

Abstract
The role of pyruvate kinase M2 isoform (PKM2) in tumor progression has been controversial. Previous studies showed that PKM2 promoted tumor growth in xenograft models; however, depletion of PKM2 in the Brca1-loss-driven mammary tumor mouse model accelerates tumor formation. Since oncogenic kinases are frequently activated in tumors and PKM2 phosphorylation promotes tumor growth, we hypothesized that phosphorylation of PKM2 by activated kinases in tumor cells confers PKM2 oncogenic function, whereas non-phosphorylated PKM2 is non-oncogenic. Indeed, PKM2 was phosphorylated at tyrosine 105 (Y105) and formed oncogenic dimers in MDA-MB-231 breast cancer cells, whereas PKM2 was largely unphosphorylated and formed non-tumorigenic tetramers in non-transformed MCF10A cells. PKM2 knockdown did not affect MCF10A cell growth but significantly decreased proliferation of MDA-MB-231 breast cancer cells with tyrosine kinase activation. Multiple kinases that are frequently activated in different cancer types were identified to phosphorylate PKM2-Y105 in our tyrosine kinase screening. Introduction of the PKM2-Y105D phospho-mimetic mutant into MCF10A cells induced colony formation and the CD44hi/CD24neg cancer stem-like cell population by increasing YAP protein nuclear localization. ErbB2, a strong inducer of PKM2-Y105 phosphorylation, boosted nuclear localization of YAP and enhanced the cancer stem-like cell population. Treatment with the ErbB2 kinase inhibitor lapatinib decreased PKM2-Y105 phosphorylation and cancer stem-like cells, impeding PKM2 tumor-promoting function. Taken together, phosphorylation of PKM2-Y105 by activated kinases exerts oncogenic functions in part via activation of YAP downstream signaling to increase cancer stem-like cell properties.
Keywords: PKM2, oncogenic kinase, breast cancer, cancer stem-like cells, cell transformation
Introduction
PKM2 is the M2 isoform of pyruvate kinase (PK), catalyzing pyruvate and adenosine 5′-triphosphate (ATP) production by transferring the phosphate from phosphoenolpyruvate (PEP) to adenosine 5′-diphosphate (ADP) (1). Among the PK genes in mammals, PKM gene encodes PKM1 and PKM2 isoforms by the mutually exclusive use of exons 9 and 10 (2). PKM2 has lower pyruvate kinase activity than PKM1, but PKM2 can be allosterically activated by its upstream metabolite fructose-1, 6-bisphosphate (FBP) (3,4). PKM1 is mainly expressed in the heart, muscle, and brain, while PKM2 is expressed in less differentiated, highly proliferating tissues, such as early fetal tissues, intestines, and especially most tumors (5).
PKM2 has been shown to promote tumor growth by inhibiting apoptosis in tumor cells (6) or switching cancer metabolism to aerobic glycolysis and channeling nutrients into biosynthesis (7,8). PKM2 also has oncogenic functions that are independent of its role in glycolysis. For example, upon EGFR activation, Erk1/2-dependent phosphorylation of PKM2 at Serine 37 (S37) promotes PKM2 translocation, which activates β-catenin to promote U87 glioma tumor cell proliferation and tumorigenesis (9–11). However, the role of PKM2 in tumor progression is complex and controversial. PKM2 deletion in the mammary glands of a BRCA1-loss-driven tumor model did not postpone tumorigenesis (12). Additionally, mice with germline loss of PKM2 developed hepatocellular carcinoma earlier compared to the age-matched wildtype (WT) mice (13). Data from these two studies indicated that PKM2 is tumor suppressive in non-transformed mouse tissues. Clearly, PKM2 has opposing functions in non-transformed tissues versus in tumor tissues. It is unknown how and when PKM2 gains the oncogenic function during tumorigenesis.
Aberrant activation of oncogenic kinases, especially tyrosine kinases, is common during tumorigenesis (14,15). Given that tyrosine phosphorylation of PKM2 promotes tumor growth (16,17), we hypothesized that PKM2 gains distinct tumor-promoting functions owing to its post-translational modification by oncogenic tyrosine kinases, which are activated in tumor tissues but not in normal tissues. Indeed, we found that in breast cancer cells, PKM2 was phosphorylated by multiple tyrosine kinases and formed dimers that promoted cancer cell growth; whereas, in non-transformed mammary epithelial cells, PKM2 was unphosphorylated and formed tetramers, which were dispensable for cell proliferation. We performed a tyrosine kinase screening and identified multiple tyrosine kinases that phosphorylated PKM2 at Y105 and promoted cell transformation. Phosphorylation of PKM2-Y105 induced cancer stem-like cell properties by enhancing YAP nuclear translocation. Inhibition of PKM2-Y105 phosphorylation by targeting ErbB2, a tyrosine kinase for PKM2-Y105, effectively reduced YAP nuclear translocation and cancer stem-like cells, and inhibited tumor growth. These data indicate that tyrosine phosphorylation of PKM2 by aberrantly activated kinases instigates oncogenic function of PKM2 during tumorigenesis.
Materials and methods
Reagents and plasmids
Antibodies to phospho-PKM2-Tyrosine (Y) 105 (3827), PKM2 (4053), Flag M2 (14793), Met (3127), Tyro3 (5585), HER2/ErbB2 (2165), phospho-HER2/ErbB2 (Y1221/1222) (2243), FAK (3285), E-Cadherin (14472), Vimentin (5741), N-Cadherin (13116), YAP (14074), phospho-YAP (Ser127) (4911), LATS1 (9153), and phospho-LATS1 (Ser909) (9157) were purchased from Cell Signaling. Antibodies against ALDH (611194), FITC Mouse Anti-Human CD44 (555478), FITC Mouse IgG2b (555057), PE Mouse Anti-Human CD24 (555428), and PE Mouse IgG2a (554648) were from BD Biosciences. Lamin A/C antibody (sc-6215) and mounting medium with DAPI (sc-24941) were from Santa Cruz. Alexa Fluor 488 goat anti-rabbit (A11008) and Alexa Fluor 594 goat anti-mouse (A11005) secondary antibodies, the HRP-linked antibodies against mouse (31430) and rabbit (31460) were from Thermo Scientific. Anti-mouse/human CD44 antibodies (103001) were purchased from BioLegend. Paraformaldehyde (PFA) (158127), PhosSTOP (4906837001), proteinase inhibitor cocktail (P8340), Verteporfin (VP) (SML0534), Protoporphyrin IX disodium salt (PPIX) (258385), β-actin antibody (A5441), and α-tubulin antibody (T6074) were from Sigma-Aldrich.
The kinase library was kindly provided by Dr. Jean Zhao’s laboratory (18). The constructs expressing kinases AXL (23945), EPHA2 (23926), FAK (23902), MET (23889) and TYRO3 (23916) in the pDONR223 vector were purchased from Addgene, and these genes were transferred into pLentiN (37444) vector by Gateway cloning. pLHCX vector, pLHCX-Flag-mPKM2, and pLHCX-Flag-mPKM2-Y105F plasmids were from Dr. Jing Chen’s laboratory (17). The mPKM2-Y105D mutation was induced using a Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) and the primers were mPKM2-Y105D FP, 5′-GACCCCATCCTCGACCGGCCCGTTG-3′, and mPKM2-Y105D RP, 5′-CAACGGGCCGGTCGAGGATGGGGTC-3′. Flag-hPKM2 was cloned into pLHCX vector using HpaI and ClaI digestion. The primers were HpaI-Flag-hPKM2 FP, 5′-GTTAACGACTACAAAGACGATGACGACAAGATGTCGAAGCCCCATAGTG-3′, and ClaI-hPKM2 RP, 5′-ATCGATTCACGGCACAGGAACAAC-3′. The primers for hPKM2-Y105F mutations were hPKM2-Y105F FP, 5′-CCTCTTCCGGCCCGTTGCTGTGG-3′, and hPKM2 YF QC RP, 5′-GCCGGAAGAGGATGGGGTCAGAAGC-3′. The primers for hPKM2-Y105D mutations were hPKM2-Y105D FP, 5′-TCCTCGACCGGCCCGTTGCTGTG-3′, and hPKM2-Y105D RP, CCGGTCGAGGATGGGGTCAGAAGC.
Cell lines and cell culture
Human cell lines (MCF10A, MCF12A, MCF7, HCC1954, MDA-MB-361, BT474, MDA-MB-231, Hs578T, and MDA-MB-435) were purchased from ATCC and further characterized by the MDACC Cell Line Characterization Core Facility. All cell lines had been tested for mycoplasma contamination. MCF10A and MCF12A cells were cultured in DMEM/F12 (Caisson No. DFL13) supplied with 5% horse serum (Thermo Fisher Scientific, 16050122), 20 ng/ml EGF, 0.5 μg/ml Hydrocortisone, 100 ng/ml Cholera toxin, 10 μg/ml Insulin, and 1× Pen/Strep Solution (Invitrogen No. 15070-063) as previously described (19). Other cancer cells were cultured in DMEM/F12 supplied with 10% fetal bovine serum (Thermo Fisher Scientific, SH3007103).
RNA interference and stable cell line generation
The shRNAs were cloned into pLKO.1 vector (Addgene, 10878). The target sequences were: shPKM2-1, 5′-CTACCACTTGCAATTATTTGA-3′; shPKM2-2, 5′-CCACTTGCAATTATTTGAGGA-3′; shYAP1-1, 5′-TTGGTTGATAGTATCACCTGT-3′; and shYAP1-2, 5′-TTAAGGAAAGGATCTGAGCTA-3′. The shRNA gene knockdown or ORF gene expression lentiviral vectors were transfected into 293T cells together with packaging plasmid (psPAX2) and envelope plasmid (pMD2G) using the LipoD293 reagent (SignaGen, SL100668) according to the manufacturer’s instructions. After 48 hours, the viruses were collected, filtered, and used to infect target cells in the presence of 8–10 μg/mL polybrene for 24 hours. The infected cells were selected by 250 μg/ml Hygromycin B Gold (InvivoGen, 31282-04-9) for 7 days or 2 μg/ml puromycin (InvivoGen, 58-58-2) for 3 days based on the drug selection genes of the introduced vectors.
Fractionation of cytoplasmic and nuclear proteins
In brief, cells were collected, resuspended in hypotonic buffer (10 mM HEPES [pH 8.0], 1.5 mM MgCl2, and 10 mM KCl) and incubated on ice for 15 min. They were then pelleted by centrifugation at 10,000 rpm at 4°C for 5 min. The pellet was resuspended in cytoplasm lysis buffer (hypotonic buffer + 0.5% NP-40) and incubated on ice for 30 min. Extracts were centrifuged at 13,000 rpm at 4°C for 10 min, and the supernatant was retained for the cytoplasmic fraction. The nuclear pellet was washed twice with hypotonic buffer and re-pelleted, then resuspended in nuclear lysis buffer (10 mM HEPES [pH 8.0], 1.5 mM MgCl2, 400 mM NaCl, 0.1 mM EDTA, and 20% Glycerol), followed by incubation on ice for 30 min. A 30-gauge syringe was used to break up the pellet and release the nuclear proteins. Extracts were centrifuged at 13,000 rpm at 4°C for 15 min, and the supernatant was retained as the nuclear fraction.
Protein cross-linking and Western blot analysis
For protein cross-linking, 10^6 cells were collected, washed with PBS, resuspended in 200 μl of 1% paraformaldehyde (PFA) in PBS for 7 min at room temperature, and the reaction was quenched with 800 μl of 125 mM Glycine in PBS for 5 min. Then, cells were lysed with Tris-free RIPA buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, adjust pH 7.4; proteinase inhibitor cocktail and phosphatase inhibitors were added before use) on ice for 30 min. The cells were centrifuged at 12,000 g for 10 min, and the lysates were collected for Western blot analysis (20).
Flow cytometry analysis
Cells were seeded at the same density and collected when cells reached 80–90% confluence. A million cells for each sample were washed twice in FACS buffer (1% BSA in PBS), resuspended in 100 μl FACS buffer, then stained with 20 μl FITC mouse anti-human CD44 and PE mouse anti-human CD24 antibodies (BD bioscience) or FITC mouse IgG2b and PE mouse IgG2a isotype controls on ice for 1 h. Cells were washed with cold PBS twice and analyzed by FACS Canto II flow cytometer.
Immunofluorescence staining and confocal microscopy
Immunofluorescence staining and confocal microscopy were performed as previously described (21). Briefly, cells were fixed by 4% PFA, permeabilized with 0.3% TritonX-100, and blocked with 10% horse serum blocking buffer. Then the samples were incubated with Flag antibody (rabbit, 1:400 dilution) and/or YAP antibody (mouse, 1:100 dilution) at 4 °C O/N, following by staining with Alexa Fluor 488 Goat Anti-Rabbit IgG and Alexa Fluor 594 Goat Anti-Mouse IgG (1:500 dilution, Invitrogen). After mounting the slides with medium containing DAPI, cells were visualized under a confocal laser scanning microscope (Zeiss LSM 880, Germany).
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, 15596026), then reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad, 1708891). To quantify gene expression, real-time PCR was conducted using SYBR Fast Universal qPCR Master Mix (Kapa Biosystems, KK4602) on a StepOnePlus real-time PCR system (Applied Biosystems). The relative expression of mRNAs was quantified by 2−ΔΔCt with logarithm transformation. The following primers were used to detect corresponding mRNAs in the qRT-PCR analyses: CTGF FQP, 5′-TCGCCTTCGTGGTCCTCC-3′; CTGF RQP, 5′-GCCGAAGTCACAGAAGAGGC-3′; Cyr61 FQP, 5′-CTCGCCTTAGTCGTCACCC-3′; Cyr61 RQP, 5′-CGCCGAAGTTGCATTCCAG-3′; YAP1 FQP, 5′-TAGCCCTGCGTAGCCAGTTA-3′; YAP1 RQP, 5′-CTCATGCTTAGTCCACTGTCTGTAC-3′; LATS1 FQP, 5′-AATTTGGGACGCATCATAAAGC-3′; LATS1 RQP, 5′-TCGTCGAGGATCTTGGTAACTC-3′; 18S FQP, 5′-AACCCGTTGAACCCCATT-3′; and 18S RQP, 5′-CCATCCAATCGGTAGTAGCG-3′.
Cell proliferation assays
Cell proliferation was measured by MTT assay as previously described (22). One thousand cells were seeded per well (six wells per sample) in 96-well plate, and the cell growth was examined by staining with MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) (Thermo Fisher Scientific, M6496). The resulting intracellular purple formazan was solubilized by DMSO and analyzed for the OD570-OD650 using Gen5 Microplate reader (BioTek, Inc.).
Soft agar colony formation assay
A soft agar assay was performed as previously described (20). In brief, 0.5 ml of 0.6% agar containing 1 × culture medium was added to each well of a 24-well plate. After the bottom agar was solidified, 0.5 ml of a mixture of 0.3% agar and 2 000 cells in 1 × culture medium around 42 °C was added on top of the 0.6% agar. One hundred microliters of medium were added twice weekly to prevent desiccation of the upper layer of agar. After 3 weeks, colonies were counted under the microscope.
Tyrosine kinases screening
The array of the kinases’ antibodies was purchased from Cell Signaling Technology (#7982) for the kinase array screening. We modified the experimental procedures as follow: i. 150 μg of cell lysates from MDA-MB-231 and MCF10A cells was added to the antibody arrays separately and incubated at 4°C, O/N; ii. purified Flag-PKM2 proteins from these two cell lines were added to the array that has been incubated with corresponding cell lysates and incubated at room temperature for 2 h; iii. Flag antibody conjugated with HRP was added for detecting the kinase-PKM2 interacting complexes; and iv. The arrays were washed and developed with HRP substrate following the manufacturer’s procedures. For the complimentary mini-screening of additional kinases, tyrosine kinase constructs from the kinase library provided by Dr. Jean Zhao (18) were introduced into 293T cells.
Metabolic assays
To measure the glucose uptake (G), cells were starved for 3 hours before treatment with 100 μM 2-NBDG (Sigma, 72987) for 30 min. Then the glucose uptake rate was analyzed by flow cytometry. For lactate production (L) measurement, cells were incubated in complete medium for 24 hours and tested by Accutrend Lactate system (Roche). Oxygen consumption (O) were measured by Agilent Seahorse XF96 analyzer according to the manufacturer’s instructions. Glycolysis index was calculated as previously reported (23).
Transgenic mouse model and treatment
All animal experiments and terminal endpoints were carried out in accordance with approved protocols from the Institutional Animal Care and Use Committee of MDACC. MMTV-NIC (Neu-IRES-Cre) mice were interbred with PTENfl/fl mice to generate ErbB2/neu-overexpressing and PTEN homozygous loss (PTEN−/−/NIC) mice (24). PTEN−/−/NIC mice were randomized to each treatment group when their palpable tumors were 3–5 mm in diameter. Mice (n=5 in each group) were treated daily with vehicle solution (0.5% wt/vol hydroxyl-propyl-methyl-cellulose with 0.1% vol/vol Tween-20) or with lapatinib (LC Laboratories, Woburn, MA, USA) at 100 mg/kg in vehicle solution via oral gavage for 3 weeks. Tumor growth was measured by caliper every five days. The mice were sacrificed after treatment, while tumors were harvested, weighted, and fixed for IHC staining. Normal mammary fat pad (MFP) tissues were from 10-week wildtype FVB mice.
Immunohistochemical (IHC) staining and score system
IHC staining was performed as previously described (20). Negative control slides without primary antibodies and positive control tissue slides were included for each staining. The immunoreactive score (IRS) was used to quantify the IHC staining, giving a range of 0–12 as a result of multiplication of positive cell proportion scores (0–4) and staining intensity scores (0–3) as previously reported (25). IHC staining and statistical analyses were performed in a double-blind manner.
Statistical analysis
All quantitative experiments were performed at least three independent biological repeats, and the results are presented as the mean ± SD. A one-way analysis of variance (ANOVA) (multiple groups) or t-test (two groups) was used to compare the means of two or more samples using the GraphPad Prism 6 software packages. P<0.05 (two-sided) was considered statistically significant. “*” means P<0.05, “**” means P<0.01, “***” means P<0.001 and “****” means P<0.0001. “n.s” means not statistically significant.
Results
PKM2 is essential for proliferation/transformation and is highly phosphorylated at Y105 in human breast cancer cells
To explore whether PKM2 promotes or suppresses breast cancer progression, we analyzed the survival data of 3951 breast cancer patients whose tumors had a high or low expression of PKM2 by Kaplan-Meier plotter (26). The results showed that patients with PKM2 high-expression tumors had poor clinical outcomes (median survival duration, 171.43 months versus 216.66 months; p-value<2.8e−5) (Fig. 1A), indicating that PKM2 is a risk factor for the poor prognosis of breast cancer.
Figure 1. PKM2 is essential for proliferation/transformation and is highly phosphorylated at Y105 in breast cancer cells. A).
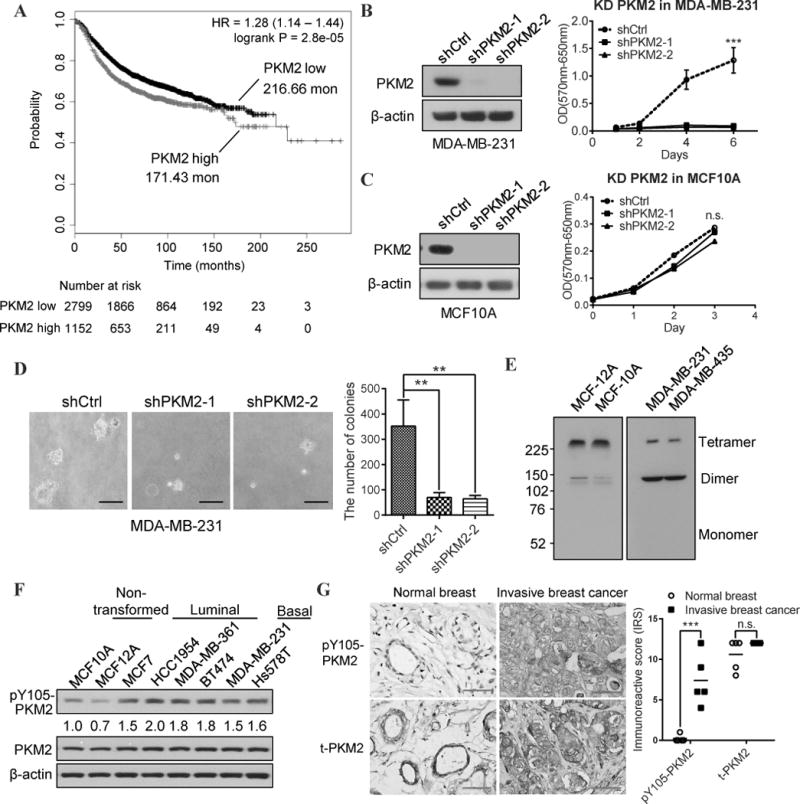
Kaplan-Meier survival curves of 3951 breast cancer patients whose tumors had a high or low PKM2 expression (DOI: 10.18632/oncotarget.10337). B) Left: Western blot analysis of PKM2 expression in MDA-MB-231 cells transfected with control shRNA (shCtrl) or two different shRNAs against PKM2 (shPKM2-1, and shPKM2-2); Right: the growth curves of the indicated MDA-MB-231 sublines by MTT assays. “***” means P<0.001. C) Left: Western blot analysis of PKM2 expression in MCF10A cells transfected with shCtrl, shPKM2-1, or shPKM2-2; Right: the growth curves of the indicated MCF10A sublines by MTT assays. “n.s” means not statistically significant. D) Representative images (left) and quantification (right) of colony formation in soft agar from the PKM2 shRNA knockdown MDA-MB-231 sublines. E) Western blot analysis using PKM2 antibodies to detect PKM2 tetramers and dimers in non-transformed mammary epithelial cell lines (MCF-12A and MCF-10A) and in triple-negative breast cancer cell lines (MDA-MB-231 and MDA-MB-435). F) Western blot analysis of pY105-PKM2 and total PKM2 protein levels in the indicated non-transformed mammary epithelial cell lines vs luminal and basal type breast cancer cell lines. Quantification of pY105-PKM2 level in different cell lines compared to that in MCF10A (normalized to β-actin) is conducted by Image J software. G) Left: Representative IHC staining of pY105-PKM2 and total PKM2 proteins in human normal breast tissues versus in human invasive breast cancer samples. Right: Quantification of the immunoreactive score (IRS) of pY105 PKM2 and total PKM2 IHC staining in mammary epithelial cells of normal human breast and breast cancer samples (n=5 in each group). “***” means P<0.001. “n.s” means not statistically significant.
Next, we examined whether PKM2 is important for cancer cell growth. Specifically knocking down PKM2 with shRNAs without the change of PKM1 (Fig. S1A) in MDA-MB-231 (231.shPKM2) and MDA-MB-435 triple negative breast cancer cells significantly inhibited cell proliferation (Fig. 1B, S1B). However, PKM2 knockdown had no discernable effect on cell growth of MCF10A and MCF12A non-transformed mammary epithelial cells (Fig. 1C, S1C). In addition, knocking down PKM2 also significantly reduced colony formation ability of MDA-MB-231 cells in soft agar (Fig. 1D), which is a key characteristic of transformed cells. These data suggested that PKM2 is critical for breast cancer cell proliferation and transformation, whereas PKM2 has no discernible effect on non-transformed cell proliferation.
PKM2 can be allosterically activated by the upstream glycolytic metabolite FBP, which was found to promote the association of PKM2 proteins into homotetramers (3). When PKM2 tetramers dissociate into dimers, the PK activity of PKM2 decreases, which are associated with increased tumorigenicity (1). Interestingly, we found that most PKM2 proteins formed homotetramers in non-transformed mammary epithelial cells (MCF10A and MCF12A), whereas a majority of PKM2 formed dimers in cancer cells (MDA-MB-231 and MDA-MB-435) (Fig 1E).
It’s known that phosphorylation of PKM2 at Y105 leads to tetramer dissociation into dimers by releasing FBP from tetramers (17). Therefore, we compared the phosphorylation of PKM2 at Y105 in various non-transformed mammary epithelial cells (MCF10A and MCF12A) versus breast cancer cells, including luminal type (MCF7, HCC1954, MDA-MB-361, and BT474) and basal type (MDA-MB-231 and Hs578T). We found PKM2 is highly phosphorylated at Y105 in breast cancer cells versus non-transformed cells, although they have similar levels of total PKM2 expressions (Fig. 1F). Importantly, the phosphorylation level of PKM2-Y105 was higher in human invasive breast cancers than in epithelial cells which compose a thin layer around the gland tubes of normal breast tissues, despite the similar total PKM2 protein levels (Fig 1G). Taken together, these data suggest that PKM2 has distinct functions: in breast cancer cells, PKM2 proteins are phosphorylated at Y105 and form dimers, which are essential for cell growth and transformation; but in non-transformed cells, most PKM2 proteins remain unphosphorylated at Y105 and form tetramers, which are dispensable for cell growth.
Multiple oncogenic kinases phosphorylate PKM2 at Y105 in human breast cancer cells
Since cancer cells accumulate significantly more phosphorylated PKM2-Y105 (pY105-PKM2), we postulated that aberrantly activated oncogenic kinases in cancer cells can induce pY105-PKM2. Activation of oncogenic tyrosine kinases is common in human breast cancers but not in normal mammary tissues (27). To identify tyrosine kinases that may bind to PKM2 and induce PKM2 phosphorylation at Y105 in cancer cells, we used an antibody array detecting 27 receptor tyrosine kinases (RTKs) and 11 important kinase signaling nodes (Table S1) for kinase screening with a modified procedure (Fig. 2A). By comparing the differences of kinase-PKM2 binding intensity between MDA-MB-231 and MCF10A cells, we identified multiple kinases with stronger binding capacity to PKM2 in MDA-MB-231 cells, including ErbB2, IRS-1, Zap-70, Src, the EphR family kinases (EphA1, EphA2, EphA3, and EphB4), the TAM family kinases (Tyro3 and AXL), and the fibroblast growth factor receptor (FGFR) family kinases (FGFR1, FGFR3, FGFR4) (Fig. 2B). In parallel, we performed a complementary mini-screen by selecting 33 tyrosine kinases (Table S1) from a kinase library (18) and transfecting them into 293T cells to identify additional tyrosine kinases that induce pY105-PKM2. Among these, nine tyrosine kinases (ITK, PTK2/FAK, YES, AXL, CLK1, EphA4, TNK2/ACK1, JAK3, and Src) induced phosphorylation of PKM2 at Y105 (Fig. 2C).
Figure 2. Multiple oncogenic kinases phosphorylate PKM2 at Y105 in human breast cancer cells. A).
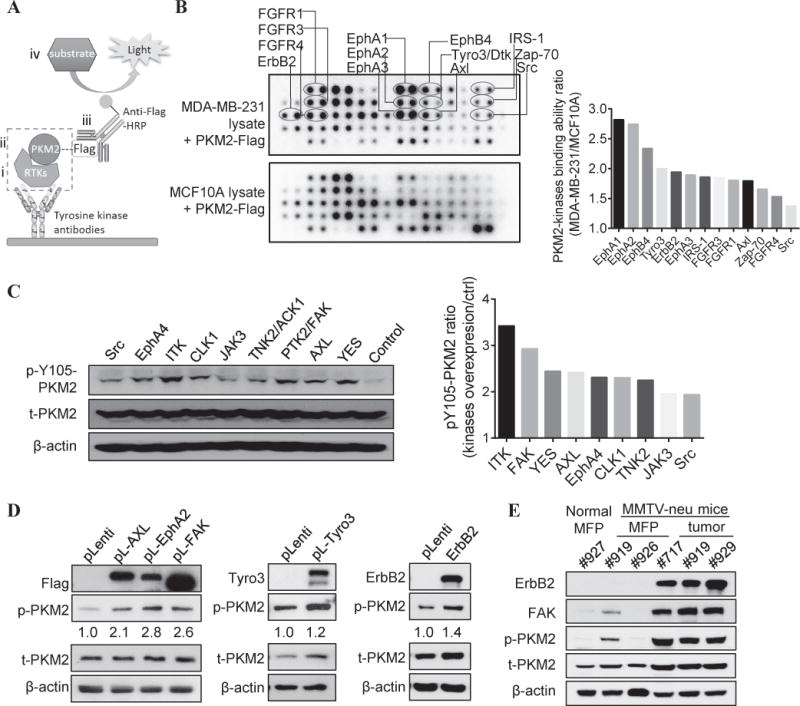
The schematics of the modified protocol using the kinase antibody array. (i) Kinases in the added cell lysates can be captured by corresponding antibodies in the kinase antibody array; (ii) if a captured kinase can phosphorylate PKM2, it can bind to the purified PKM2-Flag proteins added to the array; (iii) this interaction can be detected by anti-Flag-HRP antibodies; and (iv) the array is developed using chemiluminescence imaging. B) Left: Screening of PKM2-interacting kinases in MDA-MB-231 versus MCF10A cells was performed using kinase antibody array following the previously described procedures. Right: Quantification of PKM2-kinase binding intensity in MDA-MB-231 versus MCF10A cells (ratio) using the ImageJ software. C) Left: Western blot analysis of pY105-PKM2 and total PKM2 levels in 293T cells transfected with indicated kinase expression vectors from the kinase library. Right: Quantification of pY105-PKM2 in kinases transduced cells versus vector control cells after normalizing to β-actin with ImageJ software. D) Western blot analysis of various kinases, pY105-PKM2, and total PKM2 levels in MCF10A cells transfected with various kinase expression vectors versus pLenti-vector. ImageJ software quantification of pY105-PKM2 signals in cells transduced with kinase vectors versus pLenti-vector after normalizing to β-actin. E) Western blot analysis of ErbB2, FAK, pY105-PKM2 and total PKM2 levels in the mammary fat pads (MFPs) (from both normal mice and MMTV-neu mice) and in the mammary tumors from MMTV-neu mice.
Among the candidates from the above two screenings, we chose several kinases well-known to be frequently dysregulated in various cancer types (Table S2) (28,29) and commonly activated in human breast cancers (27,30,31). Overexpression of AXL, EphA2, FAK, Tyro3, and ErbB2 in MCF10A cells significantly increased phosphorylation of PKM2 at Y105 (Fig. 2D). Additionally, the mammary tumors of the MMTV-neu genetically engineered mice (24) had highly activated ErbB2 and FAK kinases and exhibited significantly higher pY105-PKM2 along with a moderate increase of total PKM2 proteins (Fig. 2E, S2). These data indicated that multiple oncogenic tyrosine kinases that are frequently activated in breast cancers can phosphorylate PKM2 at Y105.
Phosphorylation of PKM2-Y105 promotes transformation and induces cancer stem-like cell properties
Since pY105-PKM2 was shown to correlate with tumor progression (17) and this phosphorylation can be induced by many oncogenic kinases, we postulated that pY105-PKM2 played a role in cell transformation. To test this, we transfected MCF10A cells with pLHCX vector control (10A.pLHCX), Flag-tagged wild-type human PKM2 (10A.PKM2), a phospho-defective mutant (10A.Y105F), and a phospho-mimetic mutant (10A.Y105D) (Fig. 3A). Remarkably, the phospho-mimetic PKM2-Y105D proteins formed dimers that were predominant in cancer cells, while PKM2 WT and PKM2-Y105F proteins formed tetramers, similar to the PKM2 conformation in non-transformed cells (Fig 3B).
Figure 3. Phosphorylation of PKM2-Y105 promotes transformation and induces cancer stem-like cell properties. A).
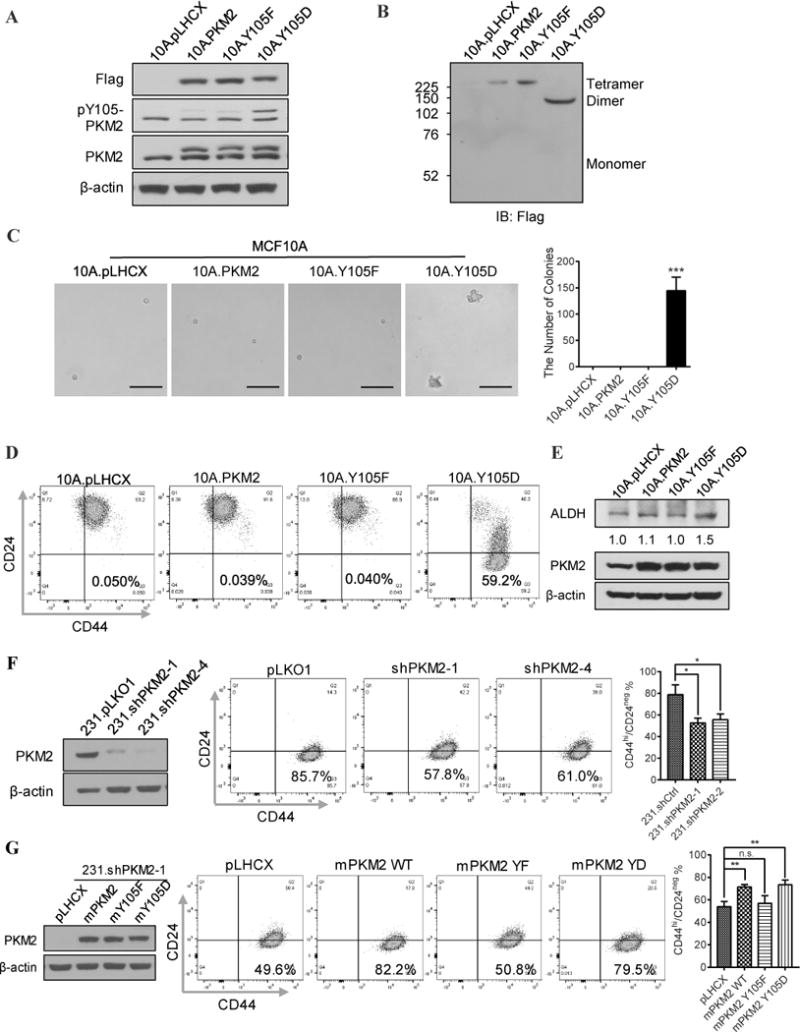
Western blot analysis of PKM2 variants expression (with Flag tag), pY105-PKM2, and total PKM2 levels in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D cells. B) Western blot analysis using anti-Flag antibodies to detect PKM2 protein tetramers and dimers in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D cell lysates. C) Left: Representative images of colony formation from the indicated MCF10A stable sublines in soft agar. Right: Quantification of colonies of the indicated MCF10A stable sublines in soft agar. “***” means P<0.001. D) Flow cytometry analysis of the cancer stem-like cell population (CD44hi/CD24neg) in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D sublines. E) Western blot analysis of ALDH and PKM2 levels in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D cells. ImageJ software was used for quantification of ALDH normalized to β-actin. F) Left: Western blot analysis of PKM2 expression in MDA-MB-231 cells transfected with shCtrl, shPKM2-1, and shPKM2-2; Right: Flow cytometry analysis and statistics of the cancer stem-like cell population (CD44hi/CD24neg) in the 231.shCtrl, 231.shPKM2-1, and 231.shPKM2-4 cells. “*” means P<0.05. G) To avoid shRNA-mediated depletion of exogenous human PKM2, mouse PKM2 WT, PKM2-Y105F, and PKM2-Y105D were introduced into PKM2 knockdown MDA.MB.231 cells. Left: Western blot analysis of mPKM2 variants expression in 231.shPKM2-1; Right: Flow cytometry analysis and statistics of the cancer stem-like cell population (CD44hi/CD24neg) in the corresponding cells. “**” means P<0.01. “n.s” means not statistically significant.
To determine whether phosphorylation of PKM2-Y105 is sufficient to promote cell transformation, we examined whether these cells can acquire anchorage-independent growth ability in soft agar (32). Strikingly, only 10A.Y105D cells formed colonies in soft agar; none of the other three MCF10A sublines grew into colonies (Fig. 3C). This finding indicates that phosphorylation of PKM2 at Y105 induces cell transformation. Previous studies showed that cell transformation was associated with increased glycolysis (23), and the glycolytic function of PKM2 promotes tumor growth (17). Consistently, 10A.Y105D cells showed an increased glycolysis (Table S3) compared to the other three MCF10A sublines.
Importantly, anchorage-independent growth is an essential characteristic of cancer stem-like cells, which can be identified by the CD44hi/CD24neg marker in human breast cancer (33). Indeed, expression of PKM2-Y105D in MCF10A cells induced a high percentage (>59%) of cancer stem-like cell population whereas expression of pLHCX vector, PKM2 WT, and PKM2-Y105F did not (Fig. 3D). The 10A.Y105D cells also showed an increased expression of ALDH (Fig. 3E), a stemness biomarker of human mammary stem cells (34). On the other hand, knocking down PKM2 in MDA-MB-231 (231.shPKM2) breast cancer cells that have high phosphorylation at Y105 of PKM2 (Fig. 1F) led to reduced cancer stem-like cell populations (Fig. 3F). Introducing mouse wild-type PKM2 (98% identical to human PKM2 protein) and PKM2-Y105D, but not PKM2-Y105F, into 231.shPKM2 cells rescued their cancer stem-like cell populations (Fig. 3G). Taken together, these data suggest that phosphorylation of PKM2-Y105 promotes cell transformation and induces cancer stem-like cell properties.
Phosphorylation of PKM2-Y105 induces cancer stem-like cell properties by activating YAP downstream signaling
Increasing evidence has shown that the Hippo pathway is tied to the acquisition of breast cancer stem cell-related traits (35). Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ) are the major downstream effectors of the Hippo pathway, regulating tissue progenitor cell growth and tumorigenesis (36). The PKM2 interaction network analyzed by the Genemania online tool revealed connections between PKM2 and YAP/TAZ (TAZ is also known as WWTR1) (Fig. S3A). Activation of YAP enhances multiple processes during tumor progression (37). Thus, we investigated whether YAP is involved in the pY105-PKM2-induced cancer stem-like cell properties. Although knocking down PKM2 in MCF10A cells did not alter the YAP protein or mRNA levels (Fig. S3B, C), immunofluorescent staining showed that the 10A.Y105D cells had less cytoplasmic YAP proteins compared to non-transformed MCF10A sublines (Fig. 4A). Furthermore, we also detected an increase of YAP proteins in the nuclear fraction of 10A.Y105D cells compared to other MCF10A sublines (Fig. 4B), whereas the cytoplasmic YAP proteins were reduced in the 10A.Y105D cells (Fig. S3D). In addition, 231.shPKM2 cells had reduced nuclear YAP (Fig. S3E), whereas re-expression of wild-type PKM2 or introducing PKM2-Y105D mutant into 231.shPKM2 cells rescued the nuclear YAP reduction (Fig. S3F).
Figure 4. Phosphorylation of PKM2-Y105 induced cancer stem-like cell properties by activating YAP downstream signaling. A).
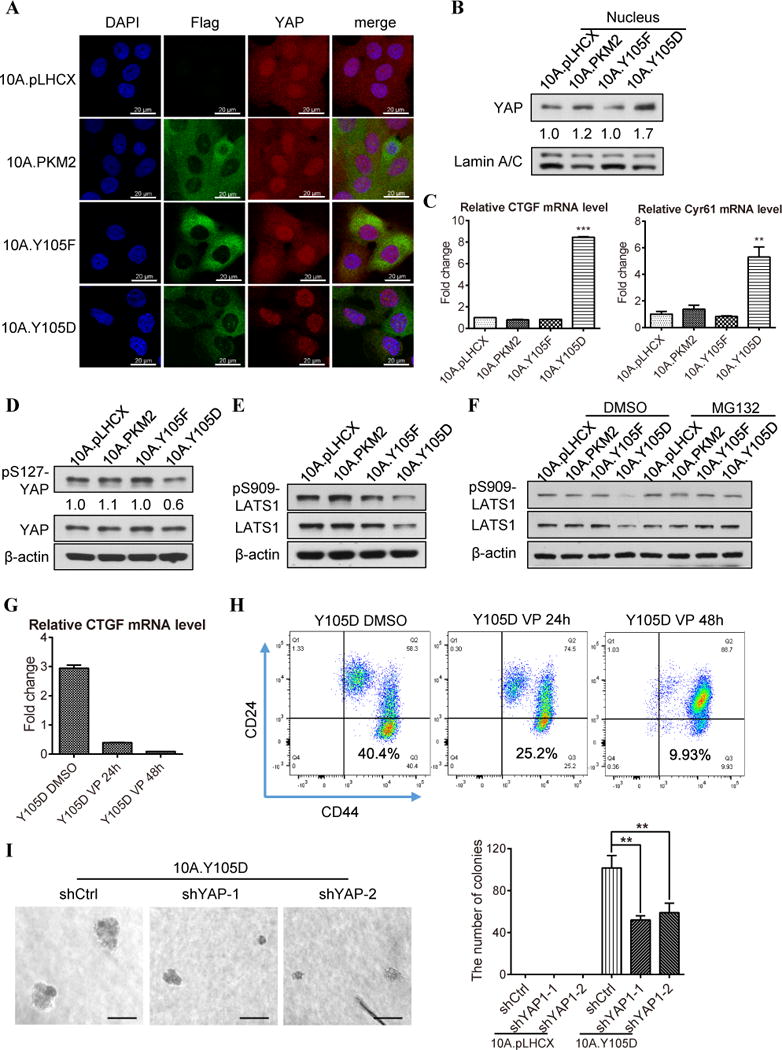
Immunofluorescence imaging of the expression and localization of exogenous PKM2 variants (indicated by Flag antibody, green), YAP (red) and the nucleus (DAPI, blue) in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D cells. B) Western blot analysis of the YAP protein levels in the nuclear fraction of the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D cells. ImageJ software was used for quantification of YAP band intensity that was normalized to Lamin A/C. C) The CTGF and Cyr61 mRNA levels in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D cells were measured by quantitative PCR. “**” means P<0.01; “***” means P<0.001. D) Western blot analysis of pS127-YAP and total YAP protein levels in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D sublines. ImageJ software was used for quantification of each sublines’ pS127-YAP level (normalized to β-actin) compared to that in the 10A.pLHCX vector control cells. E) Western blot analysis of the pS909-LATS1 and total LATS1 levels in the 10A.pLHCX, 10A.PKM2, 10A.Y105F, and 10A.Y105D sublines. F) Western blot analysis of the pS909-LATS1 and total LATS1 levels in the MCF10A sublines treated with DMSO or 10 μM MG132 for 4 h. G) The CTGF mRNA level in 10A.Y105D cells treated with DMSO (as control, first bar), 10 μM Verteporfin, for 24h or 48h. H) Flow cytometry analysis of the cancer stem-like cell population (CD44hi/CD24neg) in 10A.Y105D cells treated as in G. I) Representative images (left) and quantification (right) of the soft agar colonies from 10A.Y105D cells transfected with control shRNA (shCtrl) or two different YAP shRNAs (shYAP-1, and shYAP-2). “**” means P<0.01.
Nuclear YAP functions as a transcriptional co-factor to initiate transcription by interacting with TEA domain transcription factors 1–4 (TEAD1-4) (37). The 10A.Y105D cells with increased nuclear YAP proteins exhibited significantly higher transcription level of YAP downstream targets, such as CTGF and CYR61 (Fig. 4C). Additionally, there is a strong correlation between PKM2 and YAP downstream effector CTGF in the Esserman breast cancer dataset (Fig. S4A). These results indicate that pY105-PKM2 activates YAP downstream signaling by increasing YAP nuclear translocation.
It’s known that YAP can be retained in the cytoplasm by 14-3-3 when YAP is phosphorylated at Serine 127 (S127) (38). Consistent with the reduction of cytoplasmic YAP in the 10A.Y105D cells (Fig. 4A, S3D), pS127-YAP decreased in the 10A.Y105D cells compared to that in other MCF10A sublines (Fig 4D), but the total YAP protein or mRNA levels were similar among all four sublines (Fig. 4D, S4B).
YAP-S127 is specifically phosphorylated by a Hippo kinase LATS1 (39). Interestingly, the 10A.Y105D cells exhibited a reduction in pS909-LATS1(kinase activation marker of LATS1) and total LATS1 protein (Fig. 4E), but no decrease of LATS1 mRNA level (Fig. S4C). Thus, we tested whether the decrease of LATS1 protein in the 10A.Y105D cells was due to reduced protein stability. Indeed, after treatment with a protease inhibitor MG132, both LATS1 protein and pS909-LATS1 were rescued in the 10A.Y105D cells to a similar level as in other MCF10A sublines (Fig. 4F). These data indicate that the reduction of LATS1 protein level in the 10A.Y105D cells is due to protein destabilization.
To determine whether YAP contributed to the cancer stem-like cell population induced by pY105-PKM2, we evaluated the CD44hi/CD24neg population when YAP function was inhibited by a YAP/TAZ inhibitor Verteporfin. Verteporfin treatment effectively inhibited YAP downstream signaling (as indicated by the CTGF and Cyr61 mRNA level) (Fig. 4G) and decreased the CD44hi/CD24neg population in the 10A.Y105D cells (Fig. 4H). Knocking down YAP with siRNAs also resulted in a moderate decrease of the CD44hi/CD24neg population in the 10A.Y105D cells (Fig. S4D); this less response could partly due to the compensational increase of TAZ (Fig. S4E), as previously reported (40). Importantly, knocking down YAP with shRNAs significantly impaired the anchorage-independent growth ability of the 10A.Y105D cells (Fig. 4I) and had a more profound effect on the proliferation of the 10A.Y105D cells than that of 10A.pLHCX cells (Fig. S4F), suggesting that YAP plays an important role in the pY105-PKM2-induced transformation and cancer stem-like cell properties.
Abrogation of YAP downstream signaling inhibits oncogenic kinase-induced cancer stem-like cell population
Given that multiple kinases phosphorylate PKM2 at Y105 (Fig. 2B–D), we tested whether they can induce cancer stem-like cell properties. Indeed, Overexpression of ErbB2, AXL, EPHA2, FAK, and Tyro3 increased the CD44hi/CD24neg cancer stem-like cell population in MCF10A cells (Fig. 5A). Among these candidate kinases, ErbB2 was the strongest inducer. Inhibition of YAP/TAZ using chemical inhibitors protoporphyrin IX (PPIX) and Verteporfin (VP) (41) effectively reduced CD44hi/CD24neg population in ErbB2-transfected MCF10A cells (10A.B2) (20) (Fig. 5B), suggesting the ErbB2-induced cancer stem-like cell population was mediated by YAP activation.
Figure 5. Abrogation of YAP downstream signaling inhibited oncogenic kinase-induced cancer stem-like cell population in vitro. A).
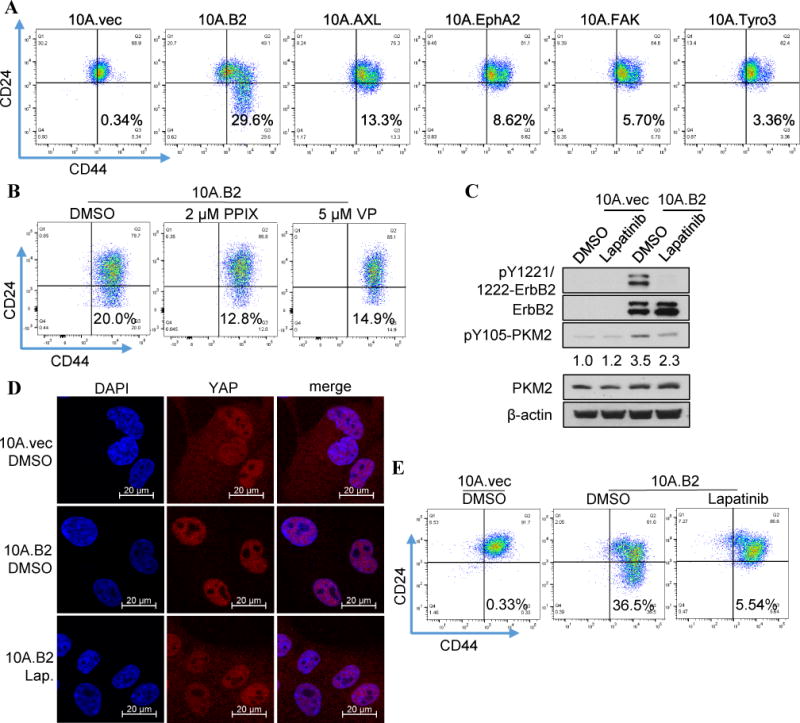
Flow cytometry analysis of the cancer stem-like cell populations (CD44hi/CD24neg) in MCF10A cells transfected with expression vectors of indicated kinases that can phosphorylate PKM2-Y105. B) Flow cytometry analysis of the cancer stem-like cell population (CD44hi/CD24neg) in the 10A.B2 cells treated with DMSO (as controls), 2 μM PPIX, or 5 μM Verteporfin. C) Western blot analysis of the pY1221/1222- ErbB2, total ErbB2 protein, pY105-PKM2, and total PKM2 protein levels in the 10A.vec and 10A.B2 cells after treatment with DMSO or lapatinib (1 μM) for 24h. Quantification of pY105-PKM2 comparing different samples with 10A.vec.DMSO (normalized by β-actin) was conducted by Image J software. D) Representative immunofluorescent staining showing the colocalization of YAP protein (red) and nuclear (DAPI, blue) in the DMSO-treated 10A.vec cells and the DMSO- or lapatinib-treated (1 μM) 10A.B2 cells. E) Flow cytometry analysis of the cancer stem-like cell population (CD44hi/CD24neg) in the DMSO-treated 10A.vec cells and the DMSO- or lapatinib-treated (1 μM) 10A.B2 cells.
Next, we tested if blocking ErbB2 kinase activity with an EGFR/ErbB2 kinase inhibitor lapatinib can inhibit YAP activation and reduce cancer stem-like cell population. Lapatinib treatment effectively inhibited ErbB2 phosphorylation and decreased pY105-PKM2 induced by ErbB2 expression (Fig. 5C). Furthermore, the increased nuclear translocation of YAP in 10A.B2 cells was inhibited by lapatinib, leading to cytoplasmic retention of YAP proteins (Fig. 5D). Importantly, lapatinib treatment dramatically reduced cancer stem-like cell population in 10A.B2 cells (Fig. 5E).
Compared to the mammary epithelial cells in wild-type FVB mice, ErbB2 high-expressing mammary tumors in PTEN−/−/NIC mice (24) exhibited significantly increased ErbB2 phosphorylation (p<0.0001), total ErbB2 proteins (p<0.0005), PKM2-Y105 phosphorylation (p<0.005), and nuclear YAP proteins (p<0.005). Remarkably, lapatinib treatment highly significantly decreased ErbB2 phosphorylation (P<0.0001), PKM2-Y105 phosphorylation (P<0.005), and nuclear YAP proteins (P<0.001) in mammary tumors with moderate reductions of total ErbB2 and PKM2 (Fig. 6A, B). Notably, ErbB2 phosphorylation significantly correlated with pY105-PKM2 and YAP nuclear localization (Fig. 6C, D, S5A, B), and there was also a strong correlation between pY105-PKM2 and YAP nuclear localization (Fig. 6E, S5C). Importantly, ErbB2-induced pY105-PKM2 was strongly correlated with cancer stem-like cell marker CD44 expression (Fig. S5D). Lapatinib treatment, which reduced PKM2-Y105 phosphorylation, significantly decreased CD44 expression (P<0.005) (Fig. 6A, B). Furthermore, lapatinib treatment led to a significant suppression of mammary tumor growth in PTEN−/−/NIC mice (Fig. 6F). Taken together, activation of oncogenic kinases, such as ErbB2, can induce phosphorylation of PKM2-Y105. Phospho-Y105-PKM2 leads to YAP nuclear translocation, enhances the CD44hi/CD24neg cancer stem-like cell population, which partly contributes to tumor progression (Fig. 7, right).
Figure 6. ErbB2 kinase inhibitor suppressed pY105-PKM2, YAP nuclear translocation, cancer stem-like cell properties, and mammary tumor growth in vivo. A).
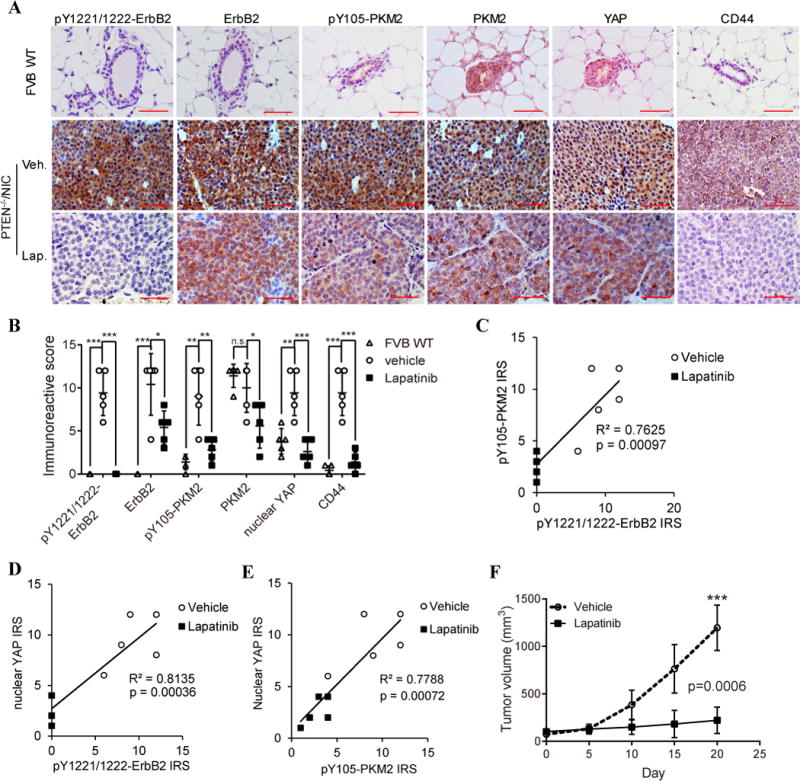
Representative IHC staining images of expression and localization of pY1221/1222- ErbB2, total ErbB2, pY105-PKM2, total PKM2, YAP proteins, and CD44 in the MFP of wildtype mice and in mammary tumors from vehicle- or lapatinib-treated (100 mg/kg, daily) PTEN−/−/NIC mice. B) Quantification of the pY1221/1222-ErbB2, ErbB2, pY105-PKM2, PKM2, nuclear YAP, and CD44 immunoreactivity scores (IRS) in the MFP of wildtype mice and mammary tumors from vehicle- or lapatinib-treated PTEN−/−/NIC mice. “*” means P<0.05, “**” means P<0.01, “***” means P<0.001, and “n.s” means not statistically significant. C) Correlation between the pY1221/1222-ErbB2 and the pY105-PKM2 immunoreactivity score (IRS) in the vehicle- or lapatinib-treated mammary tumors from PTEN−/−/NIC mice. D) Correlation between the pY1221/1222-ErbB2 and the nuclear YAP IRS in the vehicle- or lapatinib-treated mammary tumors from PTEN−/−/NIC mice. E) Correlation between the pY105-PKM2 and the nuclear YAP IRS in the vehicle- or lapatinib-treated mammary tumors from PTEN−/−/NIC mice. F) Growth curves of the vehicle- versus lapatinib-treated mammary tumors from PTEN−/−/NIC mice (n=5 for each group).
Figure 7. A model of the dichotomous functions of PKM2 in non-transformed cells versus cancer cells.
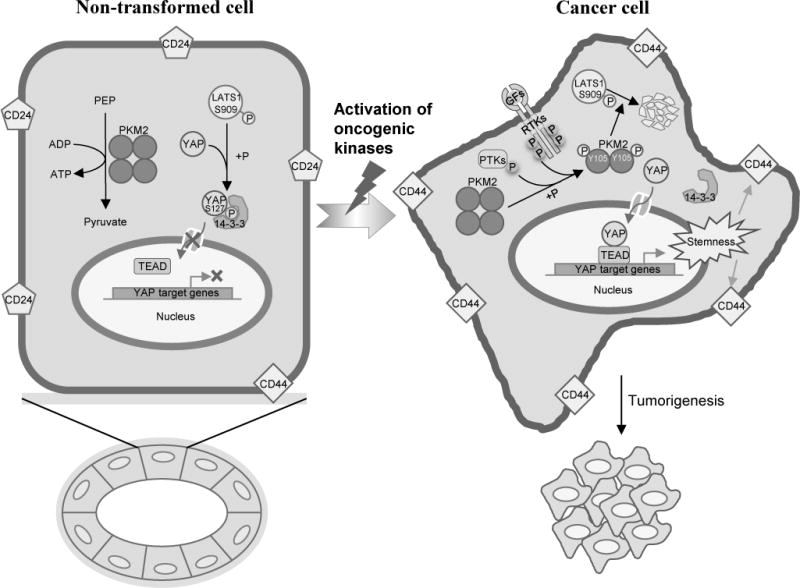
Left: In non-transformed epithelial cells, PKM2 proteins remain mostly unphosphorylated and form tetramers, which can efficiently catalyze pyruvate and ATP production. Activated hippo kinase LATS1 (indicated by pS909) can phosphorylate YAP at S127 which can be recognized and bound by 14-3-3 protein, leading to cytoplasmic retention of YAP proteins. Right: During cell transformation, multiple receptor tyrosine kinases (RTKs) and protein tyrosine kinases (PTKs) are activated, which can phosphorylate PKM2 at Y105. Phosphorylated PKM2 proteins dissociate into dimers and promote LATS1 degradation. Without hippo kinase-mediated phosphorylation, YAP cannot bind to 14-3-3 proteins, thus translocate into the nucleus and cooperates with TEAD to activate target genes transcription, induce CD44hi/CD24neg cancer stem-like population to promote tumorigenesis.
Discussion
Together, PKM2 has dichotomous functions in non-transformed cells (non-oncogenic) versus in cancer cells (tumorigenic). We found that in non-transformed mammary epithelial cells, unphosphorylated PKM2 proteins form tetramers with high PK activity (Fig. 7, left). The unphosphorylated PKM2 is dispensable for cell growth and transformation since PKM1 predominantly functions in maintaining the homeostasis of glucose metabolism in normal tissue when PKM2 is absent (12). However, in transformed tumor cells, PKM2 proteins are phosphorylated at Y105 by various oncogenic kinases and dissociate into dimers. The pY105-PKM2 promotes YAP nuclear translocation and increases cancer stem-like cells, becoming essential for tumor cell growth and transformation (Fig. 7, right). The functions of PKM2 could be altered by the different status of PKM2-Y105 phosphorylation, which is dictated by the activities of oncogenic kinases (Fig. 7).
The above findings can clarify some seemingly conflicting reports on the role of PKM2 in tumor progression. For example, PKM2 knockout inhibited leukemia initiation in mice injected with the BCR-ABL-transduced bone marrow cells, suggesting PKM2 is a tumor promoter (42). Since BCR-ABL kinase was also found to phosphorylate PKM2-Y105 (17), our explanation is that the BCR-ABL-mediated PKM2-Y105 phosphorylation promoted leukemia initiation in this case. On the other hand, depletion of PKM2 in a Brca1-loss-driven mammary tumor model did not delay tumor onset. The reason is that without activation of oncogenic kinases, PKM2 cannot contribute to tumorigenesis in Brca1-loss mammary epithelial cells. Therefore, part of the PKM2 oncogenic function is driven by its upstream oncogenic kinase activities.
In addition to metabolic functions of PKM2 in promoting tumor growth (7,17), we revealed a novel mechanism that pY105-PKM2 induced cancer stem-like cell properties by promoting YAP nuclear translocation. Activation of YAP was shown to be critical for cell transformation (43) and correlated with cancer stem-like cell properties (35). Specifically, we found that pY105-PKM2 led to the destabilization of the Hippo kinase LATS1, thus reducing phosphorylation of YAP at S127 and preventing YAP cytoplasmic retention by 14-3-3 proteins (44). Consequently, S127-unphosphorylated YAP proteins translocated into the nucleus and activated downstream targets to promote malignant transformation. It will be fascinating to explore how PKM2 crosstalks to the Hippo pathway and regulates LATS1 protein stability in future studies.
Cancer stem cells (CSCs), or cancer-initiating cells, are defined as a subset of self-renewal cancer cells. Although the function of CSCs in tumorigenicity needs further ratification (45), substantial studies indicate that an important feature of CSCs is their tumor initiation capability, frequently indicated or measured by tumor formation in xenograft model (46,47). The CSCs in human breast cancers were first marked as CD44+/CD24−/low and 1000 of CD44+/CD24−/low CSCs, but not CD44+/CD24high breast cancer cells, could induce tumors that can be serially transplanted in NOD/SCID mice (33). Another marker of CSCs in breast cancer is ALDH and 500 of ALDH+ CSCs, but not ALDH− breast cancer cells, also formed xenograft tumors (34). In this study, we found that pY105-PKM2 significantly enhanced CD44hi/CD24neg and/or ALDH+ cancer stem-like cells in MCF10A and MDA.MB.231 cell lines (Figs. 3D–G). These cancer stem-like cells also demonstrated self-renewal capacity in soft agar colony formation assay (48,49) (Fig1D, 3C). Importantly, compared to PKM2-Y105F transduced H1299 lung cancer cells, PKM2 WT-transduced H1299 cells, in which PKM2-Y105 could be phosphorylated by activated oncogenic kinases and hence have more CSCs, induced significantly bigger xenograft tumors (17). These previous studies and our new findings support the notion that pY105-PKM2-induced cancer stem-like cells contribute, at least partly, to breast cancer tumorigenicity.
The intervention of cancer metabolic dysregulations has been considered as a promising strategy for cancer therapy. However, direct modulation of PKM2 may break the balance of metabolism (50), therefore targeting oncogenic activation of PKM2 could be an alternative effective strategy. Our kinase screening identified multiple tyrosine kinases that phosphorylate PKM2 at Y105, providing multiple targets for reversing the PKM2 oncogenic functions. As a proof-of-concept study, inhibiting one of the PKM2 upstream kinases ErbB2 with lapatinib significantly reduced pY105-PKM2 and nuclear YAP, decreased cancer stem-like cell population, and inhibited tumor growth in ErbB2/neu-overexpressing mammary tumors (Fig. 6). Therefore, identifying and targeting these PKM2 upstream regulators with various kinase inhibitors can be a therapeutic strategy to inhibit PKM2 oncogenic functions in specific cancer types. As a key node of many oncogenic kinase signaling cascades, pY105-PKM2 may also serve as a biomarker for transformation and potential of tumorigenesis.
Supplementary Material
Acknowledgments
This work was supported by the MD Anderson Cancer Center Support Grant CA016672, R01-CA184836 (D. Yu), R01-CA112567 (D. Yu), R01-CA208213 (D. Yu), R21-CA223102 (D. Yu), China Medical University Research Fund (D. Yu), R21-CA211653 (Z. Songyang), CPRIT RP160462 (Z. Songyang), the Welch Foundation Q-1673 (Z. Songyang), and China Scholarship Council 201506380035 (Z. Zhou). D. Yu is the Hubert L. & Olive Stringer Distinguished Chair in Basic Science at MDACC.
We thank MD Anderson Cancer Center (MDACC) Functional Genomics Core and Flow Cytometry Core, Sequencing and Microarray Facility, Animal Core Facilities, Research Histology Core for technical support, Department of Scientific Publications of MDACC for manuscript revision, and members from Dr. Dihua Yu’s laboratory for insightful discussions.
Footnotes
Conflict of interests: The authors of this manuscript have no conflicts of interest to disclose.
References
- 1.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Takenaka M, Yamada K, Lu T, Kang R, Tanaka T, Noguchi T. Alternative splicing of the pyruvate kinase M gene in a minigene system. Eur J Biochem. 1996;235:366–71. doi: 10.1111/j.1432-1033.1996.00366.x. [DOI] [PubMed] [Google Scholar]
- 3.Ashizawa K, Willingham MC, Liang CM, Cheng SY. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J Biol Chem. 1991;266:16842–6. [PubMed] [Google Scholar]
- 4.Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–29. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 5.Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721–30. doi: 10.15252/embr.201643300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012;209:217–24. doi: 10.1084/jem.20111487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–9. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang J, Cao R, Zhang Y, Xia Y, Zheng Y, Li X, et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;7:12431. doi: 10.1038/ncomms12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. doi: 10.1016/j.cell.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, et al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016;30:1020–33. doi: 10.1101/gad.278549.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 15.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 16.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–6. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 17.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 19.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 20.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, et al. 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell. 2009;16:195–207. doi: 10.1016/j.ccr.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100–4. doi: 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang S, Huang WC, Zhang L, Zhang C, Lowery FJ, Ding Z, et al. SRC family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res. 2013;73:5764–74. doi: 10.1158/0008-5472.CAN-12-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CC, Zhang C, Zhang Q, Sahin O, Wang H, Xu J, et al. Upregulation of lactate dehydrogenase a by 14-3-3zeta leads to increased glycolysis critical for breast cancer initiation and progression. Oncotarget. 2016;7:35270–83. doi: 10.18632/oncotarget.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahin O, Wang Q, Brady SW, Ellis K, Wang H, Chang CC, et al. Biomarker-guided sequential targeted therapies to overcome therapy resistance in rapidly evolving highly aggressive mammary tumors. Cell Res. 2014;24:542–59. doi: 10.1038/cr.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fedchenko N, Reifenrath J. Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue - a review. Diagn Pathol. 2014;9:221. doi: 10.1186/s13000-014-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–31. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Zahari MS, Ma B, Liu R, Renuse S, Sahasrabuddhe NA, et al. Global phosphotyrosine survey in triple-negative breast cancer reveals activation of multiple tyrosine kinase signaling pathways. Oncotarget. 2015;6:29143–60. doi: 10.18632/oncotarget.5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ekyalongo RC, Mukohara T, Funakoshi Y, Tomioka H, Kataoka Y, Shimono Y, et al. TYRO3 as a potential therapeutic target in breast cancer. Anticancer Res. 2014;34:3337–45. [PubMed] [Google Scholar]
- 31.Xu Y, Benlimame N, Su J, He Q, Alaoui-Jamali MA. Regulation of focal adhesion turnover by ErbB signalling in invasive breast cancer cells. Br J Cancer. 2009;100:633–43. doi: 10.1038/sj.bjc.6604901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis–pathways to anchorage-independent growth in cancer. J Cell Sci. 2011;124:3189–97. doi: 10.1242/jcs.072165. [DOI] [PubMed] [Google Scholar]
- 33.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maugeri-Sacca M, De Maria R. Hippo pathway and breast cancer stem cells. Crit Rev Oncol Hematol. 2016;99:115–22. doi: 10.1016/j.critrevonc.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73–9. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109:E2441–50. doi: 10.1073/pnas.1212021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park HW, Guan KL. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci. 2013;34:581–9. doi: 10.1016/j.tips.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015;29:1271–84. doi: 10.1101/gad.262816.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang YH, Israelsen WJ, Lee D, Yu VW, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–23. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103:12405–10. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–61. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767–75. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]
- 46.Ramos EK, Hoffmann AD, Gerson SL, Liu H. New Opportunities and Challenges to Defeat Cancer Stem Cells. Trends Cancer. 2017;3:780–96. doi: 10.1016/j.trecan.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. 2012;122:2066–78. doi: 10.1172/JCI59735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klopp AH, Lacerda L, Gupta A, Debeb BG, Solley T, Li L, et al. Mesenchymal stem cells promote mammosphere formation and decrease E-cadherin in normal and malignant breast cells. PLoS One. 2010;5:e12180. doi: 10.1371/journal.pone.0012180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–47. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.