377) | N/A |
| Human Melanoma Cell Lines and TILs | Derived from patients enrolled in the clinical Trial (2004-0069, NCT00338377) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lactic Acid | Sigma-Aldrich | L1750 |
| GSK2837808A | TOCRIS | 5189 |
| Proleukin | PROMETHEUS | NDC 65483-116-07 |
| Critical Commercial Assays | ||
| XF Glycolysis Stress Test Kit | Agilent | 103020-100 |
| XF Cell Mito Stress Test Kit | Agilent | 103015-100 |
| XF Mito Fuel Flex Test Kit | Agilent | 103260-100 |
| Human Cytokine/Chemokine Panel 1 | EMD Millipore | HCYTMAG-60K-PX41 |
| Human Cytokine/Chemokine Panel 2 | EMD Millipore | HCYP2MAG-62K-PX23 |
| Human Cytokine/Chemokine Panel 3 | EMD Millipore | HCYP3MAG-63K-11 |
| HUMAN HT-12 v4 BEADCHIP Kit | Illumina | BD-103-0204 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO:GSE101644 |
| Experimental Models: Cell Lines | ||
| Murine B16 Melanoma Cells | ATCC | ATCC CRL-6475; RRID:CVCL_0159 |
| Human A375 Melanoma Cells | ATCC | ATCC CRL-1619; RRID:CVCL_0132 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6Ncr Mice | Charles River (Frederick Research Model Facility) | Strain Code 556 |
| Recombinant DNA | ||
| pCDH-CMV-IRES-RFP | System Biosciences | CD500 |
| cDNA human ALDOA | DNASU Plasmid Repository | HsCD00022119 |
| Software and Algorithms | ||
| Ingenuity Pathway Analysis | Qiagen Bioinformatics | https://www.qiagenbioinformatics.com/ |
| Wave 2.3.0 | Agilent | http://www.agilent.com/ |
| R V3.2 | R project | https://www.r-project.org/ |
| Prism v6.0 | Graphpad | https://www.graphpad.com/ |
| Tableau 10.1 | Tableau | https://www.tableau.com/ |
| SAS 9.1 | SAS Institute | https://www.sas.com/en_us/home.html |
| Other | ||
| Resources related to TCGA dataset analysis | TCGA | https://portal.gdc.cancer.gov/ |
| Resources related to PROSPECT dataset analysis | Parra et al., 2016 | N/A |
Supplementary Material
Highlights.
Therapeutic resistance to ACT in cancer patients is poorly characterized.
T cell tumor trafficking and T cell cytotoxicity are impaired in glycolytic tumors.
Glycolysis inhibition enhances antitumor activity of tumor reactive T cells.
Melanomas from ACT-refractory patients exhibit higher glycolytic activity.
Acknowledgments
This work was supported in part by the National Cancer Institute grants R01 CA187076, R01 CA184845, Melanoma SPORE grant P50 CA093459; Lung SPORE grant 5 P50 CA070907; by the Cancer Prevention and Research Institute of Texas (CPRIT RP170401, RP140106, RP170067 and RP1060183). This work was also supported in part by the Conquer Cancer Foundation of ASCO 2016 Young Investigator Award 10053 from the Lung Cancer Alliance, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, Aim at Melanoma Foundation, the Miriam and Jim Mulva research funds, Jurgen Sager & Transocean Melanoma Research Fund, El Paso Foundation for Melanoma Research, Gillson Logenbaugh Foundation, the Melanoma Moon Shots Program of MD Anderson Cancer Center, and the DoD PROSPECT grant W81XWH-07-1-0306. The flow core facility used in this work is supported by the National Cancer Institute (P30 CA16672). We thank Dr. Robert R. Langley for his editorial assistance.
Footnotes
AUTHOR CONTRIBUTIONS
T.C. coordinated the experiments, analyzed data and wrote the manuscript with input from all authors; J.A.M., R.M., S.P., Z.W., C.X., L.J.W., Z.W., L.H., S.M., C.C, S.E.L., Y.N.V.G., R.E.D., M.D.P., C.A.B, A.C., T.P.H., Y.C., H.P., P.H., and P.Y. assisted with preclinical studies; C.B, R.N.A., L.A.G., I.I.W., and J.V.H. assisted with the clinical sample analysis; L.L., T.K., J.C., J.H., J.R., S.E.W. and J.W. performed bioinformatic and biostatistical analyses for preclinical and clinical studies; J.A.M, M.A.D. and J.V.H. revised the manuscript; P.H. and W.P. led this project by designing and supervising both preclinical and clinical studies and edited the manuscript.
DECLARATION OF INTERESTS
P. Hwu is a consultant/an advisory board member for Immatics, Dragonfly, Sanofi, and GlaxoSmithKline. M.A. Davies is an advisory board member for Bristol-Myers Squibb, GlaxoSmithKline, Novartis, Roche, Genentech, Sanofi and Vaccinex. J.V. Heymach is a consultant/an advisory board member for AstraZeneca, Boehringer Ingelheim, EMD Serono, Genentech, Eli Lilly, Merck, Roche, Spectrum, Guardant, Janssen, Novartis, and Foundation Medicine. P. Hwu and W. Peng are PIs of grants to MD Anderson Cancer Center from GlaxoSmithKline. All other authors declare no competing interests. Currently, S. Malu and L.A. Garraway are employees of Eli Lilly, and R.M. Mbofung is an employee of Merck Research Laboratories.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nature reviews Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DS, Pauken KE, Huang AC, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540–1554. e1512. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi D, Cicalese A, Dellino GI, Luzi L, Riva L, D’Alesio C, Diaferia GR, Carugo A, Cavallaro E, Piccioni R, et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016;6:650–663. doi: 10.1158/2159-8290.CD-15-1200. [DOI] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardnell RJ, Behrens C, Diao L, Fan Y, Tang X, Tong P, Minna JD, Mills GB, Heymach JV, Wistuba II, et al. An Integrated Molecular Analysis of Lung Adenocarcinomas Identifies Potential Therapeutic Targets among TTF1-Negative Tumors, Including DNA Repair Proteins and Nrf2. Clin Cancer Res. 2015;21:3480–3491. doi: 10.1158/1078-0432.CCR-14-3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carugo A, Genovese G, Seth S, Nezi L, Rose JL, Bossi D, Cicalese A, Shah PK, Viale A, Pettazzoni PF, et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016;16:133–147. doi: 10.1016/j.celrep.2016.05.063. [DOI] [PubMed] [Google Scholar]
- Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, Weber J, Hwu P, Radvanyi L. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One. 2013;8:e60031. doi: 10.1371/journal.pone.0060031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Cheng KW, Lu Y, Mills GB. Assay of Rab25 function in ovarian and breast cancers. Methods in enzymology. 2005;403:202–215. doi: 10.1016/S0076-6879(05)03017-X. [DOI] [PubMed] [Google Scholar]
- Chuang CK, Wang TJ, Yeung CY, Lin DS, Lin HY, Liu HL, Ho HT, Hsieh WS, Lin SP. A method for lactate and pyruvate determination in filter-paper dried blood spots. Journal of chromatography A. 2009;1216:8947–8952. doi: 10.1016/j.chroma.2009.10.074. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwarakanath BS, Singh D, Banerji AK, Sarin R, Venkataramana NK, Jalali R, Vishwanath PN, Mohanti BK, Tripathi RP, Kalia VK, et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. Journal of cancer research and therapeutics. 2009;5(Suppl 1):S21–26. doi: 10.4103/0973-1482.55136. [DOI] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404. e399. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol. 2016;34:2389–2397. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- He L, Hakimi J, Salha D, Miron I, Dunn P, Radvanyi L. A sensitive flow cytometry-based cytotoxic T-lymphocyte assay through detection of cleaved caspase 3 in target cells. J Immunol Methods. 2005;304:43–59. doi: 10.1016/j.jim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J, Chow C, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol. 2017;28:75–82. doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, Zhang M, Cooper ZA, Frederick DT, Li Y, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–5340. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, Chen JQ, Li HS, Watowich SS, Yang Y, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SV, Giaccone G. Lung cancer: First-line immunotherapy in lung cancer - taking the first step. Nat Rev Clin Oncol. 2016;13:595–596. doi: 10.1038/nrclinonc.2016.148. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annual review of cell and developmental biology. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Ma W, Wang M, Wang ZQ, Sun L, Graber D, Matthews J, Champlin R, Yi Q, Orlowski RZ, Kwak LW, et al. Effect of long-term storage in TRIzol on microarray-based gene expression profiling. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:2445–2452. doi: 10.1158/1055-9965.EPI-10-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nature reviews Cancer. 2016;16:663–673. doi: 10.1038/nrc.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottensmeier CH, Perry KL, Harden EL, Stasakova J, Jenei V, Fleming J, Wood O, Woo J, Woelk CH, Thomas GJ, et al. Upregulated Glucose Metabolism Correlates Inversely with CD8+ T-cell Infiltration and Survival in Squamous Cell Carcinoma. Cancer Res. 2016;76:4136–4148. doi: 10.1158/0008-5472.CAN-15-3121. [DOI] [PubMed] [Google Scholar]
- Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Frontiers in immunology. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra ER, Behrens C, Rodriguez-Canales J, Lin H, Mino B, Blando J, Zhang J, Gibbons DL, Heymach JV, Sepesi B, et al. Image Analysis-based Assessment of PD-L1 and Tumor-Associated Immune Cells Density Supports Distinct Intratumoral Microenvironment Groups in Non-small Cell Lung Carcinoma Patients. Clin Cancer Res. 2016;22:6278–6289. doi: 10.1158/1078-0432.CCR-15-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, Whittington M, Yang Y, Overwijk WW, Lizee G, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res. 2010;16:5458–5468. doi: 10.1158/1078-0432.CCR-10-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mule JJ, Ibrahim-Hashim A, et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016;76:1381–1390. doi: 10.1158/0008-5472.CAN-15-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19:1236–1242. doi: 10.1093/bioinformatics/btg148. [DOI] [PubMed] [Google Scholar]
- Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, Kreutz M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Frontiers in immunology. 2017;8:248. doi: 10.3389/fimmu.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborov DW, Haverkos BM, Harris PJ. Investigational cancer drugs targeting cell metabolism in clinical development. Expert opinion on investigational drugs. 2015;24:79–94. doi: 10.1517/13543784.2015.960077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Buttner M, Aigner M, Seliger B, Walter B, Schlosser H, Hartmann A, et al. Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. International journal of cancer. 2011;128:2085–2095. doi: 10.1002/ijc.25543. [DOI] [PubMed] [Google Scholar]
- Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16:599–611. doi: 10.1038/nri.2016.80. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, Suraokar M, Corvalan A, Mao J, White MA, et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin Cancer Res. 2013;19:1577–1586. doi: 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, Mueller-Klieser W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60:916–921. [PubMed] [Google Scholar]
- Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014;19:795–809. doi: 10.1016/j.cmet.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nature reviews Cancer. 2016;16:650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nature protocols. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Wang Y, Shen Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. Journal of biochemical and biophysical methods. 2005;64:207–215. doi: 10.1016/j.jbbm.2005.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.