

Abstract
Lanthipeptides belong to the family of ribosomally-synthesized and posttranslationally-modified peptides (RiPPs) and are subdivided into four classes. The first two classes have been heavily studied, but less is known about classes III and IV. The lanthipeptide synthetases of classes III and IV share a similar organization of protein domains: A lyase domain at the N-terminus, a central kinase domain and a C-terminal cyclase domain. Here, we provide deeper insight into class IV enzymes (LanLs). A series of putative producer strains was screened to identify production conditions of four new venezuelin-like lanthipeptides and an Escherichia coli based heterologous production system was established for a fifth. The latter not only allowed production of fully modified core peptide, but was also employed as basis for mutational analysis of the precursor peptide to identify regions important for enzyme recognition. These experiments were complemented by in vitro binding studies aimed at identifying the region of the leader peptide recognized by the LanL enzymes as well as determining which domain of the enzyme is recognizing the substrate peptide. Combined, these studies revealed that the kinase domain is mediating the interaction with the precursor peptide and that a putatively α-helical stretch of residues at the center to N-terminal region of the leader peptide is important for enzyme recognition. In addition, a combination of in vitro assays and tandem mass spectrometry was used to elucidate the order of dehydration events in these systems.
Graphical Abstract
Introduction
RiPPs are a very diverse and rapidly expanding family of natural products with a wide range of structures and biological functions which include antibacterial, antifungal, insecticidal, and antiviral activities, as well as roles in signaling or as co-factors.1–25 A common feature of all RiPPs is that they start out as a gene-encoded, ribosomally-synthesized precursor peptide, which is recognized by corresponding processing enzymes for introduction of posttranslational-modifications.1,26,27 Typically, these precursor peptides are divided into an N-terminal leader region, which is needed for enzymatic recognition, and a C-terminal core region, where the modifications are incorporated. As the leader peptide is only needed for mediating the interaction of precursor peptide and processing enzymes, it is removed by proteolysis at one point during the maturation of a RiPP. Hence, a mature RiPP only consists of the fully modified core peptide.1 While the precursor peptides are limited to the use of amino acids that are tolerated by the ribosome, the enzymatic machineries in RiPP systems can introduce a wide range of chemical and structural diversity by a variety of mechanisms.1–10,12,14–24,28–41 The genetic origin of RiPP precursor peptides and the often observed promiscuity of their processing enzymes allows the use of stable RiPP scaffolds in for epitope grafting and generation of combinatorial libraries toward development of potential therapeutics.3,5,8,9,22,29,33,42–44
The defining feature of lanthipeptides is the presence of thioether macrocycles, which are comprised of (methyl)lanthionine bis-amino acids.21 These rings are formed in a two-step process. First, dehydration of serine and threonine side chains yields dehydroalanine (Dha) and dehydrobutyrine (Dhb) residues (Figure 1). Then, nucleophilic attack of cysteine thiols on these unsaturated double bonds via a conjugate addition mechanism yields the β-thioether bonds.21 In special cases, thus far only observed in class III systems, the enolate intermediate can attack another Dha residue, which results in formation of an entwined bicyclic structure called a labionin (Figure 1).45–48 Generally, lanthipeptides are subdivided into four classes based on characteristics of their corresponding synthetases.1,21,45,49,50 Class I lanthipeptides are produced by two enzymes, a LanB dehydratase and a LanC cyclase, while class II lanthipeptides are synthesized by a single LanM enzyme carrying both a dehydratase and a cyclase domain. Both class III and class IV involve single three-domain enzymes for maturation.49 These enzymes consist of an N-terminal lyase, a central kinase and a C-terminal cyclase domain. The cyclase domain is what allows differentiation between these two classes. While class IV enzymes (LanLs) feature a cyclase domain similar to the ones in class I and II systems that contain conserved Zn2+ binding sites, the cyclase domains of class III enzymes (LanKCs) are more different and lack the canonical coordination sites for Zn2+ ions (Figure 1a). In classes II–IV the dehydration is realized through ATP-dependent phosphorylation of the serine/threonine hydroxyl groups and subsequent elimination (Figure 1b). In class I, the hydroxyl groups are glutamylated prior to glutamate elimination, which is accomplished by using glutamylated tRNA as substrate.51,52
Figure 1.

Overview of (a) different lanthipeptide classes and (b) mechanisms of lanthipeptide biosynthesis.
While many studies have focused on the mechanisms of class I and II biosynthetic enzymes, including elucidation of crystal structures of representative enzymes,50,52–54 less is known about the two other classes, both first described in 2010.45,49 For class IV systems, previous studies on the biosynthetic gene cluster from Streptomyces venezuelae (Figure 2a) reported in trans activities of artificially separated lyase and kinase domains49 and identified residues that are crucial for the activity of the lyase domain.55 While the corresponding lanthipeptide (venezuelin) could not be isolated from S. venezuelae cultures, in vitro maturation of the precursor peptide VenA (and variants thereof) by VenL in combination with tandem mass spectrometry (MS) allowed determination of the cyclization pattern (Figure 2c).49
Figure 2.
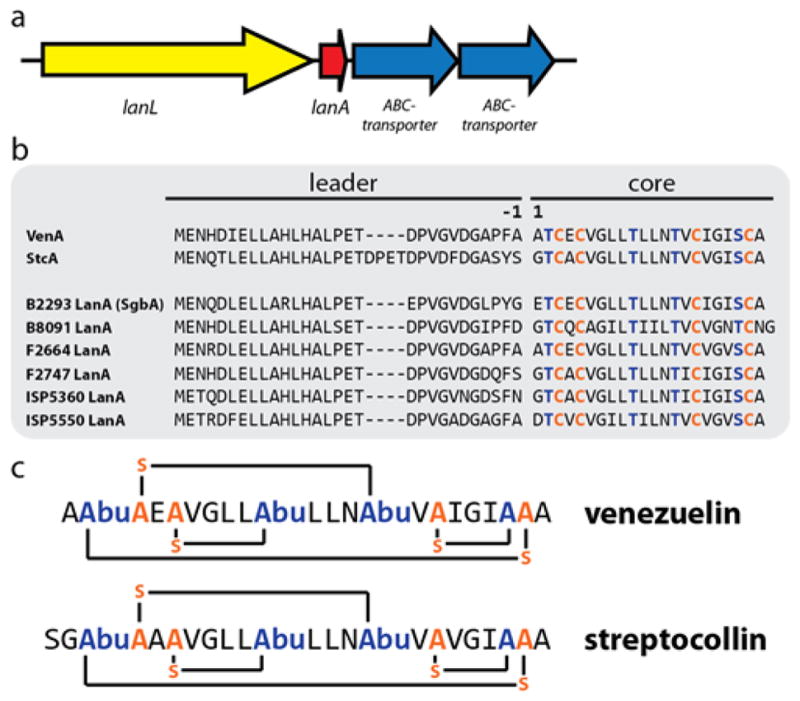
(a) General organization of class IV lanthipeptide biosynthetic gene clusters (b) Alignment of the venezuelin (VenA) and streptocollin (StcA) precursor peptides with the precursor peptides of the clusters investigated in this study. Residues undergoing dehydration are highlighted in blue, cysteines involved in ring formation in orange. According to standard RiPP nomenclature, the first amino acid of the core peptide is designated as residue 1, while the last amino acid of the leader peptide is defined as residue −1. (c) Primary structures of venezuelin49 and streptocollin.56 Abu = aminobutyric acid.
The structure of venezuelin and analogs was confirmed later by two independent studies, where production of mature class IV lanthipeptides was observed in different Streptomyces species.56,57 Genome mining in Streptomyces furthermore revealed a family of gene clusters closely related to the venezuelin system,57 and for the class IV lanthipeptide streptocollin (Figure 2c), a heterologous Streptomyces production system was established.56 Thus far, the biological function of these natural products has been elusive and only a moderate inhibitory activity against protein tyrosine phosphatase 1B (PTP1B) was described for streptocollin (33% inhibition at 50 μM).48
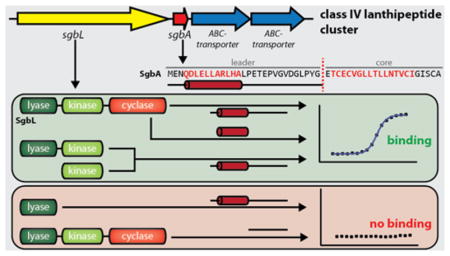
In this study, we took a closer look at the venezuelin-like gene cluster family (GCF) and selected a number of representatives to learn more about class IV lanthipeptide synthetases. First, these strains were screened under different media conditions and checked for lanthipeptide production. For the one strain that did not show production under any tested condition, a heterologous production system was established in E. coli, which allowed isolation of the fully modified core peptide. A slightly altered version of this production system was employed for mutational analysis of the precursor peptide. In this context, a block-wise alanine scan of the leader region was performed and the effect of deletions and insertions in the leader region was studied. The results of this investigation suggests that the recognition site is located on the N-terminal part of the leader peptide. To corroborate these findings and further narrow down the recognition motif, a fluorescence polarization binding study was performed. Thereby, we were able to show that only the center to N-terminal portion of the leader peptide contributes to LanL binding, while the remaining C-terminal portion of the leader peptide does not seem to interact with the enzyme in a significant manner. Secondary structure prediction of the precursor peptide suggests that the region identified as important for enzymatic recognition may be forming a short α-helix. Repetition of the binding studies with single domains of our LanL protein revealed that the central kinase domain binds the leader peptide. Finally, by combination of in vitro assays with tandem mass spectrometry (MS), the order of dehydration events in the core peptide was determined to follow a N- to C-terminal directionality. This study brings our knowledge of class IV lanthipeptide biosynthesis closer to that of the other classes.
Experimental Procedures
Bacterial Strains and Materials
E. coli DH10B cells were used for cloning and mutagenesis. E. coli BL21(DE3) cells were used for expression. All Streptomyces strains used in this study were obtained from the USDA ARS collection (Peoria, Illinois). All plasmids were validated via dideoxy sequencing (carried out by ACGT, Inc.). Gibson assembly master mix and Phusion DNA polymerase were obtained from New England Biolabs, oligonucleotide primers from Integrated DNA Technologies, synthetic peptides with >95% purity from Genscript and trypsin from Worthington Biochemical Corporation. Lysozyme, benzonase (25–29 U/μL), Millipore C18 Ziptips and fluorescein isothiocyanate (FITC) were purchased from Thermo Fisher Scientific.
Streptomyces Media Screens
Aliquots of liquid GYM medium (100 mL, see below) were inoculated from Streptomyces spore stocks and grown for 3 d at 30 °C. Six different agar media (see below) were cast into 6-well tissue plates (~2–3 mL per well) and inoculated by addition and distribution of 100 μL of Streptomyces precultures. Plates were then incubated for 14 d at 30 °C. Subsequently, colonies of each strain under each condition were transferred into 0.2 mL PCR tubes and 50 μL of MeOH was added for extraction. After overnight incubation at room temperature (RT), the extracts were centrifuged and the supernatant was transferred into a fresh tube. Of these methanol extracts, 1 μL was spotted on a mass spectrometry target and evaporated at RT. The residual dried extract was mixed with a saturated solution of sinapinic acid in 60% MeCN by pipetting up and down, crystallized at RT and analyzed by matrix-assisted laser desorption/ionization-time-of-flight MS (MALDI-TOF-MS) on an UltrafleXtreme MALDI TOFTOF (Bruker Daltonics). The media were prepared according to the following recipes (for agar media, 15 g/L agar was added prior to autoclaving the medium). ATCC172 medium: 10 g/L glucose, 20 g/L soluble starch, 5 g/L yeast extract, 5 g/L N-Z-amine type A, 1 g/L CaCO3, pH 7.2; ISP4 medium: 10 g/L soluble starch, 1 g/L K2HPO4, 1 g/L MgSO4 · 7 H2O, 1 g/L NaCl, 2 g/L (NH4)2SO4, 2 g/L CaCO3, 1 mL/L metal salt stock solution (containing 1 mg/mL of each FeSO4, MnCl2, ZnSO4), pH 7.2; SM medium: 20 g/L soy flour, 20 g/L mannitol, pH 7.2; Soy-sucrose medium: 20 g/L soy flour, 20 g/L sucrose, pH 7.2; AltMS medium: 10 g/L soy flour, 10 g/L mannitol, 10 g/L malt extract, pH 7.2; GYM medium: 4 g/L glucose, 4 g/L yeast extract, 10 g/L malt extract, pH 7.2.
Cloning
All cloning was accomplished by Gibson assembly and the primers used in this study are summarized in Supporting Information Table S1. GCF147 genes were amplified from Streptomyces gDNA (isolated employing an UltraClean Microbial DNA Isolation kit from MoBio Laboratories following their standard guidelines) and the mbp gene from an E. coli plasmid using Phusion DNA polymerase. All PCR products contained 5′ overhangs that allowed subsequent Gibson assembly. The vector backbone of pET28a was linearized by PCR with appropriate primers.
Heterologous Production of GCF147 Lanthipeptides, LC-MS Analysis and Preparative HPLC
Lysogeny broth (LB) containing kanamycin (50 μg/mL) was used for all E. coli cultures. For expression, media flasks were inoculated 1:100 with 37 °C overnight cultures. For expression at 37 °C, cells were grown continuously at 37 °C until reaching an optical density at 600 nm (OD600) of 0.5–0.7, then induced by addition of 0.2 mL of isopropyl β-D-1-thiogalactopyranoside (IPTG) stock solution (0.5 M) per 1 L of medium (= 0.1 mM final IPTG concentration) and expressed for 3 h at 37 °C. For expression at 18 °C, cells were grown at 37 °C until reaching an OD600 of 0.2–0.25. At this point, the temperature was shifted to 18 °C. Cultures were induced by IPTG addition 1 h later at an OD600 of 0.5–0.7 and grown overnight at 18 °C.
After expression, cultures were harvested by centrifugation. Cell pellets were resuspended in a small amount of water and extracted with 100 mL of MeOH per 1 L of culture by shaking overnight at 4 °C. On the next day, the extracts were centrifuged and the clear supernatant was evaporated at 40 °C under reduced pressure.
Small scale production tests were performed in volumes of 50 mL of LB medium in 250 mL Erlenmeyer flasks and the dried extracts were resuspended in 1 mL of 50% MeOH. Extracts were then cleared by centrifugation and the supernatant was either applied to MALDI-TOF-MS analysis as described above or 50 μL were used for high-resolution liquid chromatography-MS (LC-MS). The latter was accomplished on an Acquity Ultraperformance LC system (Waters) connected to a SYNAPT-MS instrument (Waters) with a C18 column (Phenomenex, Jupiter 5 μm C18 300 Å, 150×1 mm) at 35 °C with a flow rate of 0.15 mL/min using the following gradient of solvents A (0.1% formic acid in H2O) and B (0.1% formic acid in MeCN): Holding 3% B for 3 min, followed by a linear gradient from 3% to 97% B over 12 min and holding at 97% B for 3 min. For tandem MS, target ions were fragmented by collision-induced dissociation using a ramping-cone voltage setting of 25–30 kV. Tandem MS data was analyzed using the Mass Lynx software (Waters) in the following way: Raw data was smoothed, centered and then calibrated using the exact mass of the fragmented parent ion. For larger peptides, the spectra were deconvoluted using the MaxEnt3 function of the software.
Large scale production was performed in 2 L of LB medium using baffled 4 L Erlenmeyer flasks and the dried extracts were resuspended in 7 mL of 50% MeOH. After centrifugation, the cleared supernatants were applied to preparative HPLC using a Nexera HPLC system (Shimadzu) and a C18 column (Phenomenex, Luna 10 μm C18(2) 100 Å, 250×10 mm) with the following gradient at RT with a flow rate of 8 mL/min of solvents A (0.1% trifluoroacetic acid in H2O) and B (0.1% trifluoroacetic acid in MeCN): Linear increase from 8% to 80% B over 30 min, followed by a linear increase from 80% to 98% over 1 min and holding at 98% B for another 5 min. In this way, ~1.5 mg/L of the four-fold and ~1 mg/mL of the three-fold dehydrated peptides could be isolated.
Mutagenesis
Mutagenesis was accomplished by site-directed ligase-independent mutagenesis (SLIM) using primers listed in Supporting Information Table S1 and standard protocols.58,59 SLIM PCRs were carried out using Phusion DNA polymerase and the wild type co-expression plasmid as template. Number designation of the mutated residues follows standard RiPP nomenclature.1 The first residue of the core peptide is defined as residue 1 and following residues have increasing numbers. The last amino acid of the leader peptide is defined as residue −1 and numbers decrease in the direction of the N-terminus.
Expression, NiNTA Purification and TEV-Cleavage of His6-MBP-SgbA Variants
Expressions of the His6-mbp-sgbA_RBS_lanL pET28a co-expression system and mutants thereof were carried out in 0.5 L of LB medium in 2 L baffled flasks for 1 d at 18 °C or 1 h at 37 °C under the aforementioned conditions. After harvesting, cell pellets were resuspended in 10 mL of lysis buffer (300 mM NaCl, 50 mM HEPES, 10% glycerol, pH 7.5) and treated with lysozyme (10 mg) and benzonase (2.5 μL) for 1 h on ice. Cells were then lysed on ice by sonication using a Vibra Cell sonicator (Sonics & Materials) with the following settings: 40% amplitude, 5 min total sonication time; alternating between 2 s on/5 s off-pulse.
Lysates were centrifuged at 4 °C for 20 min at 75000×g and supernatants were filtered through a syringe filter (0.45 μm). Nickel nitrilotriacetic acid (NiNTA) purification was performed with an 1 mL NiNTA HisTrap column (GE Healthcare) using a peristaltic pump. After loading of the lysate, the column was washed with 5 mL of wash buffer (25 mM imidazole, 300 mM NaCl, 50 mM HEPES, 10% glycerol, pH 7.5) and then eluted with 10 mL of elution buffer (500 mM imidazole, 300 mM NaCl, 50 mM HEPES, 10% glycerol, pH 7.5). Elution fractions were buffer exchanged twice in a 1:5 ratio with Tobacco Etch Virus (TEV) cleavage buffer (300 mM NaCl, 50 mM HEPES, pH 8.0) employing an Amicon centrifugal filter unit (30 kDa cut-off) and then concentrated to final volume of ~0.5–1 mL at typically around 15–30 mg/mL. From these concentrated protein solutions, 100 μL were treated overnight at RT with 30 μL of a TEV stock solution (1 mg/mL). Aliquots (20 μL) of the TEV reactions were purified and eluted into 3 μL of 0.1% trifluoroacetic acid/80% MeCN using C18 Ziptips following the manufacturer’s protocol and prepared for MALDI-TOF-MS by mixing 1 μL with 1 μL of saturated sinapinic acid in 60% MeCN.
Protein Expression and Purification
All LanL enzymes and domains thereof (Supporting Information Figure S1) were expressed for 1 d at 18 °C in 4 L baffled flasks containing 2 L of LB medium using the conditions described above. Cell pellets from 4 L of cultures were resuspended in lysis buffer, yielding a final volume of 35–40 mL. Cells were incubated on ice with lysozyme (50 mg) and benzonase (5 μL) for 1 h and subsequently lysed by sonication on ice with the following settings: 60% amplitude, 5 min total sonication time; alternating between 2 s on/5 s off-pulse.
Lysates were centrifuged twice for 30 min at 4 °C and 75000×g (supernatants were transferred into fresh centrifuge tubes in between centrifugation steps) and filtered through a syringe filter (0.45 μm). NiNTA purification was performed using a 5 mL NiNTA HisTrap column (GE Healthcare) using a peristaltic pump. After loading, the column was washed with 25 mL of wash buffer and eluted with 15 mL of elution buffer. Elution fractions were concentrated with Amicon centrifugal filter units (at suitable MW cut-offs) to 3–4 mL and further purified by size-exclusion chromatography (SEC).
For SEC, the protein solutions were applied to a Superdex200 column on an Äkta FPLC (GE Healthcare) at a flow rate of 1 mL/min using 300 mM NaCl, 50 mM HEPES, 10% glycerol at pH 7.5 as run buffer. The purest fractions were identified by SDS-PAGE, pooled and concentrated before flash-freezing them as aliquots and then storage at −80 °C until used. For His6-SgbL(lyase-kinase 1-548) and His6-SgbL(kinase 208-548), two subsequent rounds of SEC needed to be performed to obtain the proteins in high purity due to a truncant protein eluting close to the full length target enzymes.
FITC-Labeling of Peptides
FITC-labeling of the N-termini of peptides was performed in 100 mM phosphate buffer at pH 8.4. For this, 0.4 mL of peptide stock solution (5 mg/mL in water) was diluted with 7 mL of buffer, and 125 μL of a FITC-stock solution (10 mg/mL in DMSO) was added. After mixing, the reaction was slowly shaken overnight at RT, while being protected from light. The modified peptides were purified by two rounds of preparative HPLC under the previously mentioned settings and solvents with the following gradient: Linear gradient from 16% to 72% B over 30 min, followed by a linear increase from 72% to 98% B over 1 min and keeping 98% B for another 5 min. Under these conditions, the FITC-labeled peptides would elute several minutes after the unmodified peptides.
Fluorescence Polarization (FP) Binding Studies
FP was used to determine the binding affinities of different peptides to His6-SgbL and domains thereof. Determination of the Kd is possible by this method, as the polarizability of the fluorescent moiety on the peptide probe is different in its bound compared to its unbound state. In these assays, the Kd is equal to the concentration of protein where the FP is halfway between the FP of the used FITC probe at fully bound and fully unbound states. For FP binding studies, protein stocks were prepared in buffer containing 300 mM NaCl, 50 mM HEPES and 5% glycerol at pH 7.5. Serial 1:1 dilutions of each protein were prepared in the same buffer, yielding a total of 16 different concentrations per protein. FITC-peptide stock solutions (10 mg/mL in DMSO) were diluted to 1 μM with H2O. For the assays, 5 μL of the diluted FITC-peptide solutions were mixed in separate wells of a 384-well solid black polystyrene microplate (Corning) with 45 μL of each protein concentration and plain buffer as control (yielding a final concentration of 100 nM FITC-peptide). The mixtures were equilibrated for 30 min at RT, before parallel and perpendicular fluorescence intensities were measured at an excitation wavelength of 485 nm and an emission wavelength of 528 nm with a bandwidth of 20 nm using a Synergy H4 Hybrid Reader (BioTek). FP was determined as the difference of the parallel minus the perpendicular fluorescence intensity divided by the sum of both. All measurements were performed in triplicates. Background fluorescence was assessed by mixing 45 μL of the protein stocks with 5 μL of H2O and measuring in the same way, but was negligible for every protein tested. Kd values were determined by fitting the FP against the log of the protein concentration using a non-linear dose-response fit in OriginPro2015 (OriginLab).
Acetylation of the N-terminal Amines of Peptides
For acetylation of the N-terminal amines of SgbA(−1 to −20), SgbA(−9 to −23) and SgbA(−15 to −23) the following protocol was used: 3 mg of each peptide were incubated in phosphate buffer (100 mM at pH 8.4) with a 25-fold molar excess of Sulfo-NHS acetate (added as a stock solution of 10 mM in the same buffer) for 1 h at RT. The N-terminally labeled peptides were then purified by two rounds of preparative HPLC using the same conditions as for the FITC-labeled peptides. Ac-SgbA(−1 to −14) was purchased directly as an N-terminally acetylated peptide.
Fluorescence Polarization Competition Assays
The FP competition assays were carried out at constant concentrations of FITC-SgbA(leader) (100 nM) and His6-SgbL (240 nM; which shows a strong FP change compared to the unbound state of the FITC probe, but is not yet in the saturation range) and varying concentrations of a competitor. For this, a stock solution containing 300 nM His6-SgbL and 125 nM FITC-SgbA(leader) in buffer (300 mM NaCl, 50 mM HEPES, 5% glycerol, pH 7.5) was prepared. Serial 1:1 dilutions of each competitor peptide were prepared in H2O, yielding a total of 12 different concentrations per peptide. In a well of a 384-well solid black polystyrene microplate (Corning), 10 μL of competitor peptide solution (or just water as control) was mixed with 40 μL of the His6-SgbL/FITC-SgbA(leader) stock solution (yielding a final concentration of 100 nM FITC-SgbA(leader) and 240 nM His6-SgbL). After equilibration of the mixtures for 30 min at RT, parallel and perpendicular fluorescence intensities were measured as described above and used to determine the FP for each competitor concentration. IC50 values were determined by fitting the FP versus the log of the competitor concentration using a non-linear dose-response fit in OriginPro2015 (OriginLab).
Production of SgbL Domains
All domain expression constructs were generated via SLIM-mediated deletion58,59 of the other domains (primers listed in Supporting Information Table S1) and expressed under the aforementioned conditions. For deciding on where to separate the domains, we first predicted the secondary structures in SgbL with Psipred,60 searched for homology of the domains via BLAST and aligned SgbL with VenL, for which separation of the lyase and kinase domains was accomplished previously. Based on this analysis, domain borders were first chosen at homologous positions to the ones in VenL,49,55 while avoiding disruption of secondary structure motifs. In this way, the lyase domain was anticipated to be at positions 1-218 of SgbL, while the kinase domain was initially defined to comprise residues 208-475 (there is a longer unstructured region between both domains and as residue 218 is close to a predicted β-sheet, we decided to start the kinase gene expression a few residues upstream to ensure that this region can properly fold). While His6-SgbL(lyase 1-218) expressed well, no protein could be isolated when expressing His6-SgbL(lyase-kinase 1-475) and His6-SgbL(kinase 208-475). Alignment of VenL with all other LanLs investigated in this study (Supporting Information Figure S2) shows that VenL contains a longer, putatively unstructured region following its kinase domain that is absent from the other LanLs. The area of the other LanLs surrounding this part of the alignment shows considerably low conservation (as does the region between the lyase and kinase domains), which suggests that this might be a linker region in between the domains. Therefore, two new domain boundaries (after residues 492 or 548, see Supporting Information Figure S3) were chosen for the kinase domain of SgbL, whereby one or two additional α-helices were added to the protein on its C-terminal side. While expression of His6-SgbL(lyase-kinase 1-492) also failed, His6-SgbL(lyase-kinase 1-548) could be expressed and isolated in good yields, which also held true for His6-SgbL(kinase 208-548). For the cyclase domain, expressions starting either at residues 462 or 549 and going to the end of SgbL were attempted, but no protein could be recovered. Therefore, an MBP tag was cloned into the expression construct of His6-sgbL(cyclase 549-end) pET28a, which allowed isolation of the N-terminally MBP-tagged domain. Unfortunately, no activity was observed when performing in vitro assays with this protein.
Expression and Purification of His6-SgbA
The His6-sgbA pET28a construct was obtained by SLIM-mediated deletion58,59 of the sgbL gene from the co-expression plasmid (primers listed in Supporting Information Table S1). For production of the corresponding peptide, this plasmid was expressed in E. coli BL21(DE3). Two 4 L baffled flasks, carrying 2 L of LB each, were inoculated 1:100 with an LB overnight culture and grown at 37 °C until reaching an OD600 of 0.5–0.7. Cultures were induced by addition of 400 μL of IPTG stock solution (0.5 M) per flask and harvested after expression for 1 h at 37 °C. Cells were resuspended in guanidine lysis buffer (6 M guanidine hydrochloride, 20 mM NaH2PO4, 500 mM NaCl, 0.5 mM imidazole, pH 7.5) to a final volume of 35–40 mL and lysed at RT by sonication with the following instrument settings: 60% amplitude, 5 min total sonication time; 5 s on/5 s off-pulse.
After centrifugation for 30 min at 75000×g and 4 °C, the supernatant was sonicated again for 1 min at the same settings to further shear the DNA to allow easy filtration through a syringe filter (0.45 μM). After filtering, the lysates were applied to NiNTA purification using a 5 mL NiNTA HisTrap column (GE Healthcare) and a peristaltic pump. After loading, the column was washed with 25 mL of guanidine wash buffer (4 M guanidine hydrochloride, 20 mM NaH2PO4, 500 mM NaCl, 30 mM imidazole, pH 7.5) and eluted with 15 mL of guanidine elution buffer (4 M guanidine hydrochloride, 20 mM Tris, 100 mM NaCl, 1 M imidazole, pH 7.5). The elution fraction was concentrated in an Amicon centrifugal filter unit (3 kDa cut-off) to ~0.5 mL and then diluted into 7 mL of 20% MeCN (this step was critical for successful purification, as HPLC after solid phase extraction for guanidine removal did not yield any His6-SgbA). The formed precipitate was removed by centrifugation and the clear supernatant was applied to preparative HPLC under the aforementioned settings and the following gradient: Linear gradient of 8%–80% B over 30 min, followed by a linear increase of 80–95% B over 1 min and holding 95% B for another 5 min. The elution fractions containing the target peptide were freeze-dried and redissolved in 7 mL of 20% MeCN containing 5 mM tris(2-carboxyethyl)phosphine (TCEP) and incubated overnight at RT before a second round of HPLC for further purification. In this way, His6-SgbA could be obtained with yields of ~0.3–0.7 mg/L expression culture.
In Vitro Assays
A typical enzyme assay contained 20 μL of 2× buffer (300 mM NaCl, 50 mM HEPES, pH 7.5), 2 μL of TCEP stock solution (20 mM in H2O), 2 μL of MgCl2 stock solution (200 mM in H2O), 2 μL of ATP stock solution (50 mM in H2O), 2 μL of His6-SgbA stock solution (10 mg/mL in DMSO, which yields a final concentration of 67 μM in the assay) and corresponding amounts of protein stock solution and H2O to obtain a final volume of 40 μL at a concentration of 2.5 μM for each protein. The reaction was run overnight at RT and then 20 μL of the sample was purified and eluted into 3 μL of 0.1% trifluoroacetic acid/80% MeCN using C18 Ziptips following the manufacturer’s protocol. The remaining 20 μL of the reaction mixture were used for NEM-labeling (see below). For the shortened His6-SgbL(kinase 208-548) assays, 20 μL reactions were set up as described and stopped after 1 or 3 h by C18 Ziptip purification into 3 μL of 0.1% trifluoroacetic acid/80% MeCN. In all cases, 1 μL samples were prepared for MALDI-TOF-MS by mixing with 1 μL of saturated sinapinic acid in 60% MeCN.
NEM-Labeling
Due to its selective reaction with thiol groups, N-ethylmaleimide (NEM) was used as labeling reagent to check for the presence of free thiols and thereby assessing the cyclization state of a peptide. In assay buffer, full NEM-labeling of unreacted His6-SgbA could not be achieved under standard conditions. Thus, the following modified NEM-labeling conditions were established, which accomplished complete labeling of a His6-SgbA control: 20 μL of an in vitro assay reaction were first freeze-dried and then redissolved in 20 μL of a guanidine buffer (6 M guanidine hydrochloride, 20 mM NaH2PO4, 500 mM NaCl, pH 7.5). To make sure that all free cysteines were fully reduced, 1 μL of TCEP stock solution (100 mM in H2O) was added and the mixture was incubated for 15 min at 50 °C. Then, 1 μL of NEM stock solution (400 mM in EtOH) was added and after mixing the reaction was run for 1 h at RT. Finally, the peptide was purified and eluted into 3 μL of 0.1% trifluoroacetic acid/80% MeCN using C18 Ziptips following the manufacturer’s protocol and prepared for MALDI-TOF-MS by mixing 1 μL with 1 μL of saturated sinapinic acid in 60% MeCN.
NEM-labeling of the isolated, modified SgbA truncants from the co-expression experiments was carried out in a likewise manner: 1 μL of stock solution of the three-fold dehydrated peptide (10 mg/mL in DMSO) was mixed with 19 μL of guanidine buffer and reduction, NEM-labeling and C18 Ziptip purification were carried out as described.
Determination of the Dehydration Order
For determination of the order in which His6-SgbL(lyase-kinase 1-548) dehydrates the threonine and serine residues in the core peptide, 60 μL assay reactions were set up as described above. These reactions were either stopped after 10 min (which produced mostly −1 and −2 H2O peptide) or 60 min (which produced mostly −2 and −3 H2O peptide) in a thermocycler at 25 °C by increasing and holding the temperature to 95 °C for 5 min or run overnight at RT (which completely converts the precursor peptide to the −4 H2O species). Next, 0.65 μL of CaCl2 stock solution (100 mM in H2O) and 3 μL of trypsin stock solution (3 mg/mL in 50 mM Tris, 1 mM CaCl2, pH 7.6) were added and proteolysis was carried out for 4 h at RT. This procedure was also used for a sample of non-reacted His6-SgbA to assess the unmodified state of the peptide. Afterwards, 50 μL of these reactions were applied to high-resolution LC-MS and fragmentation of the different dehydration species was performed as described earlier. By deconvolution and analysis of the respective MS2 spectra, the order of the dehydration events could be elucidated.
Results and Discussion
Heterologous Production of class IV Lanthipeptides in E. coli
A previously published genome mining study for lanthipeptide gene clusters in Streptomyces sp. described GCF147, which at the time consisted of 33 members of venezuelin-like gene clusters that are closely related to each other.57 From these, we picked five strains (S. globisporus subsp. globisporus NRRL B2293, S. virginiae subsp. virginiae NRRL B8091, S. sp. NRRL F2664, S. sp. NRRL F2747 and S. griseoluteus NRRL ISP5360) and additionally one previously reported59 class IV lanthipeptide producing strain (S. katrae NRRL ISP5550) and employed them in media screens (for simplification, we will only use the NRRL numbers for referring to these strains from here onwards). Each strain was grown on six different agar media (ATCC172, ISP4, GYM, SM, AltMS and soy-sucrose medium) for 14 days at 30 °C. Then, colonies from each condition were picked, extracted with MeOH and the extracts were analyzed by MALDI-TOF-MS for the presence of the predicted lanthipeptide masses (Supporting Information Figures S4a–S4f). Production of predicted compounds was observed in all cases except for B2293 (Supporting Information Table S2). Interestingly, besides the mass of the fully modified core peptide, species that still carried the last residue of the leader peptide were observed and sometimes these peptides were the only species detected (as was also seen for streptocollin56). This finding suggests that for class IV lanthipeptides there might not be a clear divide between leader and core region and that removal of the leader region is possibly accomplished by non-specific proteolysis, which would also explain the lack of a dedicated protease encoded in the class IV gene clusters.
A heterologous production system was then established for the lanthipeptide of the B2293 cluster, which was not produced by the native host and was named globisporin. Both the lanA (sgbA) and lanL (sgbL) gene from B2293 were cloned into a pET28a backbone. The construct was assembled such that the precursor was expressed with an N-terminal His6-tag and a thrombin cleavage site, which would allow isolation of the modified precursor peptide via NiNTA and release of the precursor via thrombin treatment. Additionally, an E. coli optimized RBS was placed in between the sgbA and sgbL genes to facilitate the expression of the SgbL processing enzyme. The final construct uses a lacI-controlled T7 promoter for co-expression of His6-SgbA and untagged SgbL (Figure 3a).
Figure 3.

(a) Schematic representation of the heterologous E. coli production system for the B2293 lanthipeptide globisporin. (b) Amino acid sequence of the last 29 residues of the SgbA precursor peptide showing the cyclization pattern known from venezuelin49 and streptocollin.56 Positions where b and y fragments were detected by tandem MS (including mass shifts due to water losses) are highlighted. (c) Tandem MS of the −4 H2O species of the extracted 29 amino acid (aa) peptide. The lack of detectable fragments in the region of Thr2-Cys21 (and overall low intensity of fragment ions) suggests formation of a methyllanthionine ring between these residues. For calculated and observed m/z values see Supporting Information Table S3. The tandem MS data only provides evidence of an overlapping ring topology that spans Dhb2 to Cys21. The topology the rings shown in (b) was predicted based on the high similarity of the globisporin system with the sequences producing venezuelin and streptocollin (Figure 2).
Surprisingly, NiNTA purification after expression in LB for 3 h at 37 °C or 1 d at 18 °C did not yield any peptide. Hence, extraction of the cell pellet was tried with MeOH after expression under the same conditions and the extracts were analyzed via high-resolution LC-MS. Indeed, we observed masses corresponding to the core peptide with some remaining residues of the leader peptide, which had lost three or four water molecules under both conditions, with the main products being 27 and 29 aa long. Generally, stronger signals were obtained for expression at 18 °C than at 37 °C, and therefore 1d at 18 °C was used for all subsequent experiments.
Application of the MeOH extracts of the pellets of larger expression cultures to preparative HPLC allowed isolation and separation of the −3 and −4 H2O precursor peptide, but the differently sized truncants of each species co-elute and therefore could not be separated. Tandem MS analysis and treatment with the thiol specific reagent N-ethylmaleimide (NEM) confirmed their cyclic nature (Figure 3 and Supporting Information Figure S5). To show that this approach also works for other LanL systems, we generated likewise expression systems for the B8091, F2664 and ISP5360 clusters, all showing the same production of −3 and −4 H2O species when expressed in E. coli under the same conditions.
Determination of the binding sites on the substrate and LanL enzyme
With a working heterologous production system in hand, we performed mutational analysis of the leader peptide to identify regions important for enzyme recognition. As our initial production system shows that the precursor peptide is prone to proteolysis during expression, we fused the start of the sgbA gene to a maltose binding protein (MBP)-encoding gene sequence with a tobacco etch virus (TEV) protease cleavage site in between for MBP removal (Figure 4a). Thereby, the N-terminus of SgbA is protected against proteolysis by aminopeptidases and the His6-MBP-SgbA fusion can be isolated by NiNTA. As expected, expression of the His6-mbp-sgbA_RBS_sgbL pET28a construct in LB at 18 °C for 1 d allowed isolation of the MBP fusion protein and TEV protease treatment of the isolated protein yielded fully modified SgbA (Supporting Information Figure S6).
Figure 4.

(a) Schematic representation of the heterologous E. coli production system for His6-MBP-SgbA co-expressed with SgbL. (b) Overview of all variants of His6-MBP-SgbA tested in the co-expression construct (see also Supporting Information Figure S6).
Encouraged by detection of modified precursor peptide, a number of mutants was generated. The sequence of the leader peptide of class IV lanthipeptides is significantly different from that of the other classes, and hence we could not rely on previous mutagenesis studies that have investigated their recognition motifs.61–70 Therefore, we first divided the leader region into blocks of four to five residues, which were individually replaced by stretches of alanines (Figure 4b). This approach would allow detection of potential small recognition motifs like the FNLD region in the nisin precursor peptide NisA; mutagenesis of this motif significantly decreases binding affinities to the NisB and NisC processing enzymes and reduces the number of observed modifications of NisA in co-expression experiments.51,52,67,68,71,72 Surprisingly, full modification was observed for all six alanine scan variants (Supporting Information Figure 6). This result suggests that an extended motif in the leader region is recognized by the processing enzyme. Therefore, a new set of four mutants was generated, in which the leader peptide was truncated in a stepwise manner from the N-terminus (Figure 4b). Full processing was observed only for His6-MBP-SgbA(Δ−21 to −29), whereas no masses were detected corresponding to His6-MBP-SgbA(Δ−16 to −29), His6-MBP-SgbA(Δ−11 to −29) and His6-MBP-SgbA(core). We attributed this absence of any detectable peptides attached to MBP to proteolytic degradation of the unmodified core peptide. To investigate this hypothesis, we repeated the co-expressions of WT His6-MBP-SgbA and the three truncated analogs with SgbL for 1 h at 37 °C, reasoning that during this short duration not all unmodified peptide would be degraded. MALDI-TOF-MS analysis of the WT MBP-SgbA fusion after TEV treatment shows major signals for fully and unmodified precursor peptide as well as weaker signals for intermediate dehydration states (Supporting Information Figure S6b), indicating that modification can still occur. In contrast, we could only detect unmodified peptides released by TEV cleavage of His6-MBP-SgbA(Δ−16 to −29), His6-MBP-SgbA(Δ−11 to −29) and His6-MBP-SgbA(core) (Supporting Information Figure S6b). This observation provides direct evidence that SgbL does process these three truncated precursor peptides, and also provides evidence that these truncated peptides are produced, but that they are likely proteolytically degraded at later time points when unmodified.
Collectively, these results suggest that the recognition motif is located in the N-terminal half of the leader peptide. To test if the distance between the recognition motif and the core peptide is important for enzymatic processing, two additional mutants were generated. One introduced a stretch of five alanines in between leader and core peptide, while the other deleted the last five residues of the leader sequence (Figure 4b). In both cases, full modification was observed, showing that at least some flexibility exists with regard to the distance between the position that the SgbL enzyme recognizes in the leader peptide and where the enzyme modifies the core peptide.
To corroborate the in vivo findings as well as further pinpoint the recognition motif in the leader peptide, an in vitro binding assay was used measuring the change in fluorescence polarization (FP) as read out. The free N-terminus of chemically synthesized SgbA leader peptide was fluorescently labeled by reaction with fluorescein isothiocyanate (FITC). Incubation of the FITC-SgbA(leader) with different concentrations of His6-SgbL demonstrated a Kd of 188 ± 5 nM (Figure 5a). Next, three N-terminally FITC-labeled truncants of the SgbA leader peptide were assayed for binding to His6-SgbL (Figure 5b). One truncant lacked the first nine residues termed SgbA(−1 to −20), while another missed the last ten residues of the leader sequence (SgbA(−11 to −28)). A third peptide consisted of only the residues identified as most conserved in an alignment of all GCF147 precursor peptides, i.e residues −9 to −23. Binding was observed for FITC-SgbA(−11 to −28) (Kd = 530 ± 17 nM) and FITC-SgbA(−9 to −23) (Kd = 744 ± 22 nM), but not for FITC-SgbA(−1 to −20). To identify a minimal binding site, we obtained an additional truncant of SgbA(−9 to −23), where another six residues were removed from the C-terminus. SgbL binding was also observed for the resulting FITC-SgbA(−15 to −23) (Kd = 2068 ± 147 nM), albeit at a roughly tenfold decreased affinity when compared to full length leader peptide (Kd = 188 ± 5 nM).
Figure 5.

(a) Schematic showing the principle of the fluorescence polarization (FP) binding studies. The representative graph was recorded at 100 nM FITC-SgbA(leader) and varying concentrations of His6-SgbL. (b) Overview of the results of the FP binding studies with the different N-terminally FITC-labeled leader peptide truncants. Predicted α-helical regions in the precursor peptide are shown in red. The corresponding graphs are depicted in Supporting Information Figures S7 and S8 (c) Overview of the results of the FP competition assays using 100 nM FITC-SgbA(leader), 240 nM His6-SgbL and varying concentrations of the respective competitor peptide.
Looking at the minimal peptide that still binds to His6-SgbL and comparing it to the results of the mutational analysis of the co-expression system, it is surprising that FITC-SgbA(−1 to −20) did not show binding, because full modification was observed for His6-MBP-SgbA(Δ−21 to −29) in the co-expression experiments. To investigate this finding further, a competition FP experiment was devised using FITC-SgbA(leader). The competitors tested were SgbA(leader), SgbA(−11 to −28), and N-terminally acetylated Ac-SgbA(−1 to −20), Ac-SgbA(−9 to −23) and Ac-SgbA(−15 to −23). The non-acetylated versions of these peptides were unable to compete with FITC-SgbA(leader), likely because the positive charge of the free N-terminus interfered with binding for these truncated peptides (see Supporting Information Figure S9). Comparison of the IC50 values of the competitors (Figure 5c) allows estimation of their affinities for His6-SgbL compared to FITC-SgbA(leader). Unsurprisingly, the best competitor was the full length leader peptide (IC50 = 4.5 ± 0.2 μM), followed by SgbA(−11 to −28) (IC50 = 16.5 ± 1.5 μM). All other peptides showed a comparable IC50 in the assays (IC50 Ac-SgbA(−9 to −23) = 197 ± 5 μM, IC50 Ac-SgbA(−15 to −23) = 147 ± 13 μM, IC50 Ac-SgbA(−1 to −20) = 174 ± 12 μM).
To understand the discrepancy of the results of the binding assay with FITC-SgbA(−1 to −20) versus the competition assay with Ac-SgbA(−1 to −20), a secondary structure prediction of SgbA was performed, which suggests the presence of two α-helical regions in the precursor peptide (Figure 5b): One spanning almost the whole core peptide, while the other one aligns well with the identified minimal binding site. Considering that this α-helical region might mediate the interaction with the SgbL enzyme, a potential explanation of the lack of binding of FITC-SgbA(−1 to −20) becomes apparent. For this peptide, only half of the α-helical region is still present and putting a bulky FITC moiety at its N-terminus probably sterically interferes with binding to the enzyme. Indeed, secondary structure predictions of all tested alanine scan variants suggest that each of them still contains an α-helical region at the identified position (see Supporting Information Figure S10).
To corroborate the suggested role of the putative α-helical region for binding to the SgbL processing enzyme, a control experiment was carried out with Ac-SgbA(−1 to −14). As expected, this peptide (that consists only of residues outside of the α-helical leader peptide region) was unable to compete with FITC-SgbA(leader). Taken together, the in vivo co-expression and in vitro FP binding experiments show that an extended motif in the center to N-terminal region of the leader peptide, which possibly may form an α-helix, is important for binding to and processing by SgbL,
In vitro Investigation of LanL Activity
Next, we investigated the binding site on SgbL. Based on alignments of SgbL with other LanLs and secondary structure predictions, we chose boundaries for the three domains of the protein. His6-SgbL(lyase-kinase 1-548), His6-SgbL(lyase 1-218), and His6-SgbL(kinase 208-548) were expressed and purified. The cyclase domain was not obtained in soluble form and therefore His6-MBP-SgbL(cyclase 549-end) was generated, which was soluble. Before using these proteins in binding assays, their activities were assessed in vitro (Supporting Information Figure S11), requiring full length SgbA. As several rounds of solid phase synthesis failed for this peptide, optimization of the expression and the subsequent purification protocol was undertaken. To prevent proteolysis, expression was carried out for only 1 h at 37 °C in LB medium, then the cell pellet was lysed and His6-SgbA was isolated via NiNTA under denaturing conditions in guanidine containing buffers. Before applying to HPLC for purification, it was essential to strongly concentrate the elution fractions and dilute them again into 20% MeCN to remove bulk guanidine. Although this procedure was successful, the yield was low (<1 mg/L, see Experimental Procedures).
With full length peptide in hand, in vitro assays were performed and combined with post-reaction NEM labeling of free thiol groups, to assess the cyclization state of the core peptide. When the assay was performed with His6-SgbL, full dehydration and cyclization of His6-SgbA was observed (Supporting Information Figure S11b). Assays carried out with His6-SgbL(lyase-kinase 1-548) alone and in trans with His6-MBP-SgbL(cyclase 549-end) revealed that four dehydrations were accomplished in both cases, and that the number of free cysteine residues at the end of each assay was identical (Supporting Information Figure S11g and S11h), meaning that the His6-MBP-SgbL(cyclase 549-end) was inactive under these conditions. Considering that expression of the cyclase domain on its own only produced insoluble protein and that the domain could only be obtained as a MBP-fusion, misfolding of the isolated cyclase domain might be a possible reason for this observation. Alternatively, we cannot rule out interference of the MBP-tag with potentially important interactions of the cyclase domain with other domains as an explanation for the lack of activity.
By carrying out the assay with His6-SgbL(lyase-kinase 1-548) and stopping the reaction at different time points by incubation at 95 °C for 5 min, all dehydration states from −1 to −4 H2O could be observed in yields that allowed tandem MS analysis (Figure 6). Treating these peptide mixtures with trypsin simplified analysis of the MS2 spectra and allowed identification of the dehydrated residues in every species. Thereby, the order of the dehydration could be determined to follow a strict N- to C-terminal directionality.
Figure 6.
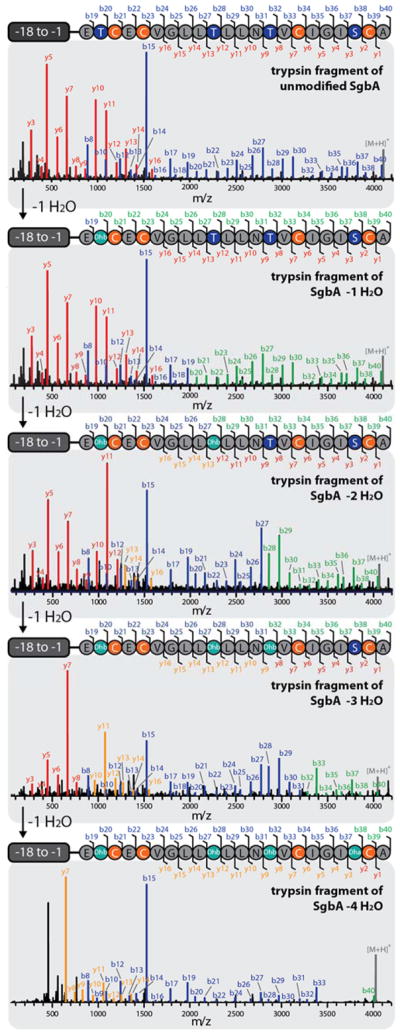
Tandem MS analysis of the different trypsin-treated, dehydrated species of His6-SgbA. If no mass change was observed for a specific b or y fragment after a single dehydration event, they are highlighted in blue or red, respectively. If a shift of −18 Da is observed for a specific b or y fragment, compared to the preceding +1 H2O species, they are colored in green or orange. It is apparent that the His6-SgbL(lyase-kinase 1-548) catalyzed dehydration starts at Thr2, proceeds to Thr10, continues to Thr14 and ends with modification of Ser20. The absence of some of the last b and first y fragments for the four-fold dehydrated peptide suggests that during the overnight incubation to obtain this species non-enzymatic cyclization of Cys16 and Dha20 occurred, which is in line with the NEM labeling results (Supporting Information Figure S11). For calculated and observed m/z values see Supporting Information Table S5.
To our surprise, overnight assays using only the kinase domain did not yield a phosphorylated peptide, but product with up to three dehydrations (Supporting Information Figure S11j). Apparently, phosphorylated His6-SgbA can undergo elimination in the absence of the lyase domain under the assay conditions. Carrying out the kinase assay for only 1 or 3 h at 25 °C instead of overnight, allowed detection of phosphorylated intermediates besides dehydrated species (Supporting Information Figure S12), which confirms this notion. These observations were surprising since mutations of active site residues in the lyase domain of the class IV lanthipeptide synthetase VenL resulted in mostly phosphorylated peptides, indicating the lyase domain clearly catalyzes phosphate elimination.55 In class II lanthipeptide synthetases, a kinase domain has recruited two additional amino acids to the active site that result in phosphate elimination inside the kinase domain.54 It is possible that the kinase domain of SgbL also has some low level catalytic activity to eliminate the phosphate group from phosphorylated SgbA.
As the in vitro assay with the kinase domain only yields up to three modifications, while use of His6-SgbL(lyase-kinase 1-548) accomplishes four dehydrations, we tested whether repeating the assay with His6-SgbL(lyase 1-218) and His6-SgbL(kinase 208-548) in trans would allow full dehydration of His6-SgbA by reestablishing potential interdomain contacts. Converse to this hypothesis, the addition of the lyase domain in trans did not change the outcome of the in vitro assay (Supporting Information Figure S11i).
The subsequent FP assays were carried out under the previously established conditions while replacing His6-SgbL with one of the protein domains (Figure 7). In these assays, binding of the FITC-labeled leader peptide was not observed for His6-SgbL(lyase 1-218), while both His6-SgbL(lyase-kinase 1-548) (Kd = 351 ± 7 nM) and His6-SgbL(kinase 208-548) (Kd = 357 ± 8 nM) did bind FITC-labeled SgbA leader peptide with almost identical Kds when compared to full length His6-SgbL (Kd = 188 ± 5 nM). Thereby, the site of leader peptide binding was pinpointed to the kinase domain. In many RiPP systems, including class I lanthipeptide dehydratases, selective binding of the leader peptide is accomplished by use of so-called RiPP Recognition Elements (RREs), which are defined by a highly conserved structural motif of three β-sheets, followed by three α-helices.18,27,31,32,52,73,74 Secondary structure prediction of the kinase domain shows that there is no part of this protein where such an arrangement of secondary structures is present (Supporting Information Figure S13), which suggests that precursor binding in class IV lanthipeptides is achieved by other means. A similar observation was recently made for the interaction of the microviridin precursor peptide MdnA and its corresponding processing enzymes MdnB and MdnC, which do not have RRE domains either. For MdnA an α-helical sequence in the leader peptide was shown to mediate recognition of the precursor peptide by the maturation machinery.75
Figure 7.

FP assays using 100 nM FITC-SgbA(leader) and varying concentrations of the respective SgbL domains.
As the precursor peptides in GCF147 are similar to each other (Figure 2b), we also probed whether the LanLs from the other strains investigated in this study could bind and modify SgbA. Isolation of the corresponding proteins succeeded in all cases except for the LanL from ISP5360, and binding of FITC-SgbA(leader) as well as full His6-SgbA modification was observed for all tested LanL homologs (Supporting Information Figures S7 and S11c–f). Closer inspection of all LanA homologs from these clusters shows that each one of them contains a predicted α-helix in the leader peptide in the same region as SgbA (Supporting Information Figure S10).
Collectively, our in vivo and in vitro experiments help to paint a picture of how LanL enzymes are interacting with their substrates. First, the kinase domain recognizes a putative α-helical region in the precursor peptide and initiates modification of the core peptide by phosphorylation of serine and threonine side chains. The phosphate is in turn eliminated by action of the lyase domain. This process follows a strict N- to C-terminal directionality. As we could not produce a soluble and active cyclase domain, we were unable to assess in vitro if the cyclization commences only after the core peptide has been fully dehydrated or already at earlier stages. Considering how GCF147 precursor peptides have very similar amino acid sequences, combined with the observed low stability of SgbA in our expression experiments, we hypothesize that the removal of the leader peptide in the native producers does not need a dedicated protease (no putative proteases are encoded in any of the GCF147 gene clusters or in close proximity thereof), but is accomplished non-specifically and facilitated by the intrinsic susceptibility for proteolysis of these particular peptides outside of the cyclized core region. This hypothesis might also explain the observed production of mature lanthipeptides with different lengths in some of the native producers. These observations are analogous to studies of several class III lanthipeptides (erythreapeptin, avermipeptin, griseopeptin, curvopeptin, stackepeptin, labyrinthopeptin and SapB), where mixtures of lanthipeptides with differently sized N-terminal overhangs on the modified core peptides were observed as well.21,45–47,76 It was suggested that aminopeptidase activity might be involved in the maturation process of these RiPPs. Furthermore, the gene cluster of the class III lanthipeptide flavipeptin encodes a prolyl oligogopeptidase-like protein (FlaP) that was shown to cleave only the fully modified FlaA precursor peptide after a proline residue at position −12, while the unmodified precursor is not accepted as substrate.77 As this cleavage occurs in the central region of the leader sequence, further aminopeptidase processing would be needed for full maturation. While class IV precursor peptides also contain a mostly conserved proline residue (Figure 2b), it remains to be seen if a likewise mechanism takes places, as no FlaP homolog is encoded in their biosynthetic gene clusters. The results of our co-expression experiments show that even in E. coli much of the leader peptide is removed in the absence of a dedicated protease.
Conclusion
In this study, we investigated class IV lanthipeptide systems from the GCF147 in actinobacteria. We uncovered new insights about the intricacies of the interactions of LanL processing enzymes and their corresponding precursor peptides. First, mutational analysis, performed in a co-expression system of His6-MBP-SgbA and untagged SgbL, provided evidence of the importance of the N-terminal leader section for enzymatic processing and furthermore hinted at the presence of an extended recognition motif that tolerates exchanges of larger portions to stretches of alanines. These findings were corroborated by FP binding studies, which did not only identify a minimal peptide that is still recognized by the lanthipeptide synthetase, but also revealed that recognition and binding of the precursor peptide is mediated by the kinase domain. Secondary structure prediction of the precursor peptide in light of these findings suggests that an α-helical region found in the center to N-terminal region of SgbA is involved in the recognition process.
Besides probing the interaction between precursor peptide and processing enzyme, new information was obtained about the biosynthesis of class IV lanthipeptides. In vitro assays with His6-SgbA and processing enzyme lacking the cyclization domain were utilized to identify that the order of the dehydration events follows an N- to C-terminal directionality. This study presents the first E. coli based class IV lanthipeptide production system, which will be helpful for future research. Not only does the rapid production of mature lanthipeptide in E. coli (expression for 1 day versus 10–14 days in native and heterologous Streptomyces producers56,57) and the easy genetic modification of the production plasmid allow mutational analysis to probe the interplay of precursor and processing machinery, it could also serve as platform for future investigations into the function of this family of compounds that is widespread in actinomycetes.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (R37 GM 058822 to W.A.V.) and the Deutsche Forschungsgemeinschaft (DFG Research Fellowship 309199717 to J.D.H.). A Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer used in this work was purchased in part with a grant from the National Institutes of Health (S10 RR027109 A).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and Supporting Figures and Tables (PDF).
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Nat Prod Rep. 2013;30:108. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Izawa M, Kawasaki T, Hayakawa Y. Appl Environ Microbiol. 2013;79:7110. doi: 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Heel AJ, Mu D, Montalban-Lopez M, Hendriks D, Kuipers OP. ACS Synth Biol. 2013;2:397. doi: 10.1021/sb3001084. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen GK, Wang S, Qiu Y, Hemu X, Lian Y, Tam JP. Nat Chem Biol. 2014;10:732. doi: 10.1038/nchembio.1586. [DOI] [PubMed] [Google Scholar]
- 5.Northfield SE, Wang CK, Schroeder CI, Durek T, Kan MW, Swedberg JE, Craik DJ. Eur J Med Chem. 2014;77:248. doi: 10.1016/j.ejmech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Nolan EM, Walsh CT. ChemBioChem. 2009;10:34. doi: 10.1002/cbic.200800438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umemura M, Nagano N, Koike H, Kawano J, Ishii T, Miyamura Y, Kikuchi M, Tamano K, Yu J, Shin-ya K, Machida M. Fungal Genet Biol. 2014;68:23. doi: 10.1016/j.fgb.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Weiz AR, Ishida K, Quitterer F, Meyer S, Kehr JC, Müller KM, Groll M, Hertweck C, Dittmann E. Angew Chem Int Ed. 2014;53:3735. doi: 10.1002/anie.201309721. [DOI] [PubMed] [Google Scholar]
- 9.Field D, Cotter PD, Hill C, Ross RP. Front Microbiol. 2015;6:1363. doi: 10.3389/fmicb.2015.01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegemann JD, Zimmermann M, Xie X, Marahiel MA. Acc Chem Res. 2015;48:1909. doi: 10.1021/acs.accounts.5b00156. [DOI] [PubMed] [Google Scholar]
- 11.Tsukui T, Nagano N, Umemura M, Kumagai T, Terai G, Machida M, Asai K. Bioinformatics. 2015;31:981. doi: 10.1093/bioinformatics/btu753. [DOI] [PubMed] [Google Scholar]
- 12.Schramma KR, Bushin LB, Seyedsayamdost MR. Nat Chem. 2015;7:431. doi: 10.1038/nchem.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding W, Liu WQ, Jia Y, Li Y, van der Donk WA, Zhang Q. Proc Natl Acad Sci USA. 2016;113:3521. doi: 10.1073/pnas.1522907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman MF, Vagstad AL, Piel J. Curr Opin Chem Biol. 2016;31:8. doi: 10.1016/j.cbpa.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Norris GE, Patchett ML. Curr Opin Struct Biol. 2016;40:112. doi: 10.1016/j.sbi.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Burkhart BJ, Schwalen C, Mann G, Naismith JH, Mitchell DA. Chem Rev. 2017;117:5389. doi: 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funk MA, van der Donk WA. Acc Chem Res. 2017;50:1577. doi: 10.1021/acs.accounts.7b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grove TL, Himes PM, Hwang S, Yumerefendi H, Bonanno JB, Kuhlman B, Almo SC, Bowers AA. J Am Chem Soc. 2017;139:11734. doi: 10.1021/jacs.7b01283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu W, Schmidt EW. Acc Chem Res. 2017;50:2569. doi: 10.1021/acs.accounts.7b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viehrig K, Surup F, Volz C, Herrmann J, Abou Fayad A, Adam S, Köhnke J, Trauner D, Müller R. Angew Chem Int Ed. 2017;56:7407. doi: 10.1002/anie.201612640. [DOI] [PubMed] [Google Scholar]
- 21.Repka LM, Chekan JR, Nair SK, van der Donk WA. Chem Rev. 2017;117:5457. doi: 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang T, Süssmuth RD. Acc Chem Res. 2017;50:1566. doi: 10.1021/acs.accounts.7b00159. [DOI] [PubMed] [Google Scholar]
- 23.Tran HL, Lexa KW, Julien O, Young TS, Walsh CT, Jacobson MP, Wells JA. J Am Chem Soc. 2017;139:2541. doi: 10.1021/jacs.6b10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith TE, Pond CD, Pierce E, Harmer ZP, Kwan J, Zachariah MM, Harper MK, Wyche TP, Matainaho TK, Bugni TS, Barrows LR, Ireland CM, Schmidt EW. Nat Chem Biol. 2018;14:179. doi: 10.1038/nchembio.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morinaka BI, Lakis E, Verest M, Helf MJ, Scalvenzi T, Vagstad AL, Sims J, Sunagawa S, Gugger M, Piel J. Science. 2018;359:779. doi: 10.1126/science.aao0157. [DOI] [PubMed] [Google Scholar]
- 26.Oman TJ, van der Donk WA. Nat Chem Biol. 2010;6:9. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11:564. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang RY, Thapa P, Espiritu MJ, Menon V, Bingham JP. Bioorg Med Chem. 2017;26:1135. doi: 10.1016/j.bmc.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 29.Camarero JA. Bioorg Med Chem Lett. 2017;27:5089. doi: 10.1016/j.bmcl.2017.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hetrick KJ, van der Donk WA. Curr Opin Chem Biol. 2017;38:36. doi: 10.1016/j.cbpa.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camarero JA. Bioorg Med Chem Lett. 2017;27:5089. doi: 10.1016/j.bmcl.2017.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Northfield SE, Wang CK, Schroeder CI, Durek T, Kan MW, Swedberg JE, Craik DJ. Eur J Med Chem. 2014;77:248. doi: 10.1016/j.ejmech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 33.Reyna-Gonzalez E, Schmid B, Petras D, Süssmuth RD, Dittmann E. Angew Chem Int Ed. 2016;55:9398. doi: 10.1002/anie.201604345. [DOI] [PubMed] [Google Scholar]
- 34.Walsh CT. ACS Chem Biol. 2014;9:1653. doi: 10.1021/cb5003587. [DOI] [PubMed] [Google Scholar]
- 35.Schwalen CJ, Hudson GA, Kosol S, Mahanta N, Challis GL, Mitchell DA. J Am Chem Soc. 2017;139:18154. doi: 10.1021/jacs.7b09899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai HC, Zakai UI, Mitchell DA. Nat Chem Biol. 2017;13:470. doi: 10.1038/nchembio.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding W, Li Y, Zhao J, Ji X, Mo T, Qianzhu H, Tu T, Deng Z, Yu Y, Chen F, Zhang Q. Angew Chem Int Ed. 2017;56:3857. doi: 10.1002/anie.201609948. [DOI] [PubMed] [Google Scholar]
- 38.LaMattina JW, Wang B, Badding ED, Gadsby LK, Grove TL, Booker SJ. J Am Chem Soc. 2017;139:17438. doi: 10.1021/jacs.7b08492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith TE, Pond CD, Pierce E, Harmer ZP, Kwan J, Zachariah MM, Harper MK, Wyche TP, Matainaho TK, Bugni TS, Barrows LR, Ireland CM, Schmidt EW. Nat Chem Biol. 2018;14:179. doi: 10.1038/nchembio.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sardar D, Hao Y, Lin Z, Morita M, Nair SK, Schmidt EW. J Am Chem Soc. 2017;139:2884. doi: 10.1021/jacs.6b12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freeman MF, Helf MJ, Bhushan A, Morinaka BI, Piel J. Nat Chem. 2017;9:387. doi: 10.1038/nchem.2666. [DOI] [PubMed] [Google Scholar]
- 42.Hegemann JD, De Simone M, Zimmermann M, Knappe TA, Xie XL, Di Leva FS, Marinelli L, Novellino E, Zahler S, Kessler H, Marahiel MA. J Med Chem. 2014;57:5829. doi: 10.1021/jm5004478. [DOI] [PubMed] [Google Scholar]
- 43.Yang X, Lennard KR, He C, Walker MC, Ball AT, Doigneaux C, Tavassoli A, van der Donk WA. Nat Chem Biol. 2018 doi: 10.1038/s41589-018-0008-5. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hetrick KJW, MC, van der Donk WA. ACS Cent Sci. 2018 doi: 10.1021/acscentsci.7b00581. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meindl K, Schmiederer T, Schneider K, Reicke A, Butz D, Keller S, Guhring H, Vertesy L, Wink J, Hoffmann H, Bronstrup M, Sheldrick GM, Süssmuth RD. Angew Chem Int Ed. 2010;49:1151. doi: 10.1002/anie.200905773. [DOI] [PubMed] [Google Scholar]
- 46.Jungmann NA, van Herwerden EF, Hügelland M, Süssmuth RD. ACS Chem Biol. 2016;11:69. doi: 10.1021/acschembio.5b00651. [DOI] [PubMed] [Google Scholar]
- 47.Völler GH, Krawczyk JM, Pesic A, Krawczyk B, Nachtigall J, Süssmuth RD. ChemBioChem. 2012;13:1174. doi: 10.1002/cbic.201200118. [DOI] [PubMed] [Google Scholar]
- 48.Wang H, van der Donk WA. ACS Chem Biol. 2012;7:1529. doi: 10.1021/cb3002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goto Y, Li B, Claesen J, Shi Y, Bibb MJ, van der Donk WA. PLoS Biol. 2010;8:e1000339. doi: 10.1371/journal.pbio.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Donk WA, Nair SK. Curr Op Struct Biol. 2014;29:58. doi: 10.1016/j.sbi.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortega MA, Hao Y, Walker MC, Donadio S, Sosio M, Nair SK, van der Donk WA. Cell Chem Biol. 2016;23:370. doi: 10.1016/j.chembiol.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ortega MA, Hao Y, Zhang Q, Walker MC, van der Donk WA, Nair SK. Nature. 2015;517:509. doi: 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B, Yu JP, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Science. 2006;311:1464. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 54.Dong SH, Tang W, Lukk T, Yu Y, Nair SK, van der Donk WA. eLife. 2015;4:e07607. doi: 10.7554/eLife.07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goto Y, Ökesli A, van der Donk WA. Biochemistry. 2011;50:891. doi: 10.1021/bi101750r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iftime D, Jasyk M, Kulik A, Imhoff JF, Stegmann E, Wohlleben W, Süssmuth RD, Weber T. ChemBioChem. 2015;16:2615. doi: 10.1002/cbic.201500377. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Q, Doroghazi JR, Zhao X, Walker MC, van der Donk WA. Appl Environ Microbiol. 2015;81:4339. doi: 10.1128/AEM.00635-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiu J, March PE, Lee R, Tillett D. Nucleic Acids Res. 2004;32:e174. doi: 10.1093/nar/gnh172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiu J, Tillett D, Dawes IW, March PE. J Microbiol Methods. 2008;73:195. doi: 10.1016/j.mimet.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 60.Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Nucleic Acids Res. 2013;41:W349. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Meer JR, Rollema HS, Siezen RJ, Beerthuyzen MM, Kuipers OP, de Vos WM. J Biol Chem. 1994;269:3555. [PubMed] [Google Scholar]
- 62.Neis S, Bierbaum G, Josten M, Pag U, Kempter C, Jung G, Sahl HG. FEMS Microbiol Lett. 1997;149:249. doi: 10.1111/j.1574-6968.1997.tb10337.x. [DOI] [PubMed] [Google Scholar]
- 63.Chen P, Qi FX, Novak J, Krull RE, Caufield PW. FEMS Microbiol Lett. 2001;195:139. doi: 10.1111/j.1574-6968.2001.tb10511.x. [DOI] [PubMed] [Google Scholar]
- 64.Patton GC, Paul M, Cooper LE, Chatterjee C, van der Donk WA. Biochemistry. 2008;47:7342. doi: 10.1021/bi800277d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagao J, Morinaga Y, Islam MR, Asaduzzaman SM, Aso Y, Nakayama J, Sonomoto K. Peptides. 2009;30:1412. doi: 10.1016/j.peptides.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 66.Müller WM, Ensle P, Krawczyk B, Süssmuth RD. Biochemistry. 2011;50:8362. doi: 10.1021/bi200526q. [DOI] [PubMed] [Google Scholar]
- 67.Plat A, Kluskens LD, Kuipers A, Rink R, Moll GN. Appl Environ Microbiol. 2011;77:604. doi: 10.1128/AEM.01503-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abts A, Montalbán-Lopez M, Kuipers OP, Smits SH, Schmitt L. Biochemistry. 2013;52:5387. doi: 10.1021/bi4008116. [DOI] [PubMed] [Google Scholar]
- 69.Khusainov R, Moll GN, Kuipers OP. FEBS Open Bio. 2013;3:237. doi: 10.1016/j.fob.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Escano J, Stauffer B, Brennan J, Bullock M, Smith L. MicrobiologyOpen. 2014;3:961. doi: 10.1002/mbo3.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khusainov R, Heils R, Lubelski J, Moll GN, Kuipers OP. Mol Microbiol. 2011;82:706. doi: 10.1111/j.1365-2958.2011.07846.x. [DOI] [PubMed] [Google Scholar]
- 72.Mavaro A, Abts A, Bakkes PJ, Moll GN, Driessen AJ, Smits SH, Schmitt L. J Biol Chem. 2011;286:30552. doi: 10.1074/jbc.M111.263210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wieckowski BM, Hegemann JD, Mielcarek A, Boss L, Burghaus O, Marahiel MA. FEBS Lett. 2015;589:1802. doi: 10.1016/j.febslet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 74.Koehnke J, Mann G, Bent AF, Ludewig H, Shirran S, Botting C, Lebl T, Houssen WE, Jaspars M, Naismith JH. Nat Chem Biol. 2015;11:558. doi: 10.1038/nchembio.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li K, Condurso HL, Li G, Ding Y, Bruner SD. Nat Chem Biol. 2016;12:973. doi: 10.1038/nchembio.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krawczyk B, Völler GH, Völler J, Ensle P, Süssmuth RD. ChemBioChem. 2012;13:2065. doi: 10.1002/cbic.201200417. [DOI] [PubMed] [Google Scholar]
- 77.Völler GH, Krawczyk B, Ensle P, Süssmuth RD. J Am Chem Soc. 2013;135:7426. doi: 10.1021/ja402296m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.