

Abstract
HER2-targeted therapies, such as trastuzumab, have increased the survival rates of HER2+ breast cancer patients. However, despite these therapies, many tumors eventually develop resistance to these therapies. Our lab previously reported an unexpected sensitivity of HER2+ breast cancer cells to poly (ADP-Ribose) polymerase inhibitors (PARPi), agents that target homologous recombination (HR) deficient tumors, independent of a DNA repair deficiency. In this study, we investigated whether HER2+ trastuzumab resistant (TR) breast cancer cells were susceptible to PARPi and the mechanism behind PARPi induced cytotoxicity. We demonstrate that the PARPi ABT-888 (veliparib) decreased cell survival in-vitro and tumor growth in vivo of HER2+ trastuzumab resistant breast cancer cells. PARP-1 siRNA confirmed that cytotoxicity was due, in part, to PARP-1 inhibition. Furthermore, PARP-1 silencing had variable effects on the expression of several NF-κB-regulated genes. In particular, silencing PARP-1 inhibited NF-κB activity and reduced p65 binding at the IL-8 promoter, which resulted in a decrease in IL-8 mRNA and protein expression. Our results provide insight in the potential mechanism by which PARPi induces cytotoxicity in HER2+ breast cancer cells and support the testing of PARPi in patients with HER2+ breast cancer resistant to trastuzumab.
Keywords: Human epidermal growth factor receptor 2, breast cancer, trastuzumab resistance, PARP inhibitor, IL-8
Introduction
Overexpression of the human epidermal growth factor receptor 2 (HER2) is observed in 20-30% of breast cancer patients and associated with poor patient survival (1). One FDA-approved agent targeted against HER2 receptors is trastuzumab (Herceptin) (2). Trastuzumab blocks HER2 signaling and ultimately leads to tumor cell lysis by multiple mechanisms (3). Despite its efficacy in the clinic, trastuzumab resistance remains a clinical challenge (4).
In the last decade, poly (ADP-Ribose) polymerase inhibitors (PARPi) have been shown to be a promising therapeutic agent, especially in patients with homologous recombination repair (HR) deficient tumors (5–8). Interestingly, we have previously found that HER2+ breast cancer cells are sensitive to the PARPi (ABT-888 (veliparib) or AZD-2281 (olaparib)) alone, independent of a basal or induced HR deficiency (9). However, cell survival was not affected after treatment with PARPi in non-HER2 overexpressing breast cancer cell lines (MCF7 and T47D). We also reported that PARPi suppressed NF-κB activity and HER2 overexpression alone conferred sensitivity to PARPi (9).
In this study, we hypothesized that trastuzumab resistant (TR) breast cancers may also be sensitive to PARPi as these tumors may rely on compensatory mechanisms to activate similar downstream effectors. Indeed, we found that trastuzumab resistant HER2+ breast cancer cells are sensitive to pharmacological or genetic inhibition of PARP-1. Furthermore, we show that PARPi diminishes NF-κB (p65/RelA) transcriptional activity. Using the PanCancer Pathway Panel on the NanoString platform, which measures the expression of 770 genes involved in the 13 canonical cancer pathways, we analyzed the effects of PARPi on expression of NF-κB target genes (10). We discovered that knockdown of PARP-1 had differential effects on the 82 NF-κB-target genes included in the PanCancer Panel, such as IL-8, BRCA2, NFKBIZ, VEGFC, PIM1, and FASLG. We also validated that PARP-1 knockdown strongly inhibited the mRNA and protein expression of IL-8, a gene involved in inflammation and angiogenesis. Furthermore, using chromatin immunoprecipitation assays, we found that silencing of PARP-1 decreased p65 recruitment to the IL-8 promoter, which resulted in reduced IL-8 mRNA and protein expression. In summary, these results provide evidence that PARP inhibitors may be used as a novel therapeutic strategy for HER2+ breast cancer patients and uncover PARP-1/NF-κB (p65) signaling as a potential mechanism behind PARPi sensitivity.
Materials and Methods
Ethics statement
The animal protocol was approved by the University of Alabama at Birmingham at Birmingham Institutional Animal Care and Use Committee (APN#: 10129). Ketamine and xylazine anesthesia was used to minimize suffering before performing surgery on the mice.
Cell culture, drugs, and reagents
BT-474, UACC-812, and SKBR3 parental and trastuzumab resistant HER2+ breast cancer cell lines were used in this study and were previously characterized in two other studies (11,12). The BT-474 and UACC-812 trastuzumab resistant breast cancer cells were also cultured with 50 μg/ ml of trastuzumab and were kindly donated by Dr. Rachel Schiff and C Kent Osborne (Department of Medicine, Baylor College of Medicine). The SKBR3 trastuzumab resistant cells were cultured with 10 μg/ ml of trastuzumab and were kindly donated by Dr. Francisco J. Esteva (Department of Medicine, NYU). All cells tested negative for mycoplasma and were validated by western analysis for HER2. However, cell authentication was not conducted. Further, the cell lines were cultured for no more than 20 passages after thawing the frozen cells. All three trastuzumab resistant breast cell lines were also verified for resistance to trastuzumab. Veliparib (ABT-888) was obtained from Enzo Life Sciences (catalog #ALX-270-444) and olaparib (AZD-2281) was purchased from LC Laboratories (catalog #O-9201). Both drugs were reconstituted in dimethyl sulfoxide (DMSO) at 10 mmol/L. ABT-888 was also obtained from AbbVie Oncology for in-vivo testing and reconstituted every five days in 0.9% saline at 100 mg/kg. Trastuzumab (Herceptin) was purchased from Besse Medical (catalog #: 23961). Recombinant human TNF-α was obtained from R&D systems (catalog #: 210-TA).
Clonogenic survival assay
The colony formation assay was utilized to determine the percent survival in both the parental and trastuzumab resistant breast cancer cell lines as previously described (13,14).
PARP-1 knockdown
PARP-1 siRNA was obtained from Santa Cruz Biotechnology and contains three to five siRNA pools specifically targeting the PARP-1 gene (sc-29437; Santa Cruz Biotechnology). Another PARP-1 siRNA from Sigma-Aldrich(#NM_001618, SASI_Hs01_00159524) was utilized to confirm siRNA studies. Control siRNA was used as a negative control (sc-37007; Santa Cruz Biotechnology). The siRNAs were transfected with Lipofectamine2000 or Lipofectamine RNAiMax according to the manufacturer’s instructions. PARP-1 knockdown was confirmed by Western Blot or Real-Time PCR analysis.
Immunoblotting
Protein expression levels were analyzed via a standard immunoblotting protocol using the M-PER Mammalian Protein Extract Reagent with protease and phosphatase inhibitors as described previously (15). The PVDF membranes were immunoblotted overnight with the following primary antibodies according to the manufacturer’s instructions: PARP-1 (Cell Signaling Technology, catalog # 9542), PARP-1 (Santa Cruz, catalog # sc-8007), PARP-2 (Abcam, catalog #ab176330), IKKα (Cell Signaling Technology, catalog #2682), and BRCA2 (Abcam, catalog #ab27976). The immunoblots were then incubated with a rabbit or mouse horseradish peroxidase-conjugated secondary antibody for an hour. β-actin expression levels were evaluated as a loading control (Santa Cruz Biotechnology, catalog # sc-47778 HRP).
Cell proliferation
Cell proliferation was also assessed after PARP1 knockdown. After four days of treatment, the cells were washed with 1× ice-cold PBS and then removed with trypsin. Subsequently, the number of cells was counted using a cell counter (Beckman Coulter, Fullerton, CA).
Apoptosis analysis
Apoptosis was measured using the Annexin V-FITC Apoptosis Detection kit (Biovison Research Products; catalog #K101-400), 96 hours after transfection with control or PARP-1 siRNA and as previously described (14).
NF-κB Luciferase Reporter Assay
The NF-κB Secreted Luciferase Reporter System was used to analyze NF-κB activity. Specifically cells were co-transfected with the NFκB-driven luciferase plasmid NFκB-MetLuc2 or its vector control MetLuc2 (Clontech; catalog # 631728) and control or PARP-1 siRNA using the Lipofectamine2000 reagent, according to the manufacturer-supplied protocol and as previously described (9).
mRNA expression
Total RNA was isolated using the Ambion PureLink RNA mini kit (catalog #12183018A) according to the manufacturer’s recommendations. Gene expression was measured using the PanCancer Pathways Panel after PARP-1 knockdown, as previously described (16). One μg of total RNA was also reverse transcribed using the SuperScript III First-Strand Synthesis System kit (Invitrogen; catalog # 18080-051) and the resulting cDNA was analyzed by semiquantitative PCR using the following primer purchased from Applied Biosystems: PARP-1 (Hs00242302_m1), IL-8 (Hs00174103_m1), BRCA2 (Hs00609073_m1). mRNA levels were determined with the ABI Prism 7000 Sequence Detection System (Applied Biosystems) as per manufacturer’s instructions. Samples were run in triplicate and then normalized to the endogenous control, GAPDH (Hs02758991_g1) relative gene expression levels was analyzed using the 2−ΔΔCt method.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed in triplicate as previously published (17). Control or PARP-1 siRNA treated cells were sonicated and lysates were immunoprecipitated using four μg of p65 (Santa Cruz; catalog # sc-372) or normal rabbit IgG (Santa Cruz; catalog #: sc-2027) antibodies.
ELISA
Supernatants were analyzed after PARP-1 knockdown or PARPi using the Human IL-8 enzyme-linked immunosorbent assay (ELISA) (BioLegend; catalog #431504).
In-vivo studies
Ten 4-6 week old female BALB/c nude mice were obtained from Charles River. The mice were allowed to acclimatize for 1 week and then supplemented with 0.36-mg 60-day-release estradiol pellets from Innovative Research. Following two to three days of recovery, BT-474 TR cells were collected and then suspended in 200 μl of growth factor-reduced Matrigel from BD Biosciences before injection. 5×106 cells were injected subcutaneously in the BALB/c nude mice. After the tumors were palpable or reached ~5-6mm in diameter, we randomized the mice into two treatment groups (n=5) : saline (vehicle control) or ABT-888. The mice were treated twice daily with 100 mg/kg of ABT-888 by oral gavage for four weeks. At this dose in mice, it has been reported to result in plasma Cmax of 35μM and is considered to be the monotherapy maximum tolerated dose (MTD) dose in mice (18). Tumor size was measured triweekly during the course of the treatment with a caliper. The tumor volume was calculated with the following formula: [(width)2 × length]/2 (Burd and Wachsberger, 2007). The animals were measured for 68 days and then sacrificed. The Institutional Animal Care and Use Committee at the University of Alabama at Birmingham approved all the animal procedures described above.
Statistical analysis
The data were analyzed via a two-tailed, student t-test or analysis of variance (ANOVA) followed by a Bonferroni post test using GraphPad Prism version 4.02 and 7.0b (GraphPad Software). Data are presented as average ± standard error of mean (SEM). P-values ≤ 0.05 were considered significant.
Results
Trastuzumab resistant HER2+ breast cancer cells exhibit in vitro and in vivo sensitivity to the PARPi ABT-888
To examine whether HER2+ trastuzumab resistant (TR) breast cancer cell lines were sensitive to PARPi, we assessed PARPi-induced cellular cytotoxicity using colony formation assays. As shown in Figure 1A-C, the BT-474 TR, UACC-812 TR, and SKBR3 TR cells all demonstrated reduced colony formation in response to increasing, clinically achievable concentrations of ABT-888 (12) and exhibited a similar profile of sensitivity as their parental counterparts. Specifically, at 10μM of ABT-888 (Veliparib), there was more than 70% reduction in the survival in all three trastuzumab resistant breast cancer cell lines. Similarly, the PARPi MK-4827(Niraparib) also reduced the survival fraction of HER2+ trastuzumab resistant and parental breast cancer cell lines (Supplementary Figure S1).
Figure 1. HER2+ trastuzumab resistant breast cancer cells are sensitive to PARP inhibition (ABT-888) in-vitro and in-vivo.
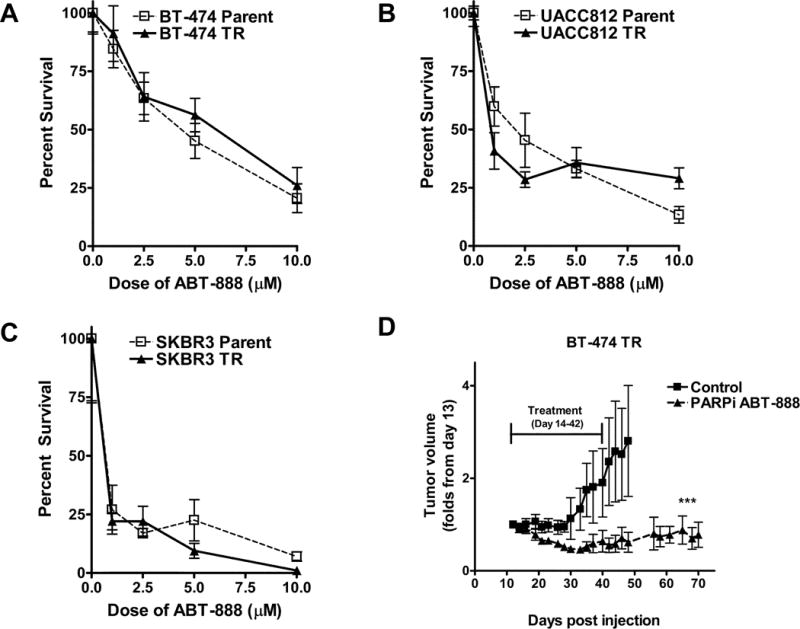
(A-C) HER2+ parental and trastuzumab resistant breast cancer cell lines were subjected to a colony formation assay after being treated with vehicle control and increasing concentrations of the PARPi ABT-888 (1-10μM). Shown is the mean percent survival (+/− SEM) from one of two independent experiments performed in triplicate. (D) BT-474 TR were injected subcutaneously in the flank of BALB/c nude mice. The mice were treated twice daily with vehicle or 100 mg/kg of ABT-888 once the tumors were palpable. Tumors were measured three times per week. Shown is the mean fold change in tumor volume +/−SEM. ***p<0.0005
To validate these results, we next established 3D microtumors (Vivo Biosciences, Supplemental Methods) using the BT-474 TR cells. As shown in Supplementary Figure S2, ABT-888 reduced the tumor viability of BT-474 TR microtumors cultured with trastuzumab. We also assessed the in vivo tumor growth effects of PARPi in the BT-474 TR xenografts. As shown in figure 1D, tumor growth was significantly inhibited after PARPi treatment in the HER2+ trastuzumab resistant xenografts. There was no significant difference in body weight observed in the ABT-888 treated group compared to the vehicle control group(Supplementary Figure S3C). To confirm that the decrease in tumor growth was due to the inhibition of PARP-1’s enzymatic activity, we examined PAR protein levels in tumors harvested from both control and ABT-888 treated animals. As shown in Supplemental Figure S3A-B, PAR levels were decreased in ABT-888 treated animals compared to control animals (Supplemental Methods, Supplementary Figure S3A-B). These results demonstrate that HER2+ trastuzumab resistant breast cancer cells are sensitive to PARPi alone both in vitro and in-vivo.
PARP-1 knockdown inhibits cell proliferation and induces apoptosis in HER2+ trastuzumab resistant breast cancer cells
To verify that our observations were indeed due to the inhibition of PARP-1 and not caused by an off-target effect, we examined cellular proliferation after silencing PARP-1 using a pooled PARP-1 siRNA, which reduced PARP-1 but not PARP2 levels (Figures 2A and 2B). Ninety-six hours after PARP-1 knockdown, cell proliferation was reduced by more than 50% in both the BT-474 TR (Figure 2C) and UACC-812 TR (Figure 2D) breast cancer cell lines. These results were confirmed with another PARP-1 siRNA from Sigma-Aldrich (Supplementary Figure S4). Next, we tested whether silencing PARP-1 induced cellular apoptosis. As shown in figures 2E and 2F, increased apoptosis was observed after 96 hours of PARP-1 suppression. Furthermore, inhibition of PARP by ABT-888 or PARP-1 siRNA treatment did not alter cell cycle progression (Supplemental Methods, Supplementary Figures S5).
Figure 2. PARP-1 siRNA reduces cell proliferation and induces apoptosis in HER2+ trastuzumab resistant breast cancer cell lines.
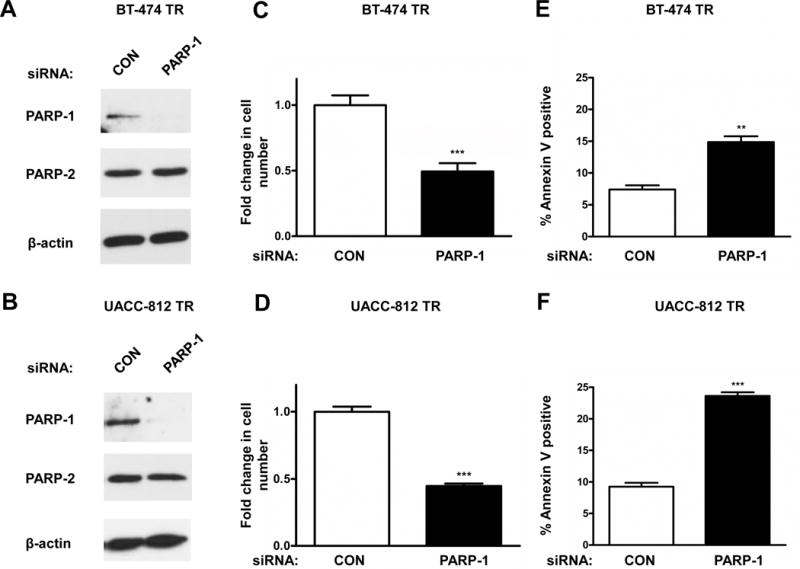
BT-474 TR and UACC-812 TR were transfected with 20 nM of control (CON) or PARP-1 siRNA for 96 hours. Knockdown of PARP-1 protein expression levels was verified via Western Blot analysis. β-actin was used as a loading control (A and B). Data shown are representative immmunoblots from one of three independent experiments. Following PARP-1 knockdown, cell counts were obtained via cellular proliferation assays (C and D). Apoptosis was assessed with FACS analysis using propidium iodide and Annexin V staining (E and F). The representative figures shown are from one of three independent experiments performed in (C and D) quadruplicate or (E and F) triplicate. ***p≤0.0005, ** p<0.005
NF-κB signaling is attenuated by inhibition and suppression of PARP-1
Previously, we reported that PARPi susceptibility correlated with inhibition of the NF-κB signaling pathway in the HER2+ parental breast cancer cells. We also observed that resistance to PARPi was stimulated after p65 overexpression while sensitivity was induced after overexpression of the NF-κB endogeneous inhibitor, IκBα (9). Subsequently, we tested NF-κB activity in the trastuzumab resistant cell lines with or without PARP inhibition using an NF-κB driven luciferase reporter assay. Consistent with our previous findings, pharmacological or genetic modulation of PARP-1 attenuated NF-κB transcriptional activity in the BT-474 TR (Figure 3A) cell line.
Figure 3. PARPi attenuates NF-κB activity and signaling in HER2+ trastuzumab resistant cell lines.
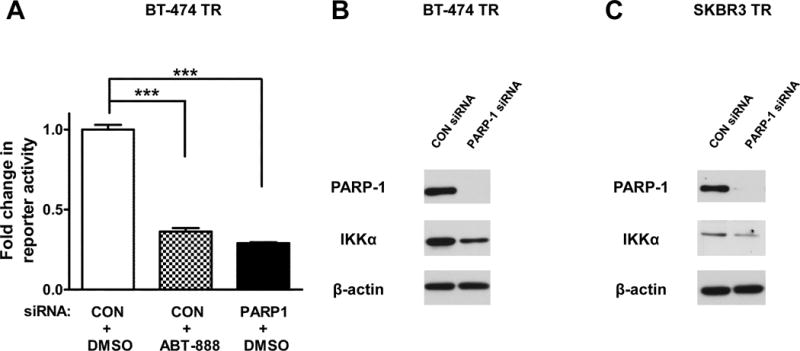
(A) Cells were co-transfected with the NFκB-driven luciferase plasmid NFκB-MetLuc2 or its vector control MetLuc2 and control or PARP-1 siRNA. The cells were then treated with vehicle or 10 μM ABT-888 four hours post transfection. NF-κB transcriptional activity was analyzed 48 hours after drug treatment using the NF-κB Secreted Luciferase Reporter System. Results shown are from one of three independent experiments performed in triplicate. (B and C) Western blot of PARP-1, IKKα, and β-actin after knockdown of PARP-1 in BT-474 TR and SKBR3 TR cells. The representative images shown are from one of two independent experiments. ***p<0.001.
It has also been reported that HER2 overexpression activates the canonical NF-κB signaling pathway via the IKKα, catalytic subunit of the IKK complex, without the stimulation of an inflammatory stimuli such as TNF-α (19). Since we observed an inhibition of NF-κB activity, we hypothesized that the expression of the IKKα protein, an NF-κB activator, would be reduced by PARP1 knockdown. Western blot analysis following PARP-1 knockdown via two different PARP-1 siRNAs revealed that the level of IKKα protein was decreased in the BT-474 TR and SKBR3 TR HER2+ breast cancer cell lines (Figure 3B and C, Supplementary Figure S6). These results indicate that PARP-1 suppression inhibits NF-κB signaling in HER2+ trastuzumab resistant breast tumors.
PARP-1 inhibits the expression of NF-κB regulated target genes
Hassa, Hottiger et al. previously reported that PARP-1 is a coactivator of NF-κB (20,21). To further examine the role of PARP-1 as a regulator of NF-κB activity, we assessed the gene expression of NF-κB target genes using the NanoString nCounter Analysis System after PARP-1 knockdown in the BT-474 TR cell line. After normalizing the data to housekeeping genes included in the panel, suppression of PARP-1 significantly altered the expression levels of a number of NF-κB regulated genes (Table 1). The gene most strongly impacted by PARP-1 knockdown was IL-8 (13 fold reduction).
Table 1.
Significant NF-κB target genes influenced by PARP-1 knockdown.
| NF-κB target genes: | CON siRNA: | PARP1 siRNA: | Fold Change: |
|---|---|---|---|
| IL-8 | 1180.83 | 89.72 | −13.16 |
| BRCA2 | 42.3 | 19.7 | −2.15 |
| NFKBIZ | 51.5 | 27.36 | −1.88 |
| VEGFC | 44.14 | 24.07 | −1.83 |
| PIM1 | 37.71 | 73.31 | 1.95 |
| FASLG | 9.2 | 22.98 | 2.5 |
The BT-474 TR cell line was treated with control (CON) or PARP-1 siRNA for 72 hours and then subjected to NanoString Analysis using the PanCancer Pathway panel. The counts were normalized to expression levels of housekeeping genes included in the probe set. Shown in the table are the normalized NanoString counts along with the fold change values
PARP-1 knockdown inhibits IL-8 gene and protein expression
To validate our NanoString data, we assessed changes in IL-8 gene expression following PARP-1 siRNA using qRT-PCR analysis. IL-8 is also an excellent gene candidate and read-out to further study the role of PARP-1 in NF-κB-mediated transcription because, unlike other genes, NF-κB plays a dominant role in its expression. Specifically, the human IL-8 promoter is highly accessible to the NF-κB transcription factor, p65 (22,23). Consistent with our NanoString data, IL-8 mRNA was significantly reduced after PARP-1 knockdown in both the BT-474 TR and UACC-812 TR breast cancer cells (Figure 4A and B). Moreover, PARP-1 siRNA significantly reduced IL-8 mRNA levels induced by TNF-α, which robustly activates the NF-κB signaling pathway and thus can be used as a positive control for NF-kB activation. IL-8 mRNA levels were similarly attenuated in the parental BT-474 breast cancer cell line (Figure 4C) after PARP-1 knockdown. Finally, ABT-888 and AZD-2281 also decreased the gene expression levels of IL-8 in the BT-474 TR cells (Figure 4D and Supplementary Figure S7D).
Figure 4. PARP-1 knockdown inhibits IL-8 expression in both HER2+ parental and trastuzumab resistant breast cancer cells.
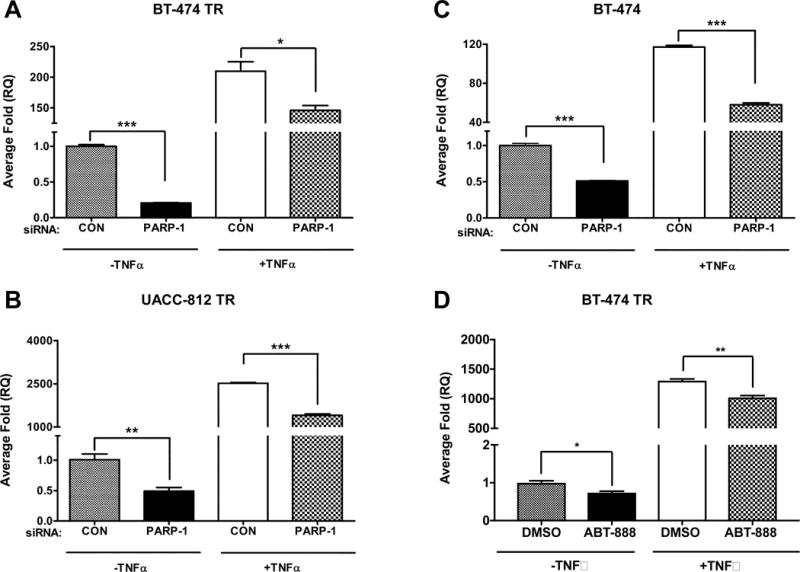
Quantitative real-time RT-PCR analysis was performed with total RNAs isolated from (A) BT-474 trastuzumab resistant, (B) UACC-812 trastuzumab resistant, and (C) BT-474 parent cells that were transfected with 20 nM of control (CON) or PARP-1 siRNA for 48 hours, serum-starved for 18 hours, and then stimulated with 10 ng/ml of tumor necrosis factor-α (TNF-α) for 2 hours. (D) BT-474 TR were serum starved and treated for four hours with DMSO or 10 μM ABT-888 and then treated with TNF-α for an additional 2 hours. One μg of isolated RNA was reverse transcribed to cDNA and then analyzed by qRT-PCR for IL-8 and GAPDH expression. The figures are representative images from one of three independent experiments performed in triplicate. A one-way ANOVA test was performed followed by a t-test to calculate the significance between groups. *p<0.05 and ***p<0.001
To determine whether these changes in mRNA expression directly correlated with similar changes in protein expression, we subjected the BT-474 TR and UACC-812 TR breast cancer cell lines to ELISA analysis after PARP-1 inhibition or knockdown. Indeed, IL-8 protein expression levels were decreased by both pharmacological or genetic modulation of PARP-1 (Figure 5A-D).
Figure 5. PARP-1 knockdown inhibits IL-8 protein expression.
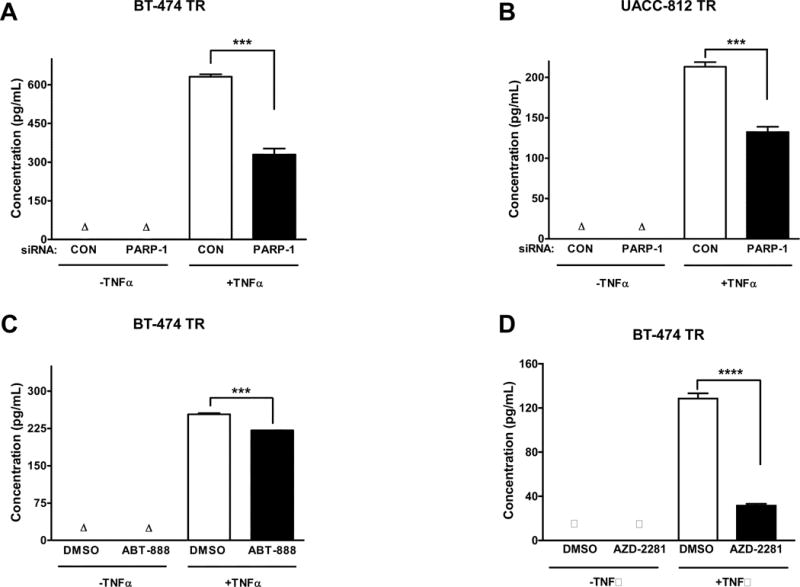
(A) BT-474 TR cells were transfected with control (CON) or PARP-1 siRNA for 48 hours and (B) UACC-812 TR cells were transfected for 24 hours. Cells were serum-starved for 18 hours, and then stimulated for 24 hours with 10 ng/mL of TNF-α. (C) BT-474 TR cells were serum-starved and treated with DMSO or 10 μM ABT-888 for 24 hours or (D) 10 μM AZD-2281 for 72 hours after seeding and then stimulated for an additional 24 hours with TNF-α. The supernatant was then collected and IL-8 protein expression was analyzed by an enzyme-linked immunosorbent assay (ELISA). Results shown are from one of three independent experiments. ***p≤0.001
PARP-1 knockdown decreases p65 binding at the IL-8 promoter
To further investigate PARP-1’s role in regulating NF-kB activity in the context of IL-8 expression, we assessed the recruitment of the NF-κB subunit p65 to the IL-8 promoter using chromatin immunoprecipitation (ChIP). As shown in Figure 6A, TNF-α stimulation increased p65 binding at the IL-8 promoter in the BT-474 TR cell line, and this effect was significantly reduced with PARP-1 knockdown. p65 and IgG (negative control) were also immunoprecipitated in untreated or TNF-α treated BT-474 TR cells. We observed that p65 was associated with the IL-8 promoter in TNF-α treated cells, and this was not detected using IgG (Figure 6B). These ChIP experiments further demonstrate that PARP-1 regulates the NF-κB subunit p65 at the IL-8 promoter.
Figure 6. PARP-1 knockdown decreases p65 recruitment to the IL-8 promoter in HER2+ trastuzumab resistant cell lines.
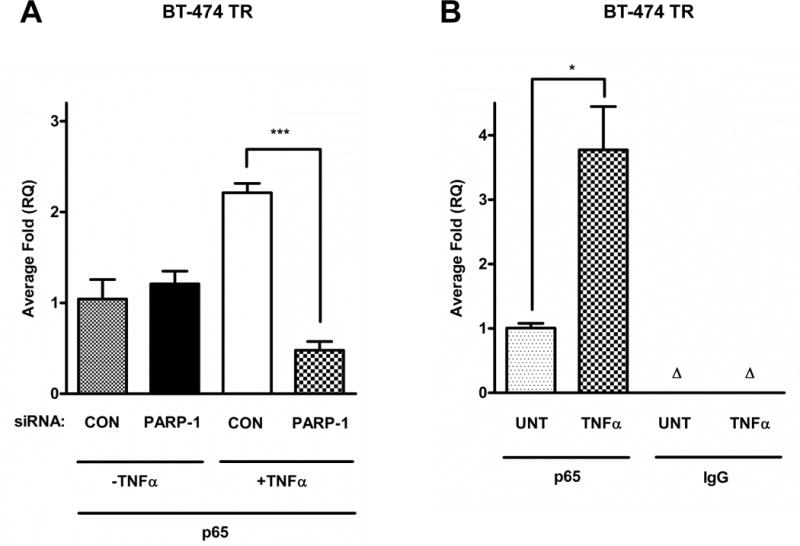
(A) BT-474 TR were transfected with control (CON) or PARP-1 siRNA for 48 hours or (B) left untreated, then serum-starved for 18 hours and stimulated without or with 10 ng/ml of TNF-α for 2 hours. The cells were then subjected to chromatin immunoprecipitation (ChIP) assay, using p65 or immunoglobulin G (IgG) control antibodies. Immunoprecipitated DNA was analyzed by qRT-PCR using human IL-8 primers. Non-immunoprecipitated DNA (input) was used as a control for total DNA levels. Data shown are from one of three independent experiments. A t-test or a one-way ANOVA test was performed followed by a t-test to calculate the significance between groups. *p<0.05 and ***p<0.001
Discussion
PARP-1’s role in DNA damage repair, specifically the base excision repair pathway, has been studied extensively. In addition to its DNA repair functions, PARP-1 also has a role in several other cellular processes (24). PARP-1 has been shown to promote chromatin decondensation by PARylating histones or binding directly to nucleosomes (25,26). Several other studies have shown that PARP-1 is involved in angiogenesis (27). Recently there has been an increased interest in targeting transcriptional factors with PARPi in various types of cancer models (28–30). In this study, we report that the susceptibility to PARPi observed in HER2+ breast cancers (sensitive or resistant to the HER2-targeted agent trastuzumab) may be dependent on the inhibition of the NF-κB signaling pathway. We also elucidated the effects of PARP-1 on the NF-κB signaling pathway in HER2+ breast cancer cells by investigating effects on IL-8 expression.
Various mechanisms of resistance to trastuzumab have been described in the literature (30). Examples include overexpression of insulin-like growth factor I receptor (11), compensatory activation of other HER receptors (12), and reactivation of the HER2 signaling pathway. Since these pathways activate similar downstream effectors, such as NF-kB, it is not surprising that TR cells retained sensitivity to PARP inhibition.
PARP inhibitors such as ABT-888 target both PARP-1 and PARP-2 members of the PARP family that are involved in DNA repair (32). PARP-1 is the most abundant nuclear member of this family and is responsible for a larger percentage of PARP activity compared to PARP-2 (33). In this study, we focused mainly on the PARP-1 protein since we recently discovered that HER2+ breast cancers express increased levels of nuclear PARP-1 and phospho-p65 compared to non-HER2 overexpressing breast cancers (34). No significant differences were found with PARP-2 protein levels (34). However, we cannot exclude PARP-2 as an essential target in this mechanism as it is still responsible for ten percent of PARylation produced in the nucleus (35). We are currently investigating the role of PARP-2 in this process but it is beyond the scope of this study.
PARP-1 has also been shown to regulate gene transcription by acting as a co-activator. Some transcription factors that are regulated by PARP-1 include the following: NF-κB (20), androgen receptor (28), and estrogen receptor (36). Our results suggest that PARP-1 may be acting as a coactivator of NF-κB (Figure 4 and 6). However, the role of PARP-1’s enzymatic activity in NF-κB dependent gene expression is controversial. Multiple studies have reported that PARP-1’s enzymatic activity is independent of its co-activator function of NF-κB (21,37). However, we observed a reduction in the gene expression levels of a NF-κB target gene, IL-8, after PARPi in TNF-α stimulated cells, suggesting here that PARP-1 enzymatic activity may indeed play a role in the its coactivation function of NF-κB (Figure 4D and Supplementary Figure S7D). These contradictory results suggest that the role of PARP-1’s enzymatic activity may be context dependent.
Conversely, the expression of PARP-1 is required and sufficient in various disease models (21,37,38). Our PARP-1 knockdown studies further confirm these results and suggest that PARP-1 regulates NF-κB activity. Specifically, PARP-1 knockdown reduced p65 binding to the IL-8 gene promoter, leading to decreased gene and protein expression in untreated or TNF-α treated cells (Figures 4–6). All together these results suggest that PARPi attenuates NF-κB signaling in HER2+ trastuzumab resistant breast cancer cells. However, we do not propose that the sole mechanism behind PARPi sensitivity is the inhibition of IL-8 expression. We would like to further stress that IL-8 is merely an excellent gene candidate to study the role of PARP-1 in NF-κB-mediated transcription.
Nevertheless, IL-8 is a HER2 regulated gene and associated with metastasis in breast cancer patients. Trastuzumab has also been shown to downregulate IL-8 gene expression levels in BT-474 breast cancer cells (39). IL-8 has also been observed to play a role in influencing trastuzumab resistance (40). For example, an inflammatory feedback loop in HER2+ breast cancers cells has also been reported to activate cancer stem cell activity and trastuzumab resistance by increasing IL-8 levels (41). These data suggest that the potential targeting of the IL-8 gene in HER2+ breast cancer cells that develop resistance to trastuzumab may be useful.
We also observed that other NF-κB-targeted genes were also regulated by PARP-1 besides IL-8. However, their expression levels were not as strongly influenced by PARP-1 suppression. This could be due to the fact that their levels are regulated by several transcription factors, whereas, IL-8 is highly dependent on NF-κB. In particular, the expression levels of BRCA2 were reduced after PARP-1 knockdown (Table 1). BRCA2 is a protein involved in the homologous recombination (HR) DNA repair pathway (42). Previous studies have shown that genetic and pharmacologic inhibition of PARP-1 reduced expression of BRCA1 and Rad51, two other proteins involved in HR. These results were also associated with a decrease in HR (43). Our results show that PARP-1 knockdown decreased the levels of BRCA2 mRNA, but did not alter BRCA2 protein levels (Supplementary Figures S8A and S8B). We have also previously shown that HER2+ breast cancer cells do not harbor a basal or induced HR deficiency (9). This suggests that the effects observed after PARP-1 knockdown were independent of an HR deficiency. Conversely, PARP-1 knockdown upregulated PIM1 and FASLG expression. PIM1 is involved in survival and cell proliferation whereas FASLG is involved in apoptosis (44,45). A possible explanation behind these findings could be NF-κB’s complex role in multiple cellular pathways (46). Several studies have also indicated that in a single cell, NF-κB can have both pro- and anti-apoptotic functions (47). These results will be investigated in future studies.
This study supports further clinical testing of PARPi in patients with HER2 positive breast tumors that are sensitive or resistant to trastuzumab. It also suggests the use of PARPi to suppress PARP-1’s transcriptional functions besides solely inhibiting its DNA repair roles.
Supplementary Material
Acknowledgments
We would like to thank Vivo Biosciences for their help with the microtumor experiments as well as Enid F. Keyser for her assistance with the apoptosis and cell cycle experiments. The authors would also like to thank Alice Weaver for her help with an experiment.
Financial information: This study was supported by grants from the American Association of Cancer Research/Genentech Career Development Award (12-30-18-YANG) to E.S. Yang), Susan G. Komen Career Catalyst Award (CCR12364491, to E.S. Yang), Breast Cancer Research Foundation of Alabama (to E.S. Yang), and the Breast SPORE (5P50CA089019 to E.S. Yang) from the University of Alabama-Birmingham Comprehensive Cancer Center. The UAB Comprehensive Flow Cytometry Core Facility is supported by Grants P30 AR048311 and Grant P30 AI027767.
Footnotes
Disclosure of Potential Conflicts of Interest
Eddy Yang has served on the advisory board of NanoString Technologies and has received honorarium from them. He also has a Materials Transfer Agreement with AbbVie, Inc. The other authors have no conflicts of interest to disclose.
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Li SG, Li L. Targeted therapy in HER2-positive breast cancer. Biomed Rep. 2013;1(4):499–505. doi: 10.3892/br.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vu T, Claret FX. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012;2:62. doi: 10.3389/fonc.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–80. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 5.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 6.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 7.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 8.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 9.Nowsheen S, Cooper T, Bonner JA, LoBuglio AF, Yang ES. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res. 2012;72(18):4796–806. doi: 10.1158/0008-5472.CAN-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26(3):317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 11.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–28. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 12.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers–role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13(6):R121. doi: 10.1186/bcr3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nowsheen S, Bonner JA, Yang ES. The poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99(3):331–8. doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowsheen S, Bonner JA, Lobuglio AF, Trummell H, Whitley AC, Dobelbower MC, et al. Cetuximab augments cytotoxicity with poly (adp-ribose) polymerase inhibition in head and neck cancer. PLoS One. 2011;6(8):e24148. doi: 10.1371/journal.pone.0024148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowsheen S, Cooper T, Stanley JA, Yang ES. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS One. 2012;7(10):e46614. doi: 10.1371/journal.pone.0046614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weaver AN, Cooper TS, Rodriguez M, Trummell HQ, Bonner JA, Rosenthal EL, et al. DNA double strand break repair defect and sensitivity to poly ADP-ribose polymerase (PARP) inhibition in human papillomavirus 16-positive head and neck squamous cell carcinoma. Oncotarget. 2015 doi: 10.18632/oncotarget.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nozell S, Laver T, Patel K, Benveniste EN. Mechanism of IFN-beta-mediated inhibition of IL-8 gene expression in astroglioma cells. J Immunol. 2006;177(2):822–30. doi: 10.4049/jimmunol.177.2.822. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, Digiammarino EL, et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol Can Res. 2015;13(11):1465–77. doi: 10.1158/1541-7786.MCR-15-0191-T. [DOI] [PubMed] [Google Scholar]
- 19.Merkhofer EC, Cogswell P, Baldwin AS. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene. 2010;29(8):1238–48. doi: 10.1038/onc.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380(7-8):953–9. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 21.Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem. 2001;276(49):45588–97. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- 22.Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13(10):6137–46. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott CL, Allport VC, Loudon JA, Wu GD, Bennett PR. Nuclear factor-kappa B is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells. Mol Hum Reprod. 2001;7(8):787–90. doi: 10.1093/molehr/7.8.787. [DOI] [PubMed] [Google Scholar]
- 24.Weaver AN, Yang ES. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front Oncol. 2013;3:290. doi: 10.3389/fonc.2013.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim MY, Mauro S, Gévry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119(6):803–14. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Huletsky A, de Murcia G, Muller S, Hengartner M, Ménard L, Lamarre D, et al. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J Biol Chem. 1989;264(15):8878–86. [PubMed] [Google Scholar]
- 27.Rajesh M, Mukhopadhyay P, Godlewski G, Bátkai S, Haskó G, Liaudet L, et al. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem Biophys Res Commun. 2006;350(4):1056–62. doi: 10.1016/j.bbrc.2006.09.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2(12):1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Legrand AJ, Choul-Li S, Spriet C, Idziorek T, Vicogne D, Drobecq H, et al. The level of Ets-1 protein is regulated by poly(ADP-ribose) polymerase-1 (PARP-1) in cancer cells to prevent DNA damage. PLoS One. 2013;8(2):e55883. doi: 10.1371/journal.pone.0055883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, et al. PARP-1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res. 2012;72(7):1608–13. doi: 10.1158/0008-5472.CAN-11-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baselga J. Treatment of HER2-overexpressing breast cancer. Ann Oncol. 2010;21(Suppl 7):vii, 36–40. doi: 10.1093/annonc/mdq421. [DOI] [PubMed] [Google Scholar]
- 32.Donawho CK, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 33.Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol Cell. 2015;58(6):925–34. doi: 10.1016/j.molcel.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanley J, Klepczyk L, Keene K, Wei S, Li Y, Forero A, et al. PARP1 and phospho-p65 protein expression is increased in human HER2-positive breast cancers. Breast Cancer Res Treat. 2015;150(3):569–79. doi: 10.1007/s10549-015-3359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amé JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274(25):17860–8. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 36.Zhang F, Wang Y, Wang L, Luo X, Huang K, Wang C, et al. Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor α-dependent gene transcription. J Biol Chem. 2013;288(16):11348–57. doi: 10.1074/jbc.M112.429134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter JE, Willmore E, Irving JA, Hostomsky Z, Veuger SJ, Durkacz BW. NF-κB mediates radio-sensitization by the PARP-1 inhibitor, AG-014699. Oncogene. 2012;31(2):251–64. doi: 10.1038/onc.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Liu L, Xia Y, Wu D. Silencing of poly(ADP-ribose) polymerase-1 suppresses hyperstretch-induced expression of inflammatory cytokines in vitro. Acta Biochim Biophys Sin (Shanghai) 2014;46(7):556–64. doi: 10.1093/abbs/gmu035. [DOI] [PubMed] [Google Scholar]
- 39.Wen XF, Yang G, Mao W, Thornton A, Liu J, Bast RC, et al. HER2 signaling modulates the equilibrium between pro- and antiangiogenic factors via distinct pathways: implications for HER2-targeted antibody therapy. Oncogene. 2006;25(52):6986–96. doi: 10.1038/sj.onc.1209685. [DOI] [PubMed] [Google Scholar]
- 40.von der Heyde S, Wagner S, Czerny A, Nietert M, Ludewig F, Salinas-Riester G, et al. mRNA profiling reveals determinants of trastuzumab efficiency in HER2-positive breast cancer. PLoS One. 2015;10(2):e0117818. doi: 10.1371/journal.pone.0117818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–84. doi: 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu K, Jiang SW, Thangaraju M, Wu G, Couch FJ. Induction of the BRCA2 promoter by nuclear factor-kappa B. J Biol Chem. 2000;275(45):35548–56. doi: 10.1074/jbc.M004390200. [DOI] [PubMed] [Google Scholar]
- 43.Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci U S A. 2010;107(5):2201–6. doi: 10.1073/pnas.0904783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, Gold MR. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-kappa B pathway. J Immunol. 2002;168(2):744–54. doi: 10.4049/jimmunol.168.2.744. [DOI] [PubMed] [Google Scholar]
- 45.Singh NP, Nagarkatti M, Nagarkatti PS. Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol Pharmacol. 2007;71(1):145–57. doi: 10.1124/mol.106.028365. [DOI] [PubMed] [Google Scholar]
- 46.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin B, Williams-Skipp C, Tao Y, Schleicher MS, Cano LL, Duke RC, et al. NF-kappaB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell Death Differ. 1999;6(6):570–82. doi: 10.1038/sj.cdd.4400528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.