

Abstract
Merkel cell carcinoma (MCC) is a rare (∼2,000 US cases/year) but aggressive neuroendocrine tumor of the skin. For advanced MCC, cytotoxic chemotherapy only infrequently (<10% of cases) offers durable clinical responses (>1 year) suggesting a great need for improved therapeutic options. In 2008, the Merkel cell polyomavirus (MCPyV) was discovered and is clonally integrated in ∼80% of MCC tumors. The remaining 20% of MCC tumors have large numbers of UV-associated mutations. Importantly, both the UV-induced-neoantigens in virus-negative tumors and the MCPyV T antigen oncogenes that are required for virus-positive tumor growth are immunogenic. Indeed, antigen-specific T cells detected in patients are frequently dysfunctional/‘exhausted’ and the inhibitory ligand, PD-L1, is often present in MCC tumors. These findings led to recent clinical trials involving PD-1 pathway blockade in advanced MCC. The combined data from these trials, involving three PD-1 pathway blocking agents (avelumab, pembrolizumab, and nivolumab) indicated a high frequency of durable responses in treated patients. Of note, prior treatment with chemotherapy was associated with decreased response rates to PD-1 checkpoint blockade. Over the past year, these striking data led to major changes in advanced MCC therapy, including the first-ever FDA drug approval for this disease. Despite these successes, ∼50% of MCC patients do not persistently benefit from PD-1 pathway blockade, underscoring the need for novel strategies to broaden anti-tumor immune responses in these patients. Here we highlight recent progress in MCC including the underlying mechanisms of immune evasion and emerging approaches to augment the efficacy of PD-1 pathway blockade.
Keywords: Merkel cell carcinoma, immunotherapy, Merkel cell polyomavirus, PD-1 checkpoint blockade
Introduction
Merkel cell carcinoma (MCC) is a rare (∼2000 US cases/ year) but aggressive skin cancer with a high risk of recurrence (27-31%) (1-3). Although MCC is rare, its incidence is rising steadily (4,5). Risk factors include advanced age, sun/UV-exposure, and chronic immunosuppression (∼8% of MCC patients have hematologic malignancy, solid organ transplant, or HIV/AIDS) (6). While 92% of MCC patients are not immunosuppressed, individuals who have chronic T cell dysfunction have an increased likelihood of developing MCC (10-30 fold) (6-8). Only 4% of MCC cases occur in patients under 50 years of age and MCC risk increases significantly with every additional decade of life (4,9), likely due in part to increased immune senescence. The disease associated mortality of MCC is 46% within 5 years (10), highlighting the need for improved therapeutic strategies.
Presentation/Diagnosis
The presentation of MCC can be challenging for physicians to recognize (Fig. 1), in part due to its rarity. In two-thirds of cases, physicians suspect a benign lesion based on clinical appearance (6). The following mnemonic summarizes features associated with MCC: Asymptomatic, Expanding rapidly, in an Immune suppressed patient Older than 50 and on UV-exposed skin (AEIOU) (6). As 89% of MCCs had 3 or more of these features (6) this mnemonic is sensitive, however it is not specific for MCC, as such lesions may often represent another non-melanoma skin cancer or a benign lesion such as an inflamed cyst. MCC diagnosis is confirmed through pathologic review of a biopsied lesion. Pathological sections of MCC exhibit small cells with little cytoplasm (Fig. 1). The histologic recognition of MCC was greatly facilitated by the determination that perinuclear, coarsely-granulated CK20 (KRT20) staining is present in 90% of MCC cases (11,12).
Figure 1. Clinical and histologic appearance of MCC.
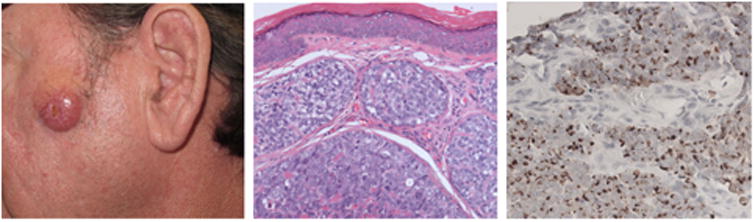
Left: Clinical appearance of an MCC arising on the left cheek of a 55 year old man. The tumor was red, firm, non-tender, and rapidly growing on sun-exposed skin. The differential diagnosis would include other types of non-melanoma skin cancer. Center: Intradermal tumor with pleiomorphic cells with large nuclei and scant cytoplasm. Right: Cytokeratin-20 (CK-20) immunohistochemical staining exhibits the characteristic peri-nuclear expression of CK-20, a highly diagnostic finding for MCC.
Virus-induced MCC
Early studies indicated that MCC can be linked to decreased immune function. One key study found that HIV patients have a 13-fold increased MCC risk compared to population controls (8). Also, case reports have described the uncommon, spontaneous regression of MCC tumors under a variety of scenarios, further indicating a link to the immune system (13-15). These data collectively suggested that MCC may be linked to a pathogen and in 2008, the Merkel cell polyomavirus (MCPyV) was discovered, and it is now clear that this virus plays a key role in the majority of MCC cases (16).
MCPyV is a member of the polyomavirus family comprised of non-enveloped, double-stranded DNA viruses, and is the first virus from this family known to cause cancer in humans. MCPyV-specific antibodies have been detected in ∼45% of children and in 80% of individuals 50 years or older, indicating that it is highly prevalent in the population (17). Interestingly, despite this high prevalence, MCPyV has not been shown to cause any disease other than when it very rarely leads to MCC. We now understand key aspects of the mystery of how a virus with an extremely high incidence leads to a cancer that is very rare.
MCPyV-related oncogenesis requires two separate events likely accounting for its rarity: 1) the circular double stranded genome must be linearized and integrated into the host-genome, perhaps after a DNA damaging event (MCPyV-positive tumors frequently occur on sun exposed skin), and 2) the virus must be mutated, with loss of expression of the C-terminus of the large T (LT) antigen that is required for viral DNA replication (Fig. 2). Virus-induced MCC is driven in part by expression of truncated large T antigen which binds to and inactivates the tumor suppressor Rb (RB1) (Fig. 2) (18), promoting cell cycle progression and uncontrolled proliferation (19,20). sT inhibits the proteasomal degradation of large T (21) as well as the oncoprotein cMyc (MYC), and cyclin E (CCNE1) (21). Both large T and small T have been demonstrated to drive transformation in mammalian cells in vitro (18,20,22), however numerous attempts to generate mouse models of MCC at best only partially emulate the disease in adult animals (23-25). The data indicate that additional, as yet undetermined factors are required for induction of MCPyV-associated MCC. While several groups have successfully generated xenografts using MCC cell lines and transfer of postoperative tumor tissue, engraftment can only be done in NOD SCID IL2Rgamma-/- (NSG) mice, which have a severely impaired immune system. These xenograft models mimic the gross pathological features of the corresponding patient's tumor but fail to recapitulate the tumor-immune interactions that are now understood greatly affect patient outcomes. In vitro experiments have demonstrated that ongoing expression of MCPyV oncoproteins is required for survival of virus-positive MCC cells (26-28). These persistently expressed non-self-antigens can potently elicit host immune recognition and the limited size of MCPyV T-antigens(<400 amino acids) has facilitated immune studies of MCPyV-specific T cell responses (29-32).
Figure 2. Comparison of virus-positive and virus-negative MCC tumors.
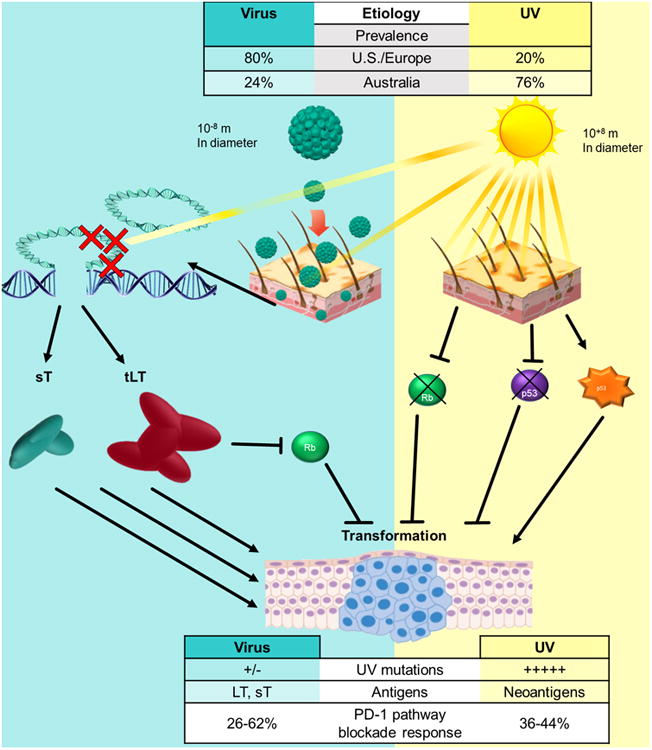
This schematic depicts the two major causes of MCC, their prevalence, differences in their potential immune targets, and frequencies of response to immune therapy. Top: Differences in MCC prevalence – US/Europe vs. Australia. Left: Virus-induced tumorigenesis – The highly prevalent Merkel cell polyomavirus (MCPyV) is often found on normal skin. Rarely, MCPyV will integrate into the host genome and through a separate rare event, large T will become truncated (tLT; depicted by red X's) prior its C-terminal. Expresssion of the sT and tLT viral oncogenes is tumorigenic through multiple pathways including inhibition of wild-type cellular Rb (see text). Right: UV-induced tumorigenesis – Sun exposure results in the generation of many UV-signature mutations (C->T mutations). The most common of which are in Rb and p53. Rb is frequently found to be inactivated in MCC tumors (67%). Mutation of p53 includes both activating and inactivating mutations. References (16, 18-21, 40-43, 71-75)
Antibodies to MCPyV T antigen correlate with tumor burden
The robust response to MCPyV-positive tumors can include both T cell and humoral components (33-35). At the time of diagnosis, approximately half of MCC patients make antibodies to MCPyV-oncoproteins. Knowing a patient's sero-status (MCPyV-positive or negative) can be helpful for their subsequent care. The prognosis of seronegative patients is less favorable (42% higher risk of recurrence than sero-positive patients) (35,36) and thus need to be followed closely with scans (36). For sero-positive patients antibody titers correlate with tumor burden (33,34), and a rising titer is an early indicator of disease recurrence (33). These findings have recently been validated in a large prospective cohort (36) and the test is now included in the 2018 National Comprehensive Cancer Network (NCCN) guidelines for MCC (37). Effective surveillance is relevant to patient care because if disease recurrence is discovered early (when tumor burden is lower), immunotherapy may be more effective (38).
UV-induced MCC
Some MCC tumors have no MCPyV detectable by either DNA-PCR or immunohistochemistry, which raised the question of whether virus-negative MCC exists or whether viral detection techniques were insufficient (39). Recent studies have demonstrated that MCPyV-negative MCC tumors do indeed exist, with variable incidence around the world (∼20% in US/Europe vs 76% in Australia, see Fig. 2) (40-43). Strikingly, virus-negative MCC is among the most mutated of all solid tumors, including melanoma and non-small cell lung cancer (40-42). These mutations are mostly UV-signature mutations (40-42). The high mutational burden (187-4707 somatic single nucleotide variants per exome) in MCC correlates to frequent amino acid changes and large numbers of UV-induced neoantigens (42). The most common mutations found in MCPyV-negative MCC are in p53 (TP53) (75% of cases) and Rb (67% of cases), commonly resulting in loss of functional protein expression (42). However, activating mutations in p53 also comprise a large proportion of the p53 mutations detected (45% of p53 mutations in MCC) (42).
Chemotherapy: The previous standard of care
Definitive treatment of primary MCC includes surgery and/or radiation. This has been quite well established, and the consensus is summarized in the 2018 NCCN guidelines (37). Historically, chemotherapy was the preferred treatment option for advanced MCC despite a lack of data rigorously assessing its benefit in this setting. Recently, several careful retrospective studies have been carried out in the US and Europe that document chemotherapy response rates and their durability (summarized in Table 1). A US academic center-based study of 62 patients with distant metastatic MCC showed a first line chemotherapy objective response rate (ORR) of 55%, however the median progression free survival (PFS) was only 94 days after chemotherapy initiation, and the median overall survival was 9.5 months (44). Second line chemotherapy was even less favorable with an ORR of 23% and a median PFS of 61 days (44). An independent study of 67 patients with metastatic MCC in the US Oncology Network also assessed responses to first and second line chemotherapy (45). This study found a first line chemotherapy ORR of only 31% with a median PFS of 4.6 months. Patients on their second or later line of chemotherapy had an ORR of 20% and a median PFS of 2.1 months (45). In a cohort of 34 patients from Europe whose disease had progressed following at least one line of chemotherapy, their next line of chemotherapy had only a 9% ORR and a median duration of response of 1.9 months (46). These studies indicate that although MCC has a relatively high response rate to chemotherapy in the first line, responses are typically short-lived and resistance develops quickly. There are multiple mechanisms likely involved with the disappointing long term benefit of chemotherapy in MCC. These may include its immunosuppressive effects in the setting of this immunogenic cancer as well as established mechanisms such as resistance to apoptosis (47).
Table 1. Selected data for chemotherapy and anti-PD1/PD-L1 in MCC.
| Chemotherapy | Nivolumab | Avelumab | Pembrolizumab | ||
|---|---|---|---|---|---|
| Line | 1st line | 2nd line | ≥1st line | ≥2nd line | 1st line |
| Cohort size | 62-67 | 20-30 | 22 | 88 | 25 |
| Agent | Etoposide & platinagentb | Topotecanb | Anti PD-1 | Anti PD-L1 | Anti PD-1 |
| ORR | 31- 55% | 9 - 23% | 68% | 32% | 56% |
| 9 month PFSa | 15-26% | 0-3% | N/Ac | 33% | 56% |
| Citations | Becker, 2017d; Cowey, 2017; Iyer, 2016; | Topalian, 2017 | Kaufman, 2016 | Nghiem, 2016 | |
Values estimated from charts
most commonly used agents
9 month PFS is not yet available, however 3 month median PFS is 82%
Data for second line chemotherapy only
Immunotherapy: A new standard of care
Over the last decade, several lines of evidence have suggested that immune status is linked to clinical outcomes in MCC, indicating that augmenting cell-mediated immunity could be beneficial. An early study focusing on tumor infiltrating lymphocytes found that patients with robust CD8+ lymphocyte infiltration into MCC tumors had 100% MCC-specific survival compared to 60% survival in those with little or no CD8+ infiltration (48). This data indicated that infiltration by CD8 T cells had profound prognostic value and that augmenting immune function could benefit patients with MCPyV-driven MCC. The specificity of CD8+ lymphocytes was then studied, and MCPyV oncoprotein-specific cells were found to be present in MCC patient blood and enriched in their tumors (29,30). Importantly, signs of dysfunction were evident in MCPyV specific CD8+ T cells from patients, as they expressed both PD-1 (PDCD1) and Tim3 (HAVCR2), the combination of which suggests functional exhaustion (29). When the tumor microenvironment was investigated, 49% of 49 tumors contained PD-L1 (CD274), (typically expressed on antigen presenting cells) and expression tended to correlate with the presence of intra-tumoral lymphocytes (49). In aggregate, these findings made a compelling case for testing PD-1 pathway blockade in MCC.
To date, three antibodies targeting the PD-1 axis have been studied in MCC with all three showing substantial response rates and impressive durability of responses (summarized in Table 1). Although the numbers of patients studied are small compared to other more prevalent cancer types, these early trials have demonstrated frequent therapeutic durability, whereas there was previously little hope for patients with advanced MCC. A National Cancer Institute-sponsored clinical trial studied pembrolizumab (anti-PD-1) in 25 patients with advanced MCC who had not received prior systemic therapy. They found an objective response rate to pembrolizumab of 56% including a 16% complete response rate. Of the 14 responsive patients, the response duration ranged from at least 2.2 months to at least 9.7 months. Overall, the trial had an estimated progression free survival of 67% at 6 months. Pembrolizumab was effective in both virus-negative and virus-positive tumors (ORR 62% and 44% respectively, not significantly different) (50). The early results of this trial led to pembrolizumab being listed as a treatment option for advanced disease in the 2017 NCCN guidelines for MCC (51).
An international, single arm, open-label trial of nivolumab (anti-PD-1) included both patients who had and those who had not received prior chemotherapy (36% and 64% respectively). In this study, 15 of 22 patients (68%) had objective responses, and PFS at 3 months was 82% with the trial still ongoing (52).
A large international clinical trial studied avelumab (anti-PD-L1) in 88 patients with distant metastatic disease who had previously received at least one line of chemotherapy. This trial found an objective response rate of 33% with a complete response rate of 11%. At 6 months, PFS was 40% and the estimated PFS at 1 year was 30%. As with pembrolizumab, avelumab was found to be effective in both virus-positive and virus-negative tumors (ORR 26% and 35% respectively, not significantly different) (53,54). In March 2017, these remarkable data in chemotherapy-refractory MCC led to the first-ever FDA approval of a drug for this cancer. Avelumab was granted accelerated approval in advanced MCC in patients at least 12 years of age, whether or not they have previously received chemotherapy (55).
Now that avelumab has been approved for treatment of advanced MCC, an important question remains. Namely, whether treatment with PD-1 pathway blockade in the adjuvant setting is appropriate and/or beneficial for treatment of this aggressive disease. As with other cancer treatments in general, catching and treating the tumor early correlates with improved prognosis. This possibility, in the context of PD-1 pathway blockade in primary MCC, will be addressed by two (one of which is double blinded and randomized) clinical trials that are now recruiting (Table 2).
Table 2. Selected immune-therapy clinical trials for Merkel cell carcinoma.
Therapies in the order listed in table: Avelumab= anti-PD-L1 (IgG1); Pembrolizumab= anti-PD-1 (IgG4); Nivolumab= anti-PD-1 (IgG4); Ipilimumab= anti-CTLA-4; GLA-SE= Glucopyranosyl Lipid A in Stable Emulsion, a TLR-4 agonist; F16-IL= Anti-tenascin C monoclonal antibody-interleukin 2 fusion protein; IL-12-EP= interleukin 12 plasmid administered with electroporation; MHC upregulation via through radiation or intratumoral Interferon ß administration; ALT-803=Interleukin 15 super-agonist complex; NK-92= activated, irradiated, allogenic natural killer cells; Bevacizumab= anti-VEGF; T-VEC=Talimogene laherparepvec, an engineered herpes oncolytic virus
| NCT identifier | Trial Arms | Recruitment Status | Phase | Targeted enrollment | Comments | Publications |
|---|---|---|---|---|---|---|
| Anti-PD-1/PD-L1 monotherapy | ||||||
|
| ||||||
| NCT02155647 | Avelumab as ≥2nd line | Active not recruiting | II | 88 | 28 of 88 Chemotherapy-refractory patients achieved a response including 8 complete responses (ORR=32%) | Kaufman, 2016 (Kaufman, Lancet Oncol, 2016) |
| NCT02155647 | Avelumab as 1st line | Recruiting | II | 112 | Preliminary results show an objective response in 20 of 29 patients (ORR=69%) | D'Angelo, 2017 (D'Angelo, ASCO, 2017) |
| NCT02267603 | Pembrolizumab as 1st line | Active, not recruiting | II | 50 | 4 of 25 patients evaluated had a complete response and 10/25 had a partial response (ORR=56%) | Nghiem, 2016 (Nghiem, NEJM, 2016) |
| NCT02488759 | Nivolumab as 1st or ≥2nd line | Active, not recruiting | I/II | 25 | 22 patients initially evaluated on nivolumab alone, 12 had a partial response and 3 had a complete response (ORR=68%) | Topalian, 2017 (Topalian, AACR, 2017) |
| NCT02196961 | Avelumab as Adjuvant versus observation following resection | Recruiting | II | 113 | Only in Europea | |
| NCT03271372 | Avelumab as Adjuvant 1st line | Not yet recruiting | III | 100 | Stage III/IIIB nodal disease, randomized, double blinded | |
|
| ||||||
| Checkpoint blockade combination therapy | ||||||
|
| ||||||
| NCT02488759 | Nivolumab ± anti-LAG3 (BMS-9861016) ± Ipilimumab (many arms) | Recruiting | I/II | 500 | Cohort of patients with virus associated cancers | |
| NCT03071406 | Ipilimumab + nivolumab, Versus ipilimumab + nivolumab + stereotactic body radiation therapy | Recruiting | II | 50 | ||
|
| ||||||
| Innate immunity agents & cytokines | ||||||
|
| ||||||
| NCT02035657 | TLR-4 agonist, GLA-SE | Completed | I | 10 | 2 of 3 patients with local nodal disease had a complete response and 2 of 7 patients with distant metastatic disease had stable disease | Bhatia, 2016 (Bhatia, ASCO, 2016) |
| NCT01440816 | IL-12-EP | Completed | II | 15 | 4 of15 patients treated with IL-12 had an objective response | Bhatia, 2015 (Bhatia, ECC, 2015) |
|
| ||||||
| Cell based therapies | ||||||
|
| ||||||
| NCT02584829 | Autologous MCPyV specific CD8 cells + avelumab + MHC upregulation Versus avelumab + MHC upregulation | Recruiting | I/II | 20 | 4 of4 patients had responses with 3/4 complete responses | Paulson, 2017 (Paulson, ASCO, 2017) |
| NCT02465957 | NK cells (activated NK-92) + ALT-803 (modified IL-15) | Closed | II | 24 | Initial 3 patients showed no major toxicities and at least one patient had a response | Bhatia, 2016 (Bhatia, SITC, 2016) |
|
| ||||||
| Oncolytic virus therapies | ||||||
|
| ||||||
| NCT02819843 | T-VEC Versus T-VEC + hypofractionated radiotherapy | Recruiting | II | 34 | Cohort of melanoma and Merkel cell carcinoma | |
| NCT02978625 | T-VEC + nivolumab | Not yet recruiting | II | 68 | Cohort of refractory lymphomas and refractory non-melanoma skin cancers | |
|
| ||||||
| Biomarker-guided combination therapy | ||||||
|
| ||||||
| NCT03167164 | Avelumab, bevacizumab, capecitabine, cisplatin, cyclophosphamide, 5-fluorouracil, leucovorin, nab-paclitaxel, omega-3-acid ethyl esters, stereotactic body radiation therapy, ALT-803, NK-92 (many arms) | Not yet recruiting | I/II | 67 | Treatment customized based on tumor specific characteristics | |
unless otherwise noted, trials include sites within the US
Anti-PD-1 checkpoint blockade therapies have proven to be well tolerated in a majority of patients. However, altering the balance of immune homeostasis can induce auto-immunity that results in grade 3 or grade 4 toxicity in 10-22% of cases (56,57). As such, informed consent of patients is critical, particularly because immune-related adverse events (irAEs) are typically idiosyncratic, making their early recognition and treatment challenging.
Importantly, the unique therapeutic benefits of these agents raise the question of whether they are indicated in patients who have a known auto-immune condition or previous irAE to ipilimumab. Indeed, MCC patients exhibit higher numbers of autoimmune conditions than the population at large. Treatment of autoimmune disease is a major known iatrogenic cause of chronic, severe immune suppression that can increase the risk of multiple cancer types, including MCC (58). A recent retrospective analysis of 52 melanoma patients with prior auto-immune disease treated with PD-1 pathway blockade found comparable objective response rates (33%) to those observed in clinical trials that have excluded patients with autoimmunity (59). While 20 (38%) of patients had a flare of autoimmune disease and another 15 (29%) developed other irAEs, only 8 patients exhibited grade 3 toxicity of a pre-existing autoimmune process or irAE, and just 2 patients permanently discontinued treatment. A separate study of 67 patients that had prior major ipilimumab toxicities exhibited a 40% objective response rate with PD-1 blockade (59). In this cohort, 25 (37%) patients experience recurrence of ipilimumab-induced irAEs or developed new/different irAEs. Although 14 (21%) patients exhibited grade 3-4 irAEs, only 8 (21%) patients discontinued therapy. In both of these cohorts, a majority of the immune toxicities could be controlled by symptom management, oral steroids and/or steroid sparing agents (>80% of all irAEs observed). Taken as a whole, this study indicates that—after appropriate informed consent discussions with the patient—PD-1 pathway blockade may be a considered, despite the presence of prior auto-immune disease or ipilimumab-induced irAEs (59).
New immunotherapy trials: A diverse pipeline
Despite the greatly improved durable responses observed through PD-1 checkpoint blockade therapy compared with chemotherapy, there remain major challenges in systemic therapy for MCC in that nearly half of patients do not derive durable benefit from these drugs. To address this issue, numerous clinical trials are underway for MCC, at least 9 of which involve immune therapy (Table 2). These trials involve 4 general strategies which will be summarized below: 1) ‘Removing an additional brake’ (i.e. CTLA-4) on the immune system; 2) ‘Stepping on the gas’ by using innate or other immune agonists; 3) ‘Adding more troops’ by infusing more of the relevant cells into the patient; and 4) ‘weaponizing viruses’ that can specifically target and kill cancer cells while preserving normal tissues.
Activated T cells express CTLA-4 (CTLA4) that suppresses their function after CTLA-4 binds its cognate receptor (CD80/CD86) on an antigen presenting cell. In this way, CTLA-4 acts as a central type of immunologic ‘brake’. Anti-CTLA-4 antibody (ipilimumab) blocks this binding, and allows the T cell to remain in a more active state. Ipilimumab's efficacy in MCC is now being determined in clinical trials (Table 2). One trial enrolling patients in Germany will assess the safety and efficacy of ipilimumab or avelumab in the adjuvant setting following surgical resection of local MCC in comparison to resection alone. The ipilimumab arm of this trial has recently closed, while the arm investigating avelumab in the adjuvant setting is currently enrolling. In those patients in which PD-1 pathway blockade is ineffective, one hypothesis is that further augmentation of the immune response is required, possibly via CTLA-4. In a US based trial, 50 patients with metastatic MCC are being enrolled to test the safety and efficacy of the combination of nivolumab (anti-PD-1) and ipilimumab with and without stereotactic body radiation that can debulk the tumor and may induce immunogenic cell death. The combination of ipilimumab with PD-1 pathway blockade is also being performed in melanoma, and safety data from these trials would expected to be similar.
Intratumoral immune infiltration and immune recognition/activation is regulated by pro- and anti-inflammatory molecules within the tumor-immune microenvironment. To increase the activity of anti-tumoral immune responses, several strategies seek to ‘step on the gas’ by adding immune agonists that can reinvigorate anti-tumor T cell responses. In a proof-of-concept trial, a Toll-like receptor-4 (TLR4) agonist, glucopyranosyl lipid-A stable emulsion (GLA-SE), was intratumorally injected into superficial MCC tumors (Table 2) (60). In this trial, two of three patients with stage IIIB MCC were recurrence-free at 23+ and 19+ months with one patient having a pathologic complete response after two injections of this TLR-4 agonist (60). In a second cohort, two of seven patients with stage IV MCC had partial responses and were progression free after 13 months at the time of publication. Encouragingly, responses correlated with increased T cell infiltration and activation of proinflammatory genes (60) providing proof-of-concept of this therapeutic approach.
Another trial of patients with superficial/accessible MCC tumors explored the utility of intratumoral electroporation of DNA encoding the potent pro-inflammatory cytokine IL-12 (IL12A) (Table 2) (61). In this study, 3 of 3 patients with local disease that received definitive surgery and/or radiation therapy at 4 weeks after one cycle of three IL-12 treatments had recurrence-free survival of 2+, 9, and 32+ months, with one patient having a pathologic complete response (61). In a second arm of this trial involving 12 patients with metastatic disease, partial responses were seen in three patients and stable disease in one patient (61). Treatment corresponded with induction of IL-12 and TNFα (TNF) expression in the tumor-microenvironment as well as enhanced T cell infiltration over baseline. Encouragingly, 40% of the injected lesions exhibited regression (30% complete, 10% partial) and another 40% were stable (61). Regression of non-injected lesions was also observed and no grade 3 or higher adverse events were reported. While very preliminary, these results highlight the potential of local IL-12 administration.
Cell-based therapy is an emerging immunotherapeutic approach, particularly in the setting of certain types of immune evasion. MCC evades immune detection through a variety of mechanisms, including down-regulation of HLA class I molecules required for antigen presentation, which occurs in 74-84% of MCC tumors (62,63). Natural killer (NK) cells typically target cells that downregulate HLA class I expression. An NK cell-based trial that accepts patients whose tumors were refractory to prior checkpoint therapy involves bi-weekly infusions of activated, irradiated, allogeneic NK-92 cells (Table 2). Thus far in this study, three patients treated with NK cells showed no major toxicities and while very preliminary, one patient, who had not responded to PD-1 pathway blockade, had a complete response (64).
MCPyV-positive MCC tumors require expression of viral T antigen oncoproteins (26-28). In patients with certain HLA types, MCPyV oncoprotein-specific T cells can be isolated and expanded ex vivo prior to therapeutic infusion. In one trial utilizing this strategy (Table 2) three of four patients given T cells plus HLA upregulation (tumor-targeted radiation or interferon) progressed, while one that had an initial complete response subsequently progressed after 14 months. It was found that the infused T cells frequently became dysfunctional/‘exhausted’ upon transfer (65,66). As such, avelumab (anti-PD-L1) has been added in combination with these autologous virus-specific T cells. In this combined-therapy cohort, all 4 patients treated with a regimen including T cells, HLA upregulation, and avelumab experienced objective responses, with 3 complete responses at last follow-up (65,66). These early results, in a limited set of patients, highlight the potential for the rational design and implementation of transgenic TCRs against virus-positive MCC tumors.
A mechanistically relevant therapy, recently approved for melanoma, is the oncolytic virotherapeutic, tamilogene laherparepvec (T-VEC) (67). The viral genes have been mutated so that the construct is replication-defective in normal cells, but constitutively active proliferative pathways in tumor cells allow the virus to replicate and kill those cells. The T-VEC design also includes a granulocyte macrophage colony-stimulating factor (GM-CSF, CSF2) expression cassette to induce a proinflammatory immune response. T-VEC is currently being investigated in two trials that include MCC (Table 2). In the first trial, T-VEC is used alone or in combination with hypofractionated radiotherapy. Another trial combines T-VEC with nivolumab (anti-PD-1) to augment the immune response in conjunction with T-VEC-mediated killing.
On the horizon
The diversity of drugs in development and currently being tested in clinical trials greatly outstrips our understanding of the cellular and molecular mechanisms at play (68). Indeed, nearly half of patients do not derive persistent benefit from PD-1 pathway blockade and neither tumor viral status nor biomarker studies accurately identify patients who will not respond (50,66). Additionally, mutation, adaptation, and selection for therapeutically resistant cells is a remarkably powerful process that continues to blunt therapeutic efficacy for all classes of drugs, including immunotherapy (68). To begin to address questions of response and non-response, a comprehensive, unbiased examination of host and tumor immune interactions in the tumor microenvironment is required.
MCC offers a particularly fertile hunting ground for studying the immune responses to cancers more broadly because:1) the unique relevance of MCC as a model for studying immunogenic cancers (e.g. viral oncoprotein vs. high UV-mutational load); 2) the robust immune evasion, likely through multiple mechanisms, required for a tumor to persist despite such a heavy viral/neoantigen burden; 3) the small size of the MCPyV T antigen oncogenes that greatly facilitates immunologic studies; and 4) the generation of tumor-specific reagents that facilitate both studies of the anti-tumor immune response and improved therapy. As such, investigations of MCC are poised to contribute to the understanding of the biology of cancer immunogenicity.
Now more than ever, we are able to delve into the cellular and molecular complexities within any given tumor. The cost of next-generation sequencing technologies is rapidly decreasing. Single cell sequencing is capable of analyzing hundreds to thousands of cells from small core biopsies making serial analysis of tumor tissues following therapy both more feasible and less invasive. Also, an ever increasing number of targets can be stained using multiplexed immunohistochemistry in combination with more sophisticated nucleic acid in situ hybridization techniques. In an attempt to combine this arsenal of molecular tools with clinical medicine, one trial will determine the genetic, transcriptomic, and proteomic details of a patient's tumor to customize therapy with immune and more traditional approaches (Table 2).
Detailed molecular analyses of the interactions within the tumor microenvironment in response to various immunotherapies will generate insights into therapeutically relevant targets (69,70). Importantly, proper assessment of therapeutic efficacy or failure requires that serial tumor biopsies be obtained from both before and after immune therapy, despite their high costs and logistical challenges. With the recent striking progress in immune therapies for MCC, the diverse pipeline of agents, and forthcoming improvements in our ability to assess the tumor microenvironment, the future for MCC immunotherapy is very encouraging.
Acknowledgments
PN and AC were supported by NIH K24-CA139052, NIH R01CA176841, Kelsey Dickson Fund of the Prostate Cancer Foundation, UW MCC Patient Gift Fund (AC), and the Bloom endowment at UW, TP was supported by 5T32GM007266
Footnotes
Conflict of Interest: Dr. Nghiem has received consulting fees from EMD Serono, Pfizer, and research grant support from Bristol-Meyers Squibb.
References
- 1.Mattavelli I, Patuzzo R, Torri V, Gallino G, Maurichi A, Lamera M, et al. Prognostic factors in Merkel cell carcinoma patients undergoing sentinel node biopsy. Eur J Surg Oncol. 2017;43(8):1536–41. doi: 10.1016/j.ejso.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Jouary T, Kubica E, Dalle S, Pages C, Duval-Modeste AB, Guillot B, et al. Sentinel node status and immunosuppression: recurrence factors in localized Merkel cell carcinoma. Acta Derm Venereol. 2015;95(7):835–40. doi: 10.2340/00015555-2099. [DOI] [PubMed] [Google Scholar]
- 3.Liang E, Brower JV, Rice SR, Buehler DG, Saha S, Kimple RJ. Merkel Cell Carcinoma Analysis of Outcomes: A 30-Year Experience. PLoS One. 2015;10(6):e0129476. doi: 10.1371/journal.pone.0129476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49(5):832–41. doi: 10.1016/s0190-9622(03)02108-x. [DOI] [PubMed] [Google Scholar]
- 5.Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surg Oncol. 2005;89(1):1–4. doi: 10.1002/jso.20167. [DOI] [PubMed] [Google Scholar]
- 6.Heath M, Jaimes N, Lemos B, Mostaghimi A, Wang LC, Penas PF, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58(3):375–81. doi: 10.1016/j.jaad.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller RW, Rabkin CS. Merkel cell carcinoma and melanoma: etiological similarities and differences. Cancer Epidemiol Biomarkers Prev. 1999;8(2):153–8. [PubMed] [Google Scholar]
- 8.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet. 2002;359(9305):497–8. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 9.Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37(1):20–7. doi: 10.1111/j.1600-0560.2009.01370.x. [DOI] [PubMed] [Google Scholar]
- 10.Lemos B, Nghiem P. Merkel cell carcinoma: more deaths but still no pathway to blame. J Invest Dermatol. 2007;127(9):2100–3. doi: 10.1038/sj.jid.5700925. [DOI] [PubMed] [Google Scholar]
- 11.Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21(2):226–34. doi: 10.1097/00000478-199702000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. 1992;140(2):427–47. [PMC free article] [PubMed] [Google Scholar]
- 13.O'Rourke MG, Bell JR. Merkel cell tumor with spontaneous regression. J Dermatol Surg Oncol. 1986;12(9):994–6. 1000. doi: 10.1111/j.1524-4725.1986.tb02143.x. [DOI] [PubMed] [Google Scholar]
- 14.Vandeven N, Nghiem P. Complete Spontaneous Regression of Merkel Cell Carcinoma Metastatic to the Liver: Did Lifestyle Modifications and Dietary Supplements Play a Role? Glob Adv Health Med. 2012;1(5):22–3. doi: 10.7453/gahmj.2012.1.5.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh NM. Complete spontaneous regression of Merkel cell carcinoma (1986-2016): a 30 year perspective. J Cutan Pathol. 2016;43(12):1150–4. doi: 10.1111/cup.12812. [DOI] [PubMed] [Google Scholar]
- 16.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolstov YL, Pastrana DV, Feng H, Becker JC, Jenkins FJ, Moschos S, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125(6):1250–6. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borchert S, Czech-Sioli M, Neumann F, Schmidt C, Wimmer P, Dobner T, et al. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J Virol. 2014;88(6):3144–60. doi: 10.1128/JVI.02916-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards KF, Guastafierro A, Shuda M, Toptan T, Moore PS, Chang Y. Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J Gen Virol. 2015;96(12):3532–44. doi: 10.1099/jgv.0.000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng J, Rozenblatt-Rosen O, Paulson KG, Nghiem P, DeCaprio JA. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J Virol. 2013;87(11):6118–26. doi: 10.1128/JVI.00385-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwun HJ, Shuda M, Feng H, Camacho CJ, Moore PS, Chang Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe. 2013;14(2):125–35. doi: 10.1016/j.chom.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. 2011;121(9):3623–34. doi: 10.1172/JCI46323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shuda M, Guastafierro A, Geng X, Shuda Y, Ostrowski SM, Lukianov S, et al. Merkel cell polyomavirus small T antigen induces cancer and embryonic Merkel cell proliferation in a transgenic mouse model. PLoS One. 2015;10(11):e0142329. doi: 10.1371/journal.pone.0142329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verhaegen ME, Mangelberger D, Harms PW, Vozheiko TD, Weick JW, Wilbert DM, et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J Invest Dermatol. 2015;135(5):1415–24. doi: 10.1038/jid.2014.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhaegen ME, Mangelberger D, Harms PW, Eberl M, Wilbert DM, Meireles J, et al. Merkel Cell Polyomavirus Small T Antigen Initiates Merkel Cell Carcinoma-like Tumor Development in Mice. Cancer Res. 2017;77(12):3151–7. doi: 10.1158/0008-5472.CAN-17-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84(14):7064–72. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrama D, Hesbacher S, Angermeyer S, Schlosser A, Haferkamp S, Aue A. Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int J Cancer. 2015 doi: 10.1002/ijc.29862. [DOI] [PubMed] [Google Scholar]
- 28.Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, Angermeyer S, et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer. 2012;130(4):847–56. doi: 10.1002/ijc.26076. [DOI] [PubMed] [Google Scholar]
- 29.Afanasiev OK, Yelistratova L, Miller N, Nagase K, Paulson K, Iyer JG, et al. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin Cancer Res. 2013;19(19):5351–60. doi: 10.1158/1078-0432.CCR-13-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyer JG, Afanasiev OK, McClurkan C, Paulson K, Nagase K, Jing L, et al. Merkel cell polyomavirus-specific CD8(+) and CD4(+) T-cell responses identified in Merkel cell carcinomas and blood. Clin Cancer Res. 2011;17(21):6671–80. doi: 10.1158/1078-0432.CCR-11-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyngaa R, Pedersen NW, Schrama D, Thrue CA, Ibrani D, Met O, et al. T-cell responses to oncogenic merkel cell polyomavirus proteins distinguish patients with merkel cell carcinoma from healthy donors. Clin Cancer Res. 2014;20(7):1768–78. doi: 10.1158/1078-0432.CCR-13-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller NJ, Church CD, Dong L, Crispin D, Fitzgibbon MP, Lachance K, et al. Tumor-Infiltrating Merkel Cell Polyomavirus-Specific T Cells Are Diverse and Associated with Improved Patient Survival. Cancer Immunol Res. 2017;5(2):137–47. doi: 10.1158/2326-6066.CIR-16-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paulson KG, Carter JJ, Johnson LG, Cahill KW, Iyer JG, Schrama D, et al. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer Res. 2010;70(21):8388–97. doi: 10.1158/0008-5472.CAN-10-2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samimi M, Molet L, Fleury M, Laude H, Carlotti A, Gardair C, et al. Prognostic value of antibodies to Merkel cell polyomavirus T antigens and VP1 protein in patients with Merkel cell carcinoma. Br J Dermatol. 2016;174(4):813–22. doi: 10.1111/bjd.14313. [DOI] [PubMed] [Google Scholar]
- 35.Touze A, Le Bidre E, Laude H, Fleury MJ, Cazal R, Arnold F, et al. High levels of antibodies against merkel cell polyomavirus identify a subset of patients with merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29(12):1612–9. doi: 10.1200/JCO.2010.31.1704. [DOI] [PubMed] [Google Scholar]
- 36.Paulson KG, Lewis CW, Redman MW, Simonson WT, Lisberg A, Ritter D, et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer. 2017;123(8):1464–74. doi: 10.1002/cncr.30475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.National Comprehensive Cancer Network, Inc, editor. NCCN Clinical Practice Guidelines in Oncology Version 1.2018. 2017. Merkel Cell Carcinoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishino M, Giobbie-Hurder A, Ramaiya NH, Hodi FS. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J Immunother Cancer. 2014;2(1):40. doi: 10.1186/s40425-014-0040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodig SJ, Cheng J, Wardzala J, DoRosario A, Scanlon JJ, Laga AC, et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J Clin Invest. 2012;122(12):4645–53. doi: 10.1172/JCI64116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong SQ, Waldeck K, Vergara IA, Schroder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75(24):5228–34. doi: 10.1158/0008-5472.CAN-15-1877. [DOI] [PubMed] [Google Scholar]
- 41.Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu YM, Dhanasekaran SM, et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma. Cancer Res. 2015;75(18):3720–7. doi: 10.1158/0008-5472.CAN-15-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative merkel cell carcinomas with implications for immunotherapy. Oncotarget. 2015 doi: 10.18632/oncotarget.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garneski KM, Warcola AH, Feng Q, Kiviat NB, Leonard JH, Nghiem P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol. 2009;129(1):246–8. doi: 10.1038/jid.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iyer JG, Blom A, Doumani R, Lewis C, Tarabadkar ES, Anderson A, et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016;5(9):2294–301. doi: 10.1002/cam4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cowey CL, Mahnke L, Espirito J, Helwig C, Oksen D, Bharmal M. Real-world treatment outcomes in patients with metastatic Merkel cell carcinoma treated with chemotherapy in the USA. Future Oncol. 2017 doi: 10.2217/fon-2017-0187. [DOI] [PubMed] [Google Scholar]
- 46.Becker JC, Lorenz E, Ugurel S, Eigentler TK, Kiecker F, Pfohler C, et al. Evaluation of real-world treatment outcomes in patients with distant metastatic Merkel cell carcinoma following second-line chemotherapy in Europe. Oncotarget. 2017 doi: 10.18632/oncotarget.19218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004;23(16):2934–49. doi: 10.1038/sj.onc.1207515. [DOI] [PubMed] [Google Scholar]
- 48.Paulson KG, Iyer JG, Tegeder AR, Thibodeau R, Schelter J, Koba S, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol. 2011;29(12):1539–46. doi: 10.1200/JCO.2010.30.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lipson EJ, Vincent JG, Loyo M, Kagohara LT, Luber BS, Wang H, et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol Res. 2013;1(1):54–63. doi: 10.1158/2326-6066.CIR-13-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med. 2016;374(26):2542–52. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.National Comprehensive Cancer Network, Inc, editor. NCCN Clinical Practice Guidelines in Oncology. Version 1.2017. 2016. Merkel Cell Carcinoma. [Google Scholar]
- 52.Topalian SL, Bhatia S, Hollebecque A, Awada A, De Boer JP, Kudchadkar RR, et al. Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): Efficacy and safety in Merkel cell carcinoma (MCC) 2017 [Google Scholar]
- 53.Kaufman HL, Russell JS, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Durable responses to avelumab (anti-PD-L1) in patients with Merkel cell carcinoma progressed after chemotherapy: 1-year efficacy update. 2017 [Google Scholar]
- 54.Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–85. doi: 10.1016/S1470-2045(16)30364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.FDA. FDA approves first treatment for rare form of skin cancer. 2017 https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm548278.htm.
- 56.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 57.El Osta B, Hu F, Sadek R, Chintalapally R, Tang SC. Not all immune-checkpoint inhibitors are created equal: Meta-analysis and systematic review of immune-related adverse events in cancer trials. Critical Reviews in Oncology/Hematology. 2017;119(Supplement C):1–12. doi: 10.1016/j.critrevonc.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 58.DePry JL, Reed KB, Cook-Norris RH, Brewer JD. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29(6):602–13. doi: 10.1016/j.clindermatol.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 59.Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol. 2017;28(2):368–76. doi: 10.1093/annonc/mdw443. [DOI] [PubMed] [Google Scholar]
- 60.Bhatia S, Ibrani D, Vandeven N, Miller N, Shinohara M, Byrd D, et al. Pilot study of intratumoral G100, toll-like receptor-4 (TLR4) agonist, therapy in patients with Merkel cell carcinoma (MCC). Presented at: The 2015 American Society for Clinical Oncology Annual Meeting; Chicago, Illinois, USA. 29 May - 2 June 2015; Abstract 3083. [Google Scholar]
- 61.Bhatia S, Iyer J, Ibrani D, Blom A, Byrd D, Parvathaneni U, et al. Intratumoral delivery of Interleukin-12 DNA via in vivo electroporation leads to regression of injected and non-injected tumors in Merkel cell carcinoma: Final Results of a phase 2 study. Abstracts of the European Cancer Congress 2015; Vienna, Austria. 25-29 September 2015; Abstract 504. [Google Scholar]
- 62.Paulson KG, Tegeder A, Willmes C, Iyer JG, Afanasiev OK, Schrama D, et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol Res. 2014;2(11):1071–9. doi: 10.1158/2326-6066.CIR-14-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ritter C, Fan K, Paschen A, Reker Hardrup S, Ferrone S, Nghiem P, et al. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci Rep. 2017;7(1):2290. doi: 10.1038/s41598-017-02608-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhatia S, Burgess M, Zhang H, Lee T, Klingemann H, Soon-Shiong P, et al. Adoptive cellular therapy (ACT) with allogeneic activated natural killer (aNK) cells in patients with advanced Merkel cell carcinoma (MCC): preliminary results of a phase II trial. National Harbor: 2016. [Google Scholar]
- 65.Chapuis AG, Afanasiev OK, Iyer JG, Paulson KG, Parvathaneni U, Hwang JH, et al. Regression of metastatic Merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-I. Cancer Immunol Res. 2014;2(1):27–36. doi: 10.1158/2326-6066.CIR-13-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paulson KG, Perdicchio M, Kulikauskas R, Wagener F, Church CD, Bui K, et al. Augmentation of adoptive T-cell therapy for Merkel cell carcinoma with avelumab. J Clin Oncol 2017 [Google Scholar]
- 67.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33(25):2780–8. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 68.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 69.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–74. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fusi A, Festino L, Botti G, Masucci G, Melero I, Lorigan P, et al. PD-L1 expression as a potential predictive biomarker. Lancet Oncol. 2015;16(13):1285–7. doi: 10.1016/S1470-2045(15)00307-1. [DOI] [PubMed] [Google Scholar]
- 71.Becker JC, Houben R, Ugurel S, Trefzer U, Pfohler C, Schrama D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol. 2009;129(1):248–50. doi: 10.1038/jid.2008.198. [DOI] [PubMed] [Google Scholar]
- 72.Kassem A, Schopflin A, Diaz C, Weyers W, Stickeler E, Werner M, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68(13):5009–13. doi: 10.1158/0008-5472.CAN-08-0949. [DOI] [PubMed] [Google Scholar]
- 73.Mangana J, Dziunycz P, Kerl K, Dummer R, Cozzio A. Prevalence of Merkel cell polyomavirus among Swiss Merkel cell carcinoma patients. Dermatology. 2010;221(2):184–8. doi: 10.1159/000315067. [DOI] [PubMed] [Google Scholar]
- 74.Schmitt M, Wieland U, Kreuter A, Pawlita M. C-terminal deletions of Merkel cell polyomavirus large T-antigen, a highly specific surrogate marker for virally induced malignancy. Int J Cancer. 2012;131(12):2863–8. doi: 10.1002/ijc.27607. [DOI] [PubMed] [Google Scholar]
- 75.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105(42):16272–7. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]