

Abstract
Mu opioid receptors (MORs) are widely distributed throughout brain reward circuits and their role in drug and social reward is well established. Substantial evidence has implicated MOR and the endogenous opioid system in alcohol reward, but circuit mechanisms of MOR-mediated alcohol reward and intake behavior remain elusive, and have not been investigated by genetic approaches. We recently created conditional knockout (KO) mice targeting the Oprm1 gene in GABAergic forebrain neurons. These mice (Dlx-MOR KO) show a major MOR deletion in the striatum, whereas receptors in midbrain (including the Ventral Tegmental Area or VTA) and hindbrain are intact. Here, we compared alcohol-drinking behavior and rewarding effects in total (MOR KO) and conditional KO mice. Concordant with our previous work, MOR KO mice drank less alcohol in continuous and intermittent two-bottle choice protocols. Remarkably, Dlx-MOR KO mice showed reduced drinking similar to MOR KO mice, demonstrating that MOR in the forebrain is responsible for the observed phenotype. Further, alcohol-induced conditioned place preference was detected in control but not MOR KO mice, indicating that MOR is essential for alcohol reward and again, Dlx-MOR KO recapitulated the MOR KO phenotype. Taste preference and blood alcohol levels were otherwise unchanged in mutant lines. Together, our data demonstrate that MOR expressed in forebrain GABAergic neurons is essential for alcohol reward-driven behaviors, including drinking and place conditioning. Challenging the prevailing VTA-centric hypothesis, this study reveals another mechanism of MOR-mediated alcohol reward and consumption, which does not necessarily require local VTA MORs but rather engages striatal MOR-dpendent mechanisms.
Keywords: mu opioid receptor, alcohol intake, forebrain GABAergic neurons
Introduction
Alcohol is the most consumed addictive substance worldwide and can lead to alcohol addiction. As for every substance use disorder, alcoholism is a progressive brain disorder involving the transition from recreational or moderate use to loss of control over drug consumption. It is well recognized that alcohol triggers opioid peptide release, and that endogenous opioid peptides and opioid receptors are involved in both acute alcohol effects and alcohol dependence (Nutt, 2014). In fact, pharmacological modulation of the opioid system is considered an effective approach to treat alcoholism, and one of the few currently available treatments for alcohol use disorders uses mu opioid receptor (MOR) blockade as a strategy. Naltrexone and more recently nalmefene, two long-acting antagonists that are shown to occupy MORs in human [11C] carfentanyl PET studies, significantly reduce drinking, craving and relapse in heavy alcohol drinkers (Drobes et al., 2003; Soyka et al., 2016) and compounds with higher MOR selectivity are currently under development (Ripley et al., 2015). A mechanism proposed to explain clinical efficacy of these compounds is that MOR blockade prevents MOR-mediated reward normally elicited by alcohol-induced endogenous opioid peptides, and this reduction of alcohol rewarding effect ultimately leads to diminished craving for alcohol and reduced risk of relapse (Nutt, 2014).
In preclinical research, gene targeting has definitely established the essential role of MORs in mediating drug (Charbogne et al., 2014) and natural (Becker et al., 2014; Moles et al., 2004) rewards. Others and we have demonstrated that rewarding properties of both opiates (Contet et al., 2004; Matthes et al., 1996; Roberts et al., 2000) and non-opioid drugs of abuse (Contet et al., 2004), including alcohol, are abolished in constitutive knockout mice lacking the Oprm1 gene (MOR KO mice). In particular, MOR KO mice do not self-administer alcohol, drink less alcohol, show lower alcohol preference and reduced alcohol place conditioning than their wild type counterparts (Hall et al., 2001; Roberts et al., 2000), indicating that MORs are necessary for rewarding properties of alcohol. Further, knockout mice lacking enkephalins and beta-endorphin show no elevation of stress-induced alcohol drinking (Racz et al., 2008), demonstrating altogether that endogenous opioid mechanisms involving MORs contribute to alcohol consumption, and thus substantiating the large pharmacological literature (Nutt, 2014). Circuit mechanisms underlying this particular MOR function, however, remain an open question and have not been investigated by genetic approaches.
MOR is broadly expressed in the nervous system (Erbs et al., 2015) including reward (Le Merrer et al., 2009) and addiction-related circuitries (Koob and Volkow, 2016). The dopamine (DA) mesolimbic circuitry is considered central to reward and motivational processes. Alcohol is self-administered in the ventral tegmental area (VTA) (Gatto et al., 1994) where DA neurons originate, and stimulates DA release in the ventral striatum (or nucleus accumbens, NAc) that receives DA inputs (Gonzales et al., 2004). Alcohol also directly regulates activity of the NAc, considered another hotspot for alcohol activity (see (Spanagel, 2009)), and MOR is present in both VTA and NAc. Hence, behavioral studies show that several sites of the mesolimbic DA system are key for the acquisition of alcohol self-administration and, interestingly, evidence also suggests that additional brain areas contribute to alcohol consummatory behaviors including the hypothalamus, amygdala and prefrontal cortex (Barson et al., 2012). In sum, MOR-mediated alcohol reward and drinking may operate at the level of the VTA through disinhibition of DA neurons (Fields and Margolis, 2015), or in the striatum that expresses high levels of endogenous opioid peptides, or even outside the DA mesolimbic system including for example the habenular circuitry where MORs are most densely expressed (Gardon et al., 2014) and modulate reward/aversion networks (Mechling et al., 2016).
Here we tested whether MORs expressed at the level of the striatum are necessary for the development of alcohol drinking behavior and alcohol reward. We took advantage of a conditional MOR knockout mouse line (Dlx-MOR KO mice), which we recently developed using a Dlx5/6-Cre driver line targeting GABAergic neurons of the forebrain (Monory et al., 2006). Dlx-MOR KO mice show a predominant and almost complete MOR deletion in the striatum, whereas MORs expressed in the VTA are intact ((Charbogne et al., 2017) and Figure 1). We therefore compared Dlx-MOR KO mice and total MOR KO mice with their respective controls for voluntary alcohol consumption in continuous and intermittent two-bottle choice paradigms, and for alcohol rewarding effect using conditioned place preference test. Our data show that Dlx-MOR KO mice fully recapitulate the behavioral phenotype of total MOR KO mice in these tests, demonstrating unambiguously that MORs expressed in GABAergic forebrain neurons are essential for alcohol reward and drinking behavior.
Figure 1. Anatomical distribution of the MOR deletion in Dlx-MOR KO mice.
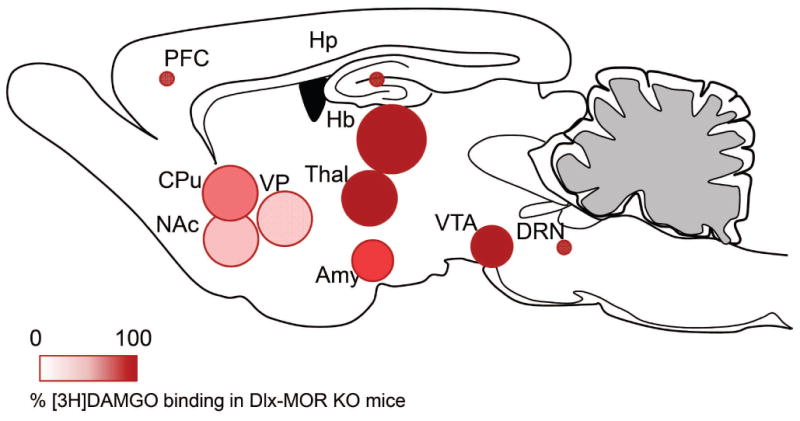
The scheme shows the quantification of MOR binding levels throughout the brain using [3H]DAMGO binding autoradiography in areas with sufficiently high signal (≥60 fmoles/mg tissue) adapted from (Charbogne et al., 2017). Circle size represents MOR density in control mice, red color intensity represents decrease of MOR agonist binding in Dlx-MOR KO mice (from dark red to white: 0% to 100% decrease). In Dlx-MOR KO mice, autoradiograms show strong receptor deletion in the NAc, VP and CPu, three structures that show abundant MOR expression in control mice. Binding is slightly decreased (Amy) or remains intact in other high-MOR (Hb, Thal, VTA) or low-MOR expressing structures (PFC, Hp, DRN). Abbreviations: Amy, Amygdala; CPu, Caudate Putamen; DRN, dorsal raphe nucleus; Hb, Habenula; Hp, Hippocampus; PFC, Prefrontal cortex; NAc, Nucleus Accumbens; Thal, Thalamus; VP, ventral pallidum; VTA, ventral tegmental area.
Material and Methods
Animals
MOR KO line. Male MOR KO mice and their wild-type controls (MOR Ctl) were bred in-house on a hybrid 50% 129SvPas - 50% C57Bl/6J background (Matthes et al., 1996).
Dlx-MOR KO line. Briefly, the floxed Oprm1 mouse line (Oprm1fl/fl) harboring exons 2 and 3 of the MOR gene flanked by loxP sites, previously reported by our group (Weibel et al., 2013), was crossed with the Dlx5/6-Cre driver transgenic Cre mouse line (obtained from Beat Lutz laboratory, Institute of Physiological Chemistry, Johannes Gutenberg University, Germany). Cre-positive conditional mutant animals (Dlx5/6-Cre-Oprm1fl/fl) were obtained (hereafter named Dlx-MOR KO) and Cre-negative animals (Oprm1fl/fl) were used as controls (hereafter named Dlx-MOR Ctl). Genetic background of Dlx-MOR KO and their corresponding controls is 63% C57BL/6J-37% 129SvPas.
Animals studied in two-bottle choice paradigm were housed individually under a 12 h reversed light / dark cycle, whereas animals used for the other experiments were group-housed (3-5 animals per cage) under a 12 h light/dark cycle. In both cases, the mice were 3-5 months old and weighed 25-35 g at the time of the experiments. Temperature and humidity were controlled and food and water were available ad libitum.
All animals procedures in this report were conducted in accordance with (i) the European Communities Council Directive of 24 November 1986 (86/609/EEC) and approved by both the Comité Régional d'Ethique en Matière d'Expérimentation Animale de Strasbourg (CREMEAS, 2003-10-08-[1]-58) and the local ethical comity (Comité d'Ethique en Experimentation Animale IGBMC-ICS, Com'Eth) or (ii) the guidelines set forth by the Canadian Council of Animal Care and by the Animal Care Committees of McGill University/Douglas Mental Health University Institute.
Drugs and treatments
Alcohol solution for the drinking experiments was prepared from absolute anhydrous alcohol diluted to 10 or 20% alcohol (v/v) in tap water, whereas alcohol solution for systemic administration was diluted to 20% alcohol (v/v) in saline.
Behavioral procedures
First set of experiments
Two-bottle choice – continuous access
Oral alcohol intake was determined using continuous access to alcohol in a two-bottle choice drinking paradigm. Drinking sessions were conducted 24h a day during 14 consecutive days, with one bottle containing tap water, while the other contained alcohol diluted to 10% alcohol (v/v) in tap water. The bottles were weighed every day and the mice were weighed at the beginning of the experiment. The position (left or right) of each solution was alternated between sessions, as a control for side preference. Possible loss of solution due to bottle handling was controlled by weighing bottles in empty cages.
Second set of experiments
Two-bottle choice – intermittent access
New groups of MOR KO and Dlx-MOR KO mice and their corresponding controls were first given continuous access to a solution of 10% alcohol for 4 days as described above (Fig. S1). Next, animals were tested for intermittent access to 20% alcohol in two-bottle choice procedure. Animals were given 24h of concurrent access to one bottle of 20% alcohol (v/v) in tap water and another bottle of water starting at 12 p.m. on Monday, Wednesday and Friday with 24 or 48h alcohol-deprivation periods (only water available) between the alcohol-drinking sessions. Water and alcohol bottles were weighed after 24 h of access.
Quinine and saccharin consumption
One week after the alcohol-drinking study, mice were tested for saccharin (0.066%) (sweet) or quinine (0.06 mM) (bitter) intake. Each solution was offered for 3 days and the amount of fluid intake and preference were recorded every day.
Third set of experiments
Conditioned place preference (CPP)
CPP was assessed in 8 place preference boxes (Panlab, Harvard Apparatus, Spain). Each box consists of two chambers (20 × 18 × 25 cm) with distinct visual and tactile cues separated by a clear acrylic rectangular corridor. On day 1, mice were given access to the entire apparatus for 20 min (preconditioning). For the conditioning phase we used a biased CPP design in which alcohol treatment was associated with the less preferred compartment during the preconditioning test. On day 2, conditioning training started with one conditioning trial per day for 6 days as follows: mice were administered (i.p.) saline solution and confined immediately to one of the compartments for 5 min (saline-paired compartment). The next day, mice were administered saline solution (saline group) or alcohol (1.8 g/Kg, 20% v/v, alcohol group) and were confined to the other compartment (drug-paired compartment). This schedule was repeated three times until day 7. On day 8, animals were allowed to explore the entire apparatus for 20 min (post-conditioning test).
Blood alcohol concentrations
An independent animal cohort was used for this experiment. Animals were given a single dose of alcohol (3.2 g/kg, 20% v/v, i.p.) and blood samples were taken from the tail at 10, 60 and 120 minutes after injection. Serum was extracted with 3.4% trichloroacetic acid followed by a 5-min centrifugation at 420 × g and assayed for alcohol content using the NAD+-NADH enzyme spectrophotometric method. BACs were determined using a standard calibration curve.
Statistical analysis
Data were analyzed using unpaired t-test or two or three-way ANOVA with or without repeated measures (RM-ANOVA). Significant main effects and interactions of the ANOVAs were further investigated with the pairwise comparisons (Bonferroni t-test). Statistical significance was set at p<0.05.
Results
Moderate alcohol drinking is similarly reduced in total MOR KO and Dlx-MOR KO mice
Our previous study showed that MOR KO mice consume less alcohol and more water than control animals using continuous access to 10% alcohol in a two bottle-choice procedure, leading to significantly lower alcohol preference (Roberts et al., 2000). Here, we used the same continuous alcohol access protocol to compare drinking behaviors of total MOR KO and Dlx-MOR KO mice, with their respective controls.
As shown in Fig. 2, both MOR KO and Dlx-MOR displayed reduced levels of alcohol intake compared to their corresponding controls. Two way ANOVA with repeated measures (RM) showed significant main effects of Genotype for (MOR KO, Fig. 2A left panel: F(1, 17)=4.5, p=0.049; Dlx-MOR KO, Fig. 2B left panel: F(1,19)=4.6, p=0.048). A significant main effect of Sessions was observed for MOR KO (F(13,221)=4.5, p<0.001) and Dlx-MOR (F(13,247)=6.99, p<0.001). No significant interaction between Genotype × Session was seen for both genotypes (MOR KO: F(13,221)=0.26, p=0.99; Dlx-MOR KO: F(13,247)=1.09, p=0.37). For each Fig. 2A and Fig. 2B, the right panel depicts mean daily alcohol intake during the experiment and showed a significant decrease of alcohol intake by 37% in MOR KO (t17=2.1, p<0.05) and 36% in Dlx-MOR KO mice (t19=2.1, p<0.05) compared to control animals. Water intake was unchanged for both genotypes (Fig. 2C and 2D). Consequently, as shown in Fig. 2E and 2F right panels, preference for alcohol was also reduced by 33% in MOR KO (Fig. 2E: t17=2.37, p=0.03) and 37% in Dlx-MOR KO mice (Fig. 2F: t19=2.3, p=0.03) compared to their control counterparts. Two-way ANOVA RM showed a significant Genotype effect (MOR KO Fig. 2E left panel: F(1,17)=5.6, p=0.03; Dlx-MOR KO Fig. 2F left panel: F(1,19)=5.5, p=0.03) and Session effect (MOR KO: F(13,221)=3.6, p<0.001; Dlx-MOR KO: F(13,247)=7.1, p<0.001) but no significant Genotype × Session interaction effect (MOR KO: F(13,221)=0.69, p=0.77; Dlx-MOR KO: F(13,247)=1.67, p=0.07).
Figure 2. MOR KO and Dlx-MOR KO mice show reduced moderate alcohol consumption.
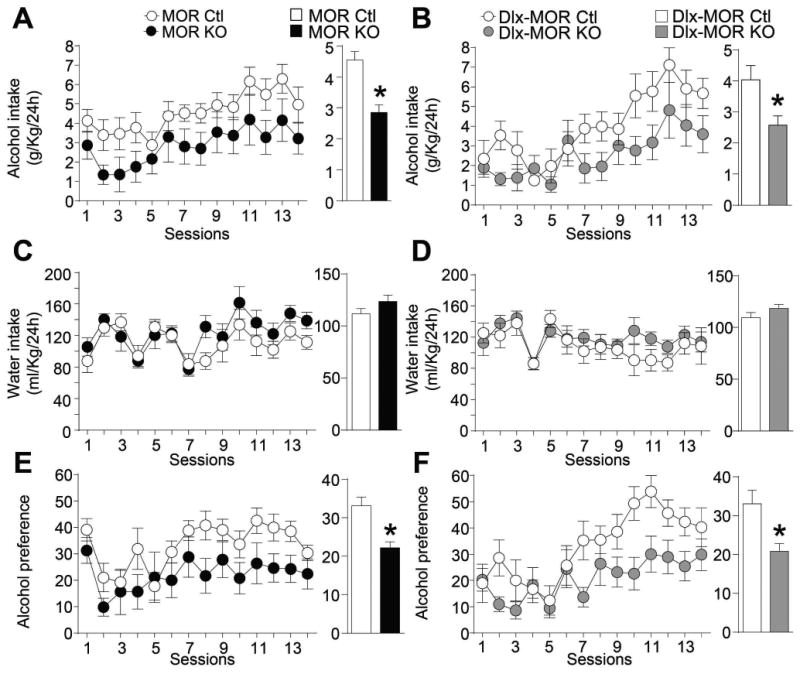
MOR KO (A) and Dlx-MOR KO (B) mice consume less alcohol than their corresponding controls in an alcohol continuous-access 2-bottle-choice drinking paradigm. Animals were offered access to alcohol (10% v/v) (A-B) and water (C-D) in their home cages for 14 consecutive days (14 sessions). Values are presented as the daily mean g/kg of alcohol intake (±SEM) and ml/kg of water intake (±SEM), respectively. Alcohol preference (E-F) was calculated by dividing the total alcohol solution consumed by total fluid (alcohol plus water) consumption. Left panels represent the mean (±SEM) of alcohol and water consumption or alcohol preference per session; Right Panels represent mean (±SEM) of daily alcohol and water consumption or alcohol preference during the entire experiment. (A, C, E) n = 9-10, (B, D, F) n=9-12 for each group. *p < 0.05.
Taken together, data from MOR KO mice first confirm our previous results (Roberts et al., 2000) demonstrating that MORs contribute to moderate alcohol consumption. Further, the strongly reduced alcohol drinking phenotype was similarly observed in both MOR KO and Dlx-MOR KO mice, and we therefore conclude that MORs expressed in forebrain GABAergic neurons are responsible for this phenotype.
Intermittent high alcohol drinking is similarly reduced in total MOR KO and Dlx-MOR KO mice
It is acknowledged that continuous 24-hour free choice access to alcohol does not lead to voluntary alcohol intake at levels sufficient to induce intoxication, or to engender dependence. A number of procedures have been designed to trigger “excessive” levels of voluntary alcohol drinking in rodents that more closely mimic heavy drinking in humans, including the modification of temporal availability of the drug from continuous to intermittent (Becker, 2013). We therefore further tested a 20% alcohol intermittent two-bottle choice drinking procedure, to determine whether the total and/or conditional MOR deletion would also affect excessive alcohol intake.
To do so, new mice (second set of experiments) were first tested in continuous access to 10% alcohol in a two bottle-choice procedure for 4 days to confirm our above described phenotype (see Fig. S1) and then switched to intermittent 20 % alcohol access over four weeks. As shown in Fig. 3A and 3B (right panels), this procedure indeed led to an escalation of mean daily alcohol intake for all the four groups, compared to continuous access procedure (MOR KO +65% and their corresponding controls +69%; Dlx-MOR KO mice +56% and their controls +54%). Importantly, and as for continuous access drinking, alcohol intake was remarkably lower in both total KO and those lacking MORs in GABAergic forebrain neurons compared to their respective controls (Fig. 3A and 3B left panels). Two way ANOVA with RM showed significant main effects of Genotype (MOR KO Fig. 3A left panel: F(1,24)=30.3, p<0.001; Dlx-MOR KO Fig. 3B left panel: F(1,45)=17.7, p<0.001) and Sessions (MOR KO: F(11,264)=3.6, p<0.001; Dlx-MOR KO : F(10,450)=3.4, p<0.001) but no significant Genotype × Session interaction effect (MOR KO: F(11,264)=0.8, p=0.61; Dlx-MOR KO : F(10,450)=0.9, p=0.58). Analyses using the method of contrasts revealed a significant difference for all of the sessions (p's <0.05) except for session 2 for MOR KO mice, as well as significant difference for sessions 3, 4, 5, 6, 7, 8, 9, 10 and 11 (p's<0.05) for Dlx-MOR KO animals. The right panels of Fig. 3A and 3B represent the average of daily alcohol intake and show a significant decrease of 55% in MOR KO (t24=5.5, p<0.001) and 39% in Dlx-MOR KO (t45=4.2, p<0.001) mice compared to their corresponding controls. As observed above in the moderate access paradigm, there was no difference in water consumption for both mouse genotypes (Fig. 3C and 3D). Consequently, preference for alcohol was reduced by 49% in MOR KO (Fig. 3E: t24=6.3, p<0.001) and by 30% in Dlx-MOR KO (Fig. 3F: t45=2.9, p=0.0052) compared to their control groups. Two way ANOVA with RM showed significant main effects of Genotype (MOR KO Fig. 3E left panel: F(11,264)=39.8, p<0.001; Dlx-MOR Fig. 3F left panel: F(1,45)=8.6, p=0.005) and Sessions (MOR KO: F(11,264)=3.01, p<0.001; Dlx-MOR KO : F(10,450)=6.99, p<0.001) but no significant Genotype × Session interaction effect (MOR KO: F(11,264)=0.4, p=0.9; Dlx-MOR KO: F(10,450)=0.5, p=0.88). Analyses using the method of contrasts showed a significant difference across all sessions for MOR KO, as well as a significant difference in session 3, 7, 9 (p's<0.05) plus a trend in sessions 6 and 11 (p's=0.06) for Dlx-MOR KO animals.
Figure 3. MOR KO and Dlx-MOR KO mice show reduced excessive alcohol consumption.
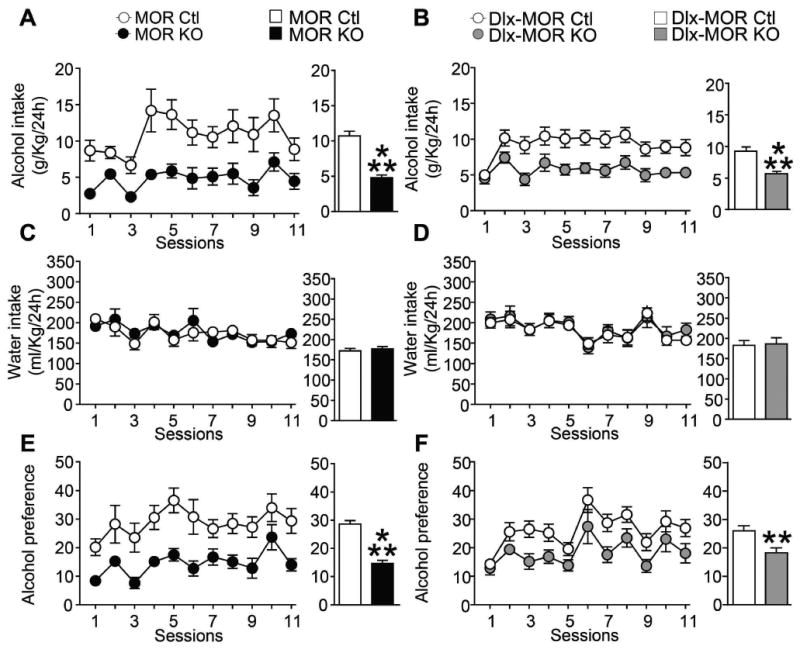
MOR KO (A) and Dlx-MOR KO (B) mice consume less alcohol than their corresponding controls in a 20% alcohol intermittent-access 2-bottle-choice drinking paradigm. Animals were offered access to alcohol (20% v/v) (A-B) and water (C-D) in their home cages for 4 weeks (11 sessions). Values are presented as the daily mean g/kg of alcohol intake (±SEM) and ml/kg of water intake (±SEM), respectively. Alcohol preference (E-F) was calculated by dividing the total alcohol solution consumed by total fluid (alcohol plus water) consumption. Left panels represent the mean (±SEM) of alcohol and water consumption or alcohol preference per session; Right Panels represent mean (±SEM) of daily alcohol and water consumption or alcohol preference during the entire experiment. (A, C, E) n = 12-14, (B, D, F) n=19-28 for each group. **p < 0.01; ***p< 0.001.
Together these results demonstrate that MORs, which contribute to moderate drinking, also significantly contribute to excessive alcohol drinking. Further, as for moderate drinking, the comparable phenotype of MOR KO and Dlx-MOR KO mice in this procedure indicates that MORs expressed in forebrain GABAergic neurons are essential for this behavior.
Taste palatability and alcohol metabolism are intact in MOR KO and Dlx-MOR mice
Diminished alcohol drinking in the two groups of mutant mice could be a consequence of altered perception of alcohol flavor (Bachmanov et al., 2003). One week after (moderate and excessive alcohol-drinking experiments were terminated, a group of animals from each genotype were tested for saccharin (0.066%) (sweet) or quinine (0.06 mM) (bitter) intake. Both MOR KO and Dlx-MOR KO mice, as well as their corresponding control animals, drank similar amounts of saccharin (MOR KO: Fig. 4A: t24=1.5, p=0.15; Dlx-MOR: Fig. 4C: t23=0.5, p=0.6) and quinine (MOR KO: Fig. 4E: t24=0.42, p=0.67; Dlx-MOR: Fig. 4G: t23=0.43, p=0.7) and expressed an equivalent high preference for saccharin (MOR KO: Fig. 4B: t24=0.9, p=0.36; Dlx-MOR: Fig. 4D: t23=0.51, p=0.62) and low preference for quinine (MOR KO: Fig. 4F: t24=0.38, p=0.7; Dl x-MOR: Fig. 4H: t23=0.74, p=0.46). This result indicates that neither the total nor the conditional MOR deletion in forebrain GABAergic neurons affects quinine and saccharin consumption, suggesting that taste palatability is unchanged.
Figure 4. MOR KO and Dlx-MOR KO mice show intact taste palatability and alcohol metabolism.
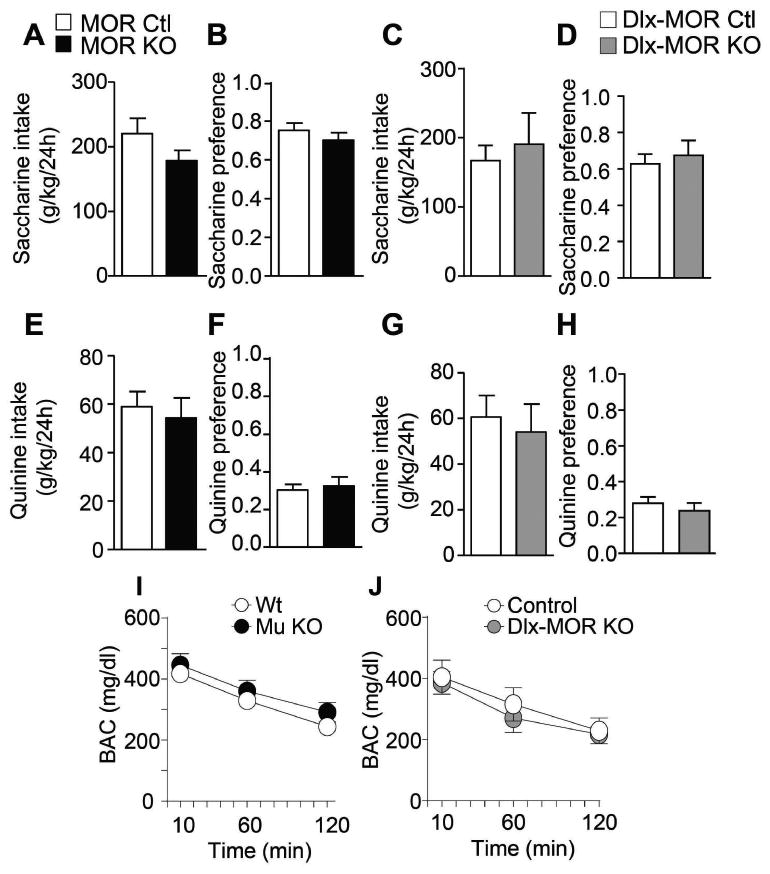
(A-H) No difference in consumption (A, C, E, G) or preference (B, D, F, H) for sweet (saccharin; A-D) or bitter (quinine, E-H) solutions between MOR KO and Dlx-MOR KO compared to their corresponding controls. Data are mean ± SEM of daily fluid intake in g/kg. (I-J) MORs KO and Dlx-MOR KO mice showed similar blood alcohol concentrations after injection of alcohol compared with WT controls. Data represent level of blood alcohol in mg/dL. (A-H) MORs KO, n = 12-14; Dlx-MOR KO, n =9-16 for each group. (I-J), n = 6-7 for each group.
Another factor influencing alcohol-drinking levels is alcohol metabolism. We thus used a separate animal cohort to measure blood alcohol levels in mutant mice and their controls after acute alcohol administration. As shown in Fig. 4I and 4J, there was no difference in blood alcohol levels between MOR KO mice and their controls, as well as Dlx-MOR KO and their corresponding controls. Two way ANOVA with RM showed no effect of Genotype (MOR KO: F(1,11)=0.86, p=0.37; Dlx-MOR KO : F(1,11)=0.01, p=0.89), a significant effect of Time (MOR KO: F(2,22)=7.9, p=0.002; Dlx-MOR KO : F(2,22)=56.62, p<0.001) and no significant interactions (MOR KO: F(2,22)=0.96, p=0.39; Dlx-MOR KO : F(2,22)=0.49, p=0.61). This result shows that alcohol clearance is not affected by the MOR deletion, either total or conditional, indicating that the lesser alcohol consumption in both MOR KO and Dlx-MOR KO mice is not due to differences in alcohol metabolism.
Conditioned place preference to alcohol is undetectable in MOR KO and Dlx-MOR mice
A most likely explanation for diminished voluntary alcohol intake in mutant mice is that rewarding effects of alcohol are reduced in these animals, as proposed in our (Roberts et al., 2000) and other (Hall et al., 2001) earlier studies. To substantiate this hypothesis, we re-tested the expression of alcohol-induced conditioned place preference (CPP) in MOR KO mice, and then compared MOR KO mice with Dlx-MOR KO mice in this test.
As shown in Fig. 5, both the total and the conditional MOR deletion resulted in a complete loss of alcohol-induced CPP. Data for MOR KO mice are shown in Fig. 5A. Three way ANOVA comparing place preference between preconditioning and post-conditioning sessions revealed significant Treatment effect (F(1,66)=8.1, p=0.006), Session effect (F(1,66)=8.2, p=0.005), Session × Treatment interaction (F(1,66)=5.5, p=0.02) and Genotype × Session × Treatment interaction (F(1,663)=4.2, p=0.04). Subsequent pairwise comparisons (Bonferroni t-test) indicated that control mice spent significantly more time in the alcohol-associated environment during post-conditioning compared to pre-conditioning phase (p<0.001). This effect was not observed in MOR KO. Data for Dlx-MOR KO mice are shown in Fig. 5B. Three way ANOVA comparing place preference between preconditioning and post-conditioning sessions revealed significant Treatment effect (F(1,160)=9.93, p=0.002), Session effect (F(1,160)=9.96, p=0.002), Genotype × Treatment interaction (F(1,160)=4.47, p=0.036) Session × Treatment interaction (F(1,160)=5.8, p=0.017), Genotype × Session interaction (F(1,160)=9.46, p=0.002) and no significant Genotype × Session × Treatment interaction (F(1,160)=1.97, p=0.16). Analyses using the method of contrasts indicated that control mice, but not Dlx-MOR KO mice, spent more time in the alcohol-associated compartment during the post-conditioning phase vs. pre-conditioning (p<0.001).
Figure 5. MOR KO and Dlx-MOR mice lack alcohol-induced conditioned place preference (CPP).
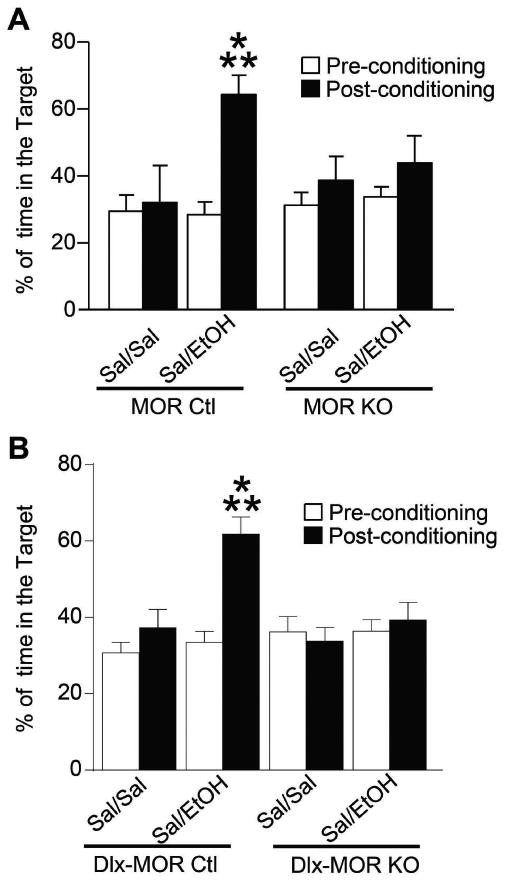
MOR KO (A) and Dlx-MOR KO (B) mice do not develop alcohol-induced CPP. The habituation day (pre-conditioning; day 1) was designed to evaluate drug-free baseline preference for the compartments. Animals were placed in the neutral compartment and had free access to the entire apparatus for 20 min. Next, during the conditioning phase (6 days), animals were daily administered (i.p.) alcohol (1.8 g/kg) or saline solution and were then confined in the drug- or non-drug-paired compartment for 5 min. 24 hours after the last conditioning day, mice went through the post-conditioning test which was similar to the habituation phase. Data represent percentage of total test time spent in the alcohol- or saline- paired compartment during the pre-conditioning and the post-conditioning tests. Data represent mean (± SEM). (A) n = 7-12, (B) n=19-23 for each group. ***p < 0.001.
These data first confirm that MOR is essential for the expression of place preference to alcohol, and the lack of detectable alcohol CPP under our experimental conditions suggests that reduced moderate and excessive alcohol drinking in these mice both result from lower alcohol reward MOR (see discussion). Second, the lack of detectable alcohol CPP in both MOR KO and Dlx-MOR KO mice demonstrates that MORs expressed in GABAergic forebrain neurons are responsible for this behavioral alcohol phenotype.
Discussion
In our study, MOR KO mice show reduced moderate and excessive alcohol drinking, and conditioned place preference to alcohol was undetectable under our experimental conditions, strengthening previous evidence that the MOR is essential for alcohol reward-driven behavioral responses. Further, our data show that Dlx-MOR KO and MOR KO mice show a comparable phenotype in all the behavioral experiments, demonstrating for the first time that MORs expressed in GABAergic forebrain neurons, i. e. predominantly in the striatum (Fig. 1), are responsible for MOR-mediated alcohol reward. This study therefore identifies a circuit mechanism underlying alcohol reward and consumption, which engages endogenous MOR-mediated neurotransmission at the level of striatal networks.
The reduction of moderate alcohol drinking in MOR KO mice, in this study, confirms previous evidence showing that MOR KO mice do not self-administer alcohol under several conditions, including oral self-administration and the continuous two-bottle choice procedure (Roberts et al., 2000). Our further observation that MOR KO mice also show reduced alcohol drinking in the intermittent alcohol access procedure is novel, and indicates that MOR-driven alcohol intake remains a prominent mechanism after animals have shifted from moderate to excessive alcohol consumption. While neurobiological mechanisms driving increases in free-choice alcohol consumption in this procedure are not completely understood, evidence for neuroadaptations at the level of several signaling pathways and at distinct sites of the reward system have been described (see (Carnicella et al., 2014). It is interesting to note that MOR remains a key player for alcohol intake even when neurochemical and circuit modifications have developed in response to higher levels of alcohol exposure. MOR therefore plays a major role in several aspects of alcohol consumption and dependence, including the transition from the controlled/moderate drinking to excessive/compulsive use. This is concordant with human data reporting that naltrexone, a fairly low selective MOR antagonist, reduces heavy drinking (Balldin et al., 2003; Guardia et al., 2002; Gueorguieva et al., 2010; Pettinati et al., 2006) and shows significant efficacy to treat alcohol use disorders (Nutt, 2014), and supports on-going strategies to develop specific MOR antagonists (Ripley et al., 2015).
Our study reveals that MORs expressed in GABAergic forebrain neurons are responsible for MOR-mediated alcohol reward-driven behavior. Brain sites associated with alcohol reward have been identified throughout the corticomesolimbic dopaminergic pathway originating in the VTA and projecting to the NAc, prefrontal cortex and amygdala. The VTA, in return, is regulated by GABAergic and enkephalinergic projections from the ventral pallidum and NAc, as well as glutamatergic afferents from the prefrontal cortex (Sesack and Grace, 2010). MORs and opioid peptides are weakly present in cortical areas, but are strongly expressed at striatal and VTA levels (Erbs et al., 2015; Le Merrer et al., 2009), and can therefore regulate alcohol reward and drinking at both sites. Our previous molecular characterization of Dlx-MOR KO mice indicate that the conditional mutants lack receptors predominantly in striatal GABAergic neurons, both at the level of medium spiny neuron cell bodies and at their terminals within the ventral pallidum and VTA, while MORs expressed locally in the VTA are preserved (Charbogne et al., 2017). Thus, our finding that this particular MOR population is responsible for MOR-driven alcohol reward is remarkable for two reasons, first because we identify a main circuit mechanism for alcohol reward, and second because the data suggest that MORs in the VTA do not substantially contribute to this MOR function.
The latter conclusion is at odds with the classical disinhibition hypothesis of alcohol reward (see Rev (Spanagel, 2009). In this model, alcohol exposure would trigger endogenous opioid release in the VTA, which in turn would activate MORs expressed in VTA local GABAergic interneurons, remove an inhibitory tone on dopamine neurons and promote enhanced dopamine release in the NAc, contributing to rewarding properties of alcohol (see (Spanagel and Weiss, 1999). In support of this model, acute alcohol administration increased beta-endorphin release in the VTA (Hall et al., 2001; Jarjour et al., 2009). Further, VTA electrophysiology in acute midbrain slices from rats indicated that alcohol enhances MOR agonist-induced facilitation of dopamine neuron firing and interacts with MOR agonists/antagonists to regulate GABAergic transmission and plasticity (see (Guan and Ye, 2010) and references therein). Finally, MOR signaling and levels of the GRK2 protein were increased in the lower midbrain upon alcohol treatment (Shibasaki et al., 2013). Our study suggests that, despite evidence for reciprocal MOR/alcohol effects locally in the VTA, the mechanism that would involve MOR-mediated disinhibition of dopamine neurons in the VTA is not the only mechanism contributing to rewarding effects of alcohol and the development of alcohol drinking behavior.
Our demonstration that striatal MORs are required for alcohol reward and drinking is otherwise consistent with previous pharmacological evidence that the NAc is critical site for the reinforcing and rewarding property of alcohol (reviewed in (Spanagel, 2009). Notably, and with regards to involvement of the opioid system, a moderate alcohol dose elevates levels of the MOR-preferring opioid peptide beta-endorphin (Lam et al., 2010) and Met-enkephalin (Marinelli et al., 2006) targeting the kappa opioid receptor and aversive responses. Further, intra-accumbal infusions of MOR agonists and antagonists enhances or decreases alcohol consumption intake and preference, respectively, in rat studies (Barson et al., 2009; Heyser et al., 1999; Hyytia and Kiianmaa, 2001; Nealey et al., 2011). In humans, several fMRI studies indicate that brain responses to alcohol cues are associated to the OPRM1 A118G variant in ventral and dorsal striatum, the ventromedial prefrontal and the orbitofrontal cortex (Bach et al., 2015; Filbey et al., 2008; Ray et al., 2014). Further, a recent report using [11C]carfentanyl PET imaging showed correlated MOR occupancy/opioid release in both NAc and orbitofrontal cortex upon alcohol drinking, and changes in [11C]carfentanyl binding in the cortex correlated with heavy drinking (Mitchell et al., 2012). These studies reveal an intriguing striato-cortical MOR implication in alcohol use disorder, consistent with the importance of MORs in the forebrain in our study. Together therefore, and beyond prior evidence, our data establish the causality between MOR activation in GABAergic forebrain neurons of the striatum and alcohol consummatory behaviors. Whether this primary event further recruits dopaminergic or non-dopaminergic mesocorticostriatal pathways to drive alcohol intake will require additional studies.
In our previous study, Dlx-MOR mice showed intact morphine and heroin conditioned place preference (Charbogne et al., 2017), suggesting that striatal MORs are not required for opioid reward. This finding is consistent with the notion that MOR-mediated disinhibition of dopamine neurons in the VTA is essential for opioid reward (Fields and Margolis, 2015), and the fact that MORs are intact in the VTA of Dlx-MOR KO mice. In contrast, the present study shows that alcohol place preference is not expressed in these mice, indicating that the VTA-centric disinhibition model is not the only mechanism underlying MOR-dependent alcohol reward. Circuit mechanisms subserving opioid and alcohol reward therefore differ. First, this is in accordance with a recent study showing that MOR blockade in the VTA prevents morphine- but not alcohol-stimulated dopamine release in the NAc (Valenta et al., 2013). Second, this conclusion substantiates the notion of divergent reward mechanisms across distinct drugs of abuse, which is notably well documented when comparing opiates and psychostimulants (Badiani et al., 2011; Becker et al., 2016). Although the exact explanation for the alcohol/opioid difference is yet to be determined, it is well accepted that alcohol, in contrast to opiates and most other drugs of abuse, hits numerous molecular targets distributed throughout brain circuits, including ionotropic receptors and ion channels, and therefore acts via multiple and much more complex mechanisms than other abused substances (Spanagel, 2009). To this respect, our genetic approach enabled the identification of a mechanism essential for alcohol reward-driven behaviors, which is mediated by a striatal MOR population and does not operate for opioid reward.
In conclusion, endogenous opioids have been implicated in pharmacological effects of alcohol and the development of alcohol use disorders (Drews and Zimmer, 2010). Here we show for the first time that rewarding effects of alcohol and alcohol consummatory behaviors are driven by MORs expressed in forebrain GABAergic neurons. Future experiments will identify the exact nature of striatal MOR-positive neurons responsible for these behaviors, and also the origin of endogenous opioid peptides responsible for this critical MOR function within neural circuits of alcohol use disorders.
Supplementary Material
Acknowledgments
We thank the Mouse Clinical Institute and the animal core facility at the Institut de Génétique et de Biologie Moléculaire et Cellulaire for technical support (Illkirch, France), and Neurophenotyping Center of the Douglas Mental Health University Institute for animal care. This work was supported by the Centre National de la Recherche Scientifique (CNRS), Institut National de la Santé et de la Recherche Médicale (INSERM) and Université de Strasbourg. SBH acknowledges postdoctoral fellowship from the French Academy of Sciences/Institut de France. We also thank the National Institutes of Health (NIAAA #16658 and NIDA # 005010) and the Canada Research Chairs for financial support.
Footnotes
Authors Contribution: SBH and BLK designed the experiments. SBH and LJB performed and analyzed behavioral experiments. SBH, LJB and BLK interpreted the results and wrote the article.
Disclosure/Conflict of Interest: The authors have no conflict of interest to disclose
References
- Bach P, Vollsta Dt-Klein S, Kirsch M, Hoffmann S, Jorde A, Frank J, Charlet K, Beck A, Heinz A, Walter H, Sommer WH, Spanagel R, Rietschel M, Kiefer F. Increased mesolimbic cue-reactivity in carriers of the mu-opioid-receptor gene OPRM1 A118G polymorphism predicts drinking outcome: a functional imaging study in alcohol dependent subjects. Eur Neuropsychopharmacol. 2015;25:1128–1135. doi: 10.1016/j.euroneuro.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Bachmanov AA, Kiefer SW, Molina JC, Tordoff MG, Duffy VB, Bartoshuk LM, Mennella JA. Chemosensory factors influencing alcohol perception, preferences, and consumption. Alcohol Clin Exp Res. 2003;27:220–231. doi: 10.1097/01.ALC.0000051021.99641.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Belin D, Epstein D, Calu D, Shaham Y. Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci. 2011;12:685–700. doi: 10.1038/nrn3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balldin J, Berglund M, Borg S, Mansson M, Bendtsen P, Franck J, Gustafsson L, Halldin J, Nilsson LH, Stolt G, Willander A. A 6-month controlled naltrexone study: combined effect with cognitive behavioral therapy in outpatient treatment of alcohol dependence. Alcohol Clin Exp Res. 2003;27:1142–1149. doi: 10.1097/01.ALC.0000075548.83053.A9. [DOI] [PubMed] [Google Scholar]
- Barson JR, Carr AJ, Soun JE, Sobhani NC, Leibowitz SF, Hoebel BG. Opioids in the nucleus accumbens stimulate ethanol intake. Physiol Behav. 2009;98:453–459. doi: 10.1016/j.physbeh.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson JR, Morganstern I, Leibowitz SF. Neurobiology of consummatory behavior: mechanisms underlying overeating and drug use. ILAR J. 2012;53:35–58. doi: 10.1093/ilar.53.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC. Animal models of excessive alcohol consumption in rodents. Curr Top Behav Neurosci. 2013;13:355–377. doi: 10.1007/7854_2012_203. [DOI] [PubMed] [Google Scholar]
- Becker JA, Clesse D, Spiegelhalter C, Schwab Y, Le Merrer J, Kieffer BL. Autistic-like syndrome in mu opioid receptor null mice is relieved by facilitated mGluR4 activity. Neuropsychopharmacology. 2014;39:2049–2060. doi: 10.1038/npp.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JA, Kieffer BL, Le Merrer J. Differential behavioral and molecular alterations upon protracted abstinence from cocaine versus morphine, nicotine, THC and alcohol. Addict Biol. 2016 doi: 10.1111/adb.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicella S, Ron D, Barak S. Intermittent ethanol access schedule in rats as a preclinical model of alcohol abuse. Alcohol. 2014;48:243–252. doi: 10.1016/j.alcohol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbogne P, Gardon O, Martín-García E, Keyworth HL, Matsui A, Mechling AE, Bienert T, Robé A, Moquin L, Robledo P, Matifas A, Befort K, Gavériaux-Ruff C, Harsan LA, Von Everfeldt D, Hennig J, Gratton G, Kitchen I, Bailey A, Alvarez VA, Maldonado R, Kieffer BL. Mu opioid receptors in GABAergic forebrain neurons regulate heroin and food seeking. Biol Psychiatry. 2017 doi: 10.1016/j.biopsych.2016.12.022. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbogne P, Kieffer BL, Befort K. 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology. 2014;76 Pt B:204–217. doi: 10.1016/j.neuropharm.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Drews E, Zimmer A. Modulation of alcohol and nicotine responses through the endogenous opioid system. Prog Neurobiol. 2010;90:1–15. doi: 10.1016/j.pneurobio.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Drobes DJ, Anton RF, Thomas SE, Voronin K. A clinical laboratory paradigm for evaluating medication effects on alcohol consumption: naltrexone and nalmefene. Neuropsychopharmacology. 2003;28:755–764. doi: 10.1038/sj.npp.1300101. [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, Koch M, Kessler P, Hentsch D, Birling MC, Koutsourakis M, Vasseur L, Veinante P, Kieffer BL, Massotte D. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct. 2015;220:677–702. doi: 10.1007/s00429-014-0717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Margolis EB. Understanding opioid reward. Trends Neurosci. 2015;38:217–225. doi: 10.1016/j.tins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbey FM, Ray L, Smolen A, Claus ED, Audette A, Hutchison KE. Differential neural response to alcohol priming and alcohol taste cues is associated with DRD4 VNTR and OPRM1 genotypes. Alcohol Clin Exp Res. 2008;32:1113–1123. doi: 10.1111/j.1530-0277.2008.00692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardon O, Faget L, Chu Sin Chung P, Matifas A, Massotte D, Kieffer BL. Expression of mu opioid receptor in dorsal diencephalic conduction system: new insights for the medial habenula. Neuroscience. 2014;277:595–609. doi: 10.1016/j.neuroscience.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto GJ, McBride WJ, Murphy JM, Lumeng L, Li TK. Ethanol self-infusion into the ventral tegmental area by alcohol-preferring rats. Alcohol. 1994;11:557–564. doi: 10.1016/0741-8329(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol Ther. 2004;103:121–146. doi: 10.1016/j.pharmthera.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Guan YZ, Ye JH. Ethanol blocks long-term potentiation of GABAergic synapses in the ventral tegmental area involving mu-opioid receptors. Neuropsychopharmacology. 2010;35:1841–1849. doi: 10.1038/npp.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia J, Caso C, Arias F, Gual A, Sanahuja J, Ramirez M, Mengual I, Gonzalvo B, Segura L, Trujols J, Casas M. A double-blind, placebo-controlled study of naltrexone in the treatment of alcohol-dependence disorder: results from a multicenter clinical trial. Alcohol Clin Exp Res. 2002;26:1381–1387. doi: 10.1097/01.ALC.0000030561.15921.A9. [DOI] [PubMed] [Google Scholar]
- Gueorguieva R, Wu R, Donovan D, Rounsaville BJ, Couper D, Krystal JH, O'Malley SS. Naltrexone and combined behavioral intervention effects on trajectories of drinking in the COMBINE study. Drug Alcohol Depend. 2010;107:221–229. doi: 10.1016/j.drugalcdep.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Heyser CJ, Roberts AJ, Schulteis G, Koob GF. Central administration of an opiate antagonist decreases oral ethanol self-administration in rats. Alcohol Clin Exp Res. 1999;23:1468–1476. [PubMed] [Google Scholar]
- Hyytia P, Kiianmaa K. Suppression of ethanol responding by centrally administered CTOP and naltrindole in AA and Wistar rats. Alcohol Clin Exp Res. 2001;25:25–33. doi: 10.1111/j.1530-0277.2001.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Jarjour S, Bai L, Gianoulakis C. Effect of acute ethanol administration on the release of opioid peptides from the midbrain including the ventral tegmental area. Alcohol Clin Exp Res. 2009;33:1033–1043. doi: 10.1111/j.1530-0277.2009.00924.x. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3:760–773. doi: 10.1016/S2215-0366(16)00104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MP, Nurmi H, Rouvinen N, Kiianmaa K, Gianoulakis C. Effects of acute ethanol on beta-endorphin release in the nucleus accumbens of selectively bred lines of alcohol-preferring AA and alcohol-avoiding ANA rats. Psychopharmacology (Berl) 2010;208:121–130. doi: 10.1007/s00213-009-1733-y. [DOI] [PubMed] [Google Scholar]
- Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev. 2009;89:1379–1412. doi: 10.1152/physrev.00005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli PW, Lam M, Bai L, Quirion R, Gianoulakis C. A microdialysis profile of dynorphin A(1-8) release in the rat nucleus accumbens following alcohol administration. Alcohol Clin Exp Res. 2006;30:982–990. doi: 10.1111/j.1530-0277.2006.00112.x. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Mechling AE, Arefin T, Lee HL, Bienert T, Reisert M, Ben Hamida S, Darcq E, Ehrlich A, Gaveriaux-Ruff C, Parent MJ, Rosa-Neto P, Hennig J, von Elverfeldt D, Kieffer BL, Harsan LA. Deletion of the mu opioid receptor gene in mice reshapes the reward-aversion connectome. Proc Natl Acad Sci U S A. 2016;113:11603–11608. doi: 10.1073/pnas.1601640113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JM, O'Neil JP, Janabi M, Marks SM, Jagust WJ, Fields HL. Alcohol consumption induces endogenous opioid release in the human orbitofrontal cortex and nucleus accumbens. Sci Transl Med. 2012;4:116ra116. doi: 10.1126/scitranslmed.3002902. [DOI] [PubMed] [Google Scholar]
- Moles A, Kieffer BL, D'Amato FR. Deficit in attachment behavior in mice lacking the mu-opioid receptor gene. Science. 2004;304:1983–1986. doi: 10.1126/science.1095943. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wolfel B, Dodt HU, Zieglgansberger W, Wotjak CT, Mackie K, Elphick MR, Marsicano G, Lutz B. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nealey KA, Smith AW, Davis SM, Smith DG, Walker BM. kappa-opioid receptors are implicated in the increased potency of intra-accumbens nalmefene in ethanol-dependent rats. Neuropharmacology. 2011;61:35–42. doi: 10.1016/j.neuropharm.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt DJ. The role of the opioid system in alcohol dependence. J Psychopharmacol. 2014;28:8–22. doi: 10.1177/0269881113504017. [DOI] [PubMed] [Google Scholar]
- Pettinati HM, O'Brien CP, Rabinowitz AR, Wortman SP, Oslin DW, Kampman KM, Dackis CA. The status of naltrexone in the treatment of alcohol dependence: specific effects on heavy drinking. J Clin Psychopharmacol. 2006;26:610–625. doi: 10.1097/01.jcp.0000245566.52401.20. [DOI] [PubMed] [Google Scholar]
- Racz I, Schurmann B, Karpushova A, Reuter M, Cichon S, Montag C, Furst R, Schutz C, Franke PE, Strohmaier J, Wienker TF, Terenius L, Osby U, Gunnar A, Maier W, Bilkei-Gorzo A, Nothen M, Zimmer A. The opioid peptides enkephalin and beta-endorphin in alcohol dependence. Biol Psychiatry. 2008;64:989–997. doi: 10.1016/j.biopsych.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Squeglia LM, Ashenhurst JR, Anton RF. Interactive effects of OPRM1 and DAT1 genetic variation on subjective responses to alcohol. Alcohol Alcohol. 2014;49:261–270. doi: 10.1093/alcalc/agt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley TL, Sanchez-Roige S, Bullmore ET, Mugnaini M, Maltby K, Miller SR, Wille DR, Nathan P, Stephens DN. The novel mu-opioid antagonist, GSK1521498, reduces ethanol consumption in C57BL/6J mice. Psychopharmacology (Berl) 2015;232:3431–3441. doi: 10.1007/s00213-015-3995-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki M, Watanabe K, Takeda K, Itoh T, Tsuyuki T, Narita M, Mori T, Suzuki T. Effect of chronic ethanol treatment on mu-opioid receptor function, interacting proteins and morphine-induced place preference. Psychopharmacology (Berl) 2013;228:207–215. doi: 10.1007/s00213-013-3023-y. [DOI] [PubMed] [Google Scholar]
- Soyka M, Friede M, Schnitker J. Comparing Nalmefene and Naltrexone in Alcohol Dependence: Are there any Differences? Results from an Indirect Meta-Analysis. Pharmacopsychiatry. 2016;49:66–75. doi: 10.1055/s-0035-1565184. [DOI] [PubMed] [Google Scholar]
- Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci. 1999;22:521–527. doi: 10.1016/s0166-2236(99)01447-2. [DOI] [PubMed] [Google Scholar]
- Valenta JP, Job MO, Mangieri RA, Schier CJ, Howard EC, Gonzales RA. mu-opioid receptors in the stimulation of mesolimbic dopamine activity by ethanol and morphine in Long-Evans rats: a delayed effect of ethanol. Psychopharmacology (Berl) 2013;228:389–400. doi: 10.1007/s00213-013-3041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.