

Structured Abstract
Background
An influx of lipid-loaded macrophages characterizes visceral adipose tissue (VAT) inflammation, which is an important factor in the development of insulin resistance (IR) in obesity. Depletion of macrophage lipids accompanies increased whole body insulin sensitivity, but the underlying mechanism is unknown. Deficiency of autophagy protein ATG16L1 is associated with increases in inflammatory diseases and lipid metabolism, but the connection between ATG16L1, IR, and obesity remains elusive. We hypothesize that myeloid ATG16L1 contributes to lipid loading in macrophages and to IR.
Methods
Wild-type (WT) bone marrow derived macrophages (BMDMs) were treated with fatty acids and assessed for markers of autophagy. Myeloid-deficient Atg16l1 and littermate control male mice were fed high fat diet (HFD) or low fat diet (LFD) for 3 months starting at 8 weeks of age. Mice were assessed for body mass, fat and lean mass, glucose and insulin sensitivity, food consumption and adipose inflammation. Fluorescence-activated cell sorted VAT macrophages were assessed for lipid content and expression of autophagy related genes.
Results
VAT and VAT macrophages from HFD-fed WT mice did not show differences in autophagy protein and gene expression compared to tissue from LFD-fed mice. Fatty acid-treated BMDMs increased neutral lipid content but did not change autophagy protein expression. HFD-fed Atg16l1 myeloid-deficient and littermate mice demonstrated no differences in body mass, glucose or insulin sensitivity, food consumption, fat or lean mass, macrophage lipid content, or adipose tissue inflammation.
Conclusion
ATG16L1 does not contribute to obesity, IR, adipose tissue inflammation or lipid loading in macrophages in mice fed HFD.
Keywords: autophagy, obesity, inflammation, adipose tissue
1. Introduction
Chronic low-grade inflammation in visceral adipose tissue (VAT) accompanies the development of insulin resistance (IR) in humans and mice [1, 2]. An important component of VAT inflammation is macrophage infiltration into the VAT. Macrophages comprise approximately 10% of VAT cellular content in low fat diet (LFD)-fed mice, but this increases to approximately 50% of VAT cellular content in high fat diet (HFD)-fed mice [3]. These adipose tissue macrophages (ATMs) produce the majority of pro-inflammatory cytokines, which occurs before the development of IR [3, 4]. ATMs from mice with obesity have increased lipid content, which is associated with the expression of the cell surface marker, CD11c [5, 6]. Lipid depletion from ATMs is accompanied by increases in whole body insulin sensitivity [5], suggesting a link between macrophage lipid content and IR. However, the mechanism (or mechanisms) by which these lipids affect whole body IR is unknown.
Autophagy is a cellular degradation process that has been tested as a possible mechanism to connect lipid metabolism and inflammation to whole body IR. The results of these studies have yet to fully clarify the possible contribution of autophagy to obesity and metabolic dysfunction, as they appear to be condition-dependent. Factors such as the model of obesity (dietary vs. genetic) and the specific autophagy gene deleted, in part underlie contradictory findings. For example, in mice with genetically induced obesity (Lepob), myeloid-specific deletion of Atg7 increases VAT inflammation and exacerbates whole body IR [7, 8]. However, in a diet-induced obesity (DIO) model, myeloid deletion of Atg7 does not affect any metabolic endpoint, nor does it change ATM lipid content [9]. Myeloid-specific deletion of Atg5 in a lipopolysaccharide (LPS)-stimulated and HFD-fed obesity model in C57BL/6 mice increases hepatic inflammation but does not affect VAT inflammation [10]. Neither of these models, Lepob or LPS-mediated induced inflammation with HFD feeding, is representative of the majority of the clinical population affected by obesity-related T2D. Furthermore, in vitro, deletion of Atg5, Atg7, or Atg16l1 in bone marrow-derived macrophages (BMDMs) does not affect lipotoxic cell death [11, 12].
In human populations, a common polymorphism of ATG16L1 confers partial loss of function and increased risk of developing inflammatory Crohn’s disease [13, 14]. Yet, ATG16L1 has not been studied as extensively as ATG5 and ATG7 in the context of obesity and T2D. While both ATG7 and ATG5 are important in catalyzing LC3 lipidation, which is required for autophagosomal membrane expansion, ATG16L1 is important in initiation of autophagosomal biogenesis and in the localization of the ATG7 and ATG5 catalytic complexes [15, 16]. ATG16L1 is specifically connected to inflammation and lipid metabolism, as myeloid deletion of Atg16l1 increases colitis in a murine model of Crohn’s disease model [17] and mice hypomorphic for Atg16l1 have increased expression of lipid metabolism genes in Paneth cells [18].
Hence, on account of the distinct role of ATG16L1 in the autophagy cascade and the connection of ATG16L1 to inflammation and lipid metabolism, we hypothesized that myeloid–specific deletion of Atg16l1 connects VAT inflammation and IR in DIO. To test this hypothesis, we fed mice with myeloid-specific deletion of Atg16l1 HFD verses LFD to probe the effect of this specific component of the autophagy pathway on lipid metabolism, obesity and IR.
2. Material and Methods
2.1 Animal Care and Diets
2.1.1 Animal Care
C57BL/6 male mice (WT; Jackson Laboratory, Bar Harbor ME) were delivered at 5 – 7 weeks of age and acclimated for a minimum of 3 days before experimental use. LysMcre(+/−).Atg16l1flox/flox and LysMcre(−/−).Atg16l1flox/flox mice [19, 20] breeder pairs were generously shared by Dr. Ken Cadwell (New York University Langone Medical Center) and Dr. Herbert “Skip” Virgin (Washington University School of Medicine) and bred in house. Male littermates were used for all studies. All mice were housed in the same vivarium with 12 h light:dark cycle for the duration of breeding, weaning, and experimental time course through euthanasia. All mouse housing and procedures were approved by the New York University Institutional Animal Care and Use Committee and conducted according to the National Institute of Health Animal Care Guidelines.
2.1.2 Diet
At age 7–9 weeks, weight-matched male mice were fed a 60% kcal/fat high fat diet (D12492, Research Diets Inc., New Brunswick, NJ) or a 13% kcal/fat low fat diet (5053; PicoLab Rodent Diet 20; Lab Diet, Brentwood, MO) for 11–13 weeks. In total, 23 WT, 15 LysMcre(+/−).Atg16l1flox/flox and 13 LysMcre(−/−).Atg16l1flox/flox were fed HFD, and 39 WT, 16 LysMcre(+/−).Atg16l1flox/flox and 12 LysMcre(−/−).Atg16l1flox/flox were fed LFD. Body weights of all the LysMcre(+/−).Atg16l1flox/flox and LysMcre(−/−).Atg16l1flox/flox mice were measured once a week for the duration of the experimental time course.
2.1.3 Food Consumption
To determine food intake, pre-weighed food pellets were administered to 9 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 8 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 7 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and 7 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. Pellets were re-weighed 24 h later. All mice were single-housed and edible bedding was removed during this time period. The number of grams consumed was multiplied by the kcal per gram in each diet (LFD, 3.03 kcal/gram; HFD, 5.24 kcal/gram) to report food consumption in kcal.
2.2 DEXA Scanning
Body composition (fat and lean mass) of 10 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 12 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 12 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and 11 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice was measured using a calibrated LUNAR PIXI dual-energy X-ray absorptiometry (DEXA) scanning machine (LUNAR Corp., Madison, WI). Data were recorded using the LUNAR PIXImus 2.10 software.
2.3 Glucose and Insulin Tolerance Tests, and Fasting Blood Glucose
7 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 7 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 5 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and 6 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice were fasted for 4–5 h. After checking baseline fasting blood glucose, 20 mg D-glucose/kg body weight (Sigma, G8270) or 1 unit insulin/kg body weight (Lilly, 0002-8215-01) was injected via the intraperitoneal route for all mice regardless of diet or genotype. Blood glucose in awake mice was assessed via tail vein at 15, 30, 60, 90, 120, 150, and 180 min post injection. GTT was analyzed as incremental area under curve [21].
2.4 Primary BMDM Isolation and Culture
2.4.1 BMDM Isolation
Bone marrow from the tibiae and femora of 8 – 10 week old 16 WT LFD-fed mice was extracted by centrifugation and made into a single cell suspension by resuspending in BMDM differentiation media (DMEM (ThermoFisher, 11885) containing 10% L929 cell media [22], 5mM glucose, 10% fetal bovine serum (FBS, Corning, 35-010-CV) and 1% penicillin/streptomycin (P/S; ThermoFisher, 15140-122)) with a syringe and 18.5 gauge needle, and cultured at 37°C for 7–8 days. Fresh differentiation media was administered on day 5 of differentiation.
2.4.2 BMDM Treatments
At the end of day 6 of BMDM differentiation, cells were placed in OptiMem reduced serum media (ThermoFisher, 31985) overnight. OptiMem was replaced with DMEM containing palmitate (100 μM) (Sigma, P9767) vs dPBS control or a mixture of palmitate and oleate (Sigma, 07501) (500 μM each) vs fatty acid free BSA (0.17mM) (Roche, 03117057001) conjugate control, 5mM glucose, and 1% P/S for 24 h. 10 μM Bafilomycin (Sigma, B1793) was added to a portion of the palmitate (100 μM) vs dPBS treated cells for the last 3 h of treatment. Cells were washed with dPBS before collection.
2.5 Flow Cytometry
2.5.1 Whole Epididymal White Adipose Tissue Preparation
Epididymal white adipose tissue (eWAT) was extracted from 19 LFD-fed WT mice, 19 HFD-fed WT mice, 7 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 7 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 5 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and 6 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. eWAT was washed once in FACS Buffer (dPBS buffer containing 0.2% BSA (Sigma, A2153) 1% EDTA) and centrifuged at 500g x 5 min to remove peripheral red blood cells. Tissue was digested in collagenase (Sigma, C6885) at 37°C for 30 min – 1 h to obtain a single cell suspension. Cells were filtered (100 μm) and centrifuged (500g x 5 min at 4°C) to separate the floating adipocyte fraction from the stromal vascular fraction (SVF). The floating adipocyte fraction from 8 LFD-fed WT mice and 8 HFD-fed WT mice were removed and placed into QIAzol lysis reagent (QIAGEN, 79306) for RNA extraction or 1X cell lysis buffer (Cell Signaling, 9803) for protein extraction. SVFs were incubated with RBC lysis buffer solution (eBioscience, 00-4333-57) for 3 min at room temperature and then centrifuged at 500g x 5 min at 4°C.
2.5.2 Primary Cell Culture Preparation
Differentiated BMDMs from 8–10 week old 4 LFD-fed WT mice treated as in 2.4.2 excluding the bafilomycin were gently scraped in FACS Buffer and washed once by centrifugation (500g x 5 min at 4°C).
2.5.3 BODIPY analysis
SVFs from 7 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 7 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, 5 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and 6 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice were blocked with CD16/32 Antibody (BioLegend, 101302) for 30 min at 4°C, centrifuged at 500g x 5 min at 4°C, and then incubated with CD45 - Alexa Fluor 700 (BioLegend, 103128), F4/80 - PerCPCy5.5 (eBioscience, 45-4801-82), CD11b - APC-Cy7 (BD Pharmingen, 557657), CD11c - PE-Cy7 (eBioscience, 25-0114-82), DAPI (ThermoFisher, D1306), and BODIPY 493/503 (ThermoFisher, D3922) for 30 min at 4°C. Cells were then washed and centrifuged (500g x 5 min at 4°C) and resuspended in FACS buffer for quantification on the Sony SY3200 (HAPS) sorter. FCS files were analyzed with FlowJo 10.1 software.
2.5.4 LipidTox™ analysis
SVFs from 15 LFD-fed WT mice and 15 HFD-fed WT mice and primary BMDMs as described in 2.5.2 were incubated with Fc block and CD45, F4/80, CD11b, and CD11c as described in 2.5.3. Live/Dead® Fixable Blue Dead Cell Stain Kit (ThermoFisher, L34962) was used instead of DAPI. Then cells were fixed with 4% paraformaldehyde (Affymetrix, 19943) for 10–15 min at 4°C or with the Cytofix/Cytoperm kit (BD Biosciences, 51-2090KZ). Cells were then incubated with HCS LipidTox™ Red Neutral Lipid (ThermoFisher, H34476) for 30 min at 4°C, then washed, centrifuged (500g x 5 min at 4°C) and resuspended in FACS buffer for quantification on the BD LSR-II UV analyzer. FCS files were analyzed with FlowJo 10.1 software.
2.5.5 Fluorescent Activated Cell Sorting
SVFs from 4 LFD-fed WT mice and 4 HFD-fed WT mice were prepared as described in 2.5.3 without BODIPY labeling and sorted on the BD FACSAria IIu SORP into two populations: FB (CD45+, F4/80+, CD11b+, CD11c−) and FBC (CD45+, F4/80+, CD11b+, CD11c+) (Supplemental Fig. 1).
2.6 Western Blot Analysis
2.6.1
Tissues or cells were homogenized in cell lysis buffer, vortexed for 30 seconds and frozen at −80°C. Samples were thawed, vortexed for 30 seconds, and then centrifuged at 14,000 rpm for 10–20 min at 4°C. Supernatants containing the protein were collected and protein concentration was measured using the Pierce BCA Protein Assay kit (ThermoFisher, 23225). 50 μg of protein were loaded into 12% Mini-Protean TGX gels (BioRad, 456-1043) and subjected to electrophoresis at 80V using 1X Tris/Glycine/SDS Buffer (BioRad, 161-0732) for 1.5 – 2 h on ice. Proteins were then transferred at 30V for 90 min to a 2 μm nitrocellulose membrane (BioRad, 162-0212) using 1X Tris/Glycine Buffer (BioRad, 161-0734) with 20% methanol. Membranes were blocked at room temperature for 20–30 min using dPBS Odyssey® Blocking Buffer (LI-COR, 927-40000) and then incubated with primary antibody overnight at 4°C. Membranes were washed with 1X TBST, 3 times for 5 min each at room temperature, incubated with secondary antibody for 1 h at 4°C, and then washed again using dPBS without tween for the final wash. Membranes were scanned using the LI-COR Odyssey® Imager and analyzed using Odyssey 3.0 and Image Studio Lite 5.2.5 software. Primary antibodies used were SQSTM1/p62 (Cell Signaling, 5114, 1:1000) LC3B polyclonal (Cell Signaling, 2775, 1:1000), and tubulin (Cell Signaling, 3873, 1:1000). Secondary antibodies used were anti-mouse (LI-COR, 926-68070, 1:50,000) and anti-rabbit (LI-COR, 925-32211, 1:10,000).
2.7 Quantitative PCR Analysis
2.7.1
Tissues or cells were homogenized in QIAzol or RLT buffer (QIAGEN, RNeasy Micro Kit) with 0.1% β-mercaptoethanol (Sigma, M6250) and either frozen in −80°C or processed immediately. RNA extraction was done with the RNeasy Mini Kit (QIAGEN, 74106) or the RNeasy Micro Kit (QIAGEN, 74004). RNA concentration was assessed by the NanoDrop® Spectrophotometer (ND-1000) and cDNA was made with iScript cDNA synthesis kit (BioRad, 170-8891). qPCR was performed with TaqMan® probes (Atg2b - Mm00620760_m1, Atg16l1 - Mm00513085_m1, Atg5 - Mm00504340_m1, and Atg7 - Mm00512209_m1, Adgre1 - Mm00802529_m1, Itgax - Mm00498698_m1, Tnf - Mm00443258_m1, Ccl2 - Mm00441242_m1, Nos2 - Mm00440502_m1, Il-10 - Mm01288386_m1, Arg-1 - Mm00475988_m1, CD163 - Mm00474091, Mrc-1 - Mm01329362_m1) using the comparative CT method (ΔΔCT) on the Applied Biosystems StepOne™ Real-Time PCR machine. The relative abundance of mRNA was normalized to the CT of 18s rRNA (Applied Biosystems, 4318839) or Actb (Applied Biosystems, 4351315) for each sample.
2.8 Statistical Analyses
Data are presented as mean ± SEM. All statistical analyses were performed using GraphPad Prism 7.0b. Independent two-sample t-tests (two tails) were used to assess significance between 2 groups of samples. Two way ANOVA with Tukey HSD post hoc test was used to evaluate the difference among 2 groups with 2 factors. P-values <0.05 were used to denote statistical significance.
3. Results
3.1 No significant effect of HFD on autophagy in whole eWAT, eWAT floating adipocyte fractions or eWAT ATMs in mice
To determine the effect of HFD feeding on autophagy, C57BL/6 WT male mice were fed a LFD or HFD for 3 months beginning at 8 weeks of age. Whole eWAT, eWAT floating adipocyte fractions, and eWAT ATMs were collected at the end of the time course (following 3 months of diet) and assessed for markers of autophagy. In whole eWAT, there was no significant difference between LC3 protein expression in HFD- vs LFD-fed mice, as assessed by the ratio of LC3-II:LC3-I (Fig. 1A – 1B). To determine if there was evidence of lysosomal dysfunction, we measured expression of lysosomal protein, p62. There was a trend toward increased expression of the p62 protein in eWAT from HFD-fed vs. LFD-fed mice, but this difference is not significant (Fig. 1A, 1C). Since we did not observe differences in autophagy in whole eWAT from HFD-fed vs LFD-fed mice, we examined autophagy in two distinct eWAT cell populations: floating adipocyte fractions and ATMs. In eWAT floating adipocytes from HFD-fed vs. LFD-fed WT mice, there was no difference in the LC3-II:LC3-I protein ratio and a trend toward decreased expression of p62 protein, which did not reach statistical significance (Fig 1D – 1F). Similarly, no differences in mRNA expression of autophagy genes Atg2b, Atg16l1, Atg5, and Atg7 were observed in the floating adipocyte fractions (Fig. 1G). In two subsets of eWAT ATMs, FB (F4/80+, CD11b+, CD11c−; less prone to accumulate lipids) and FBC (F4/80+, CD11b+, CD11c+; more prone to accumulate lipids) [6], we did not observe significant differences in the autophagy genes Atg2b, Atg16l1, Atg5, and Atg7 (Fig. 1H), nor was there a significant interaction effect. Taken together, HFD feeding does not induce significant changes in autophagy in whole eWAT, in eWAT floating adipocyte fractions, or in eWAT ATMs.
Figure 1. No significant effect of HFD on autophagy in whole eWAT, eWAT floating adipocyte fraction or eWAT ATMs in mice.
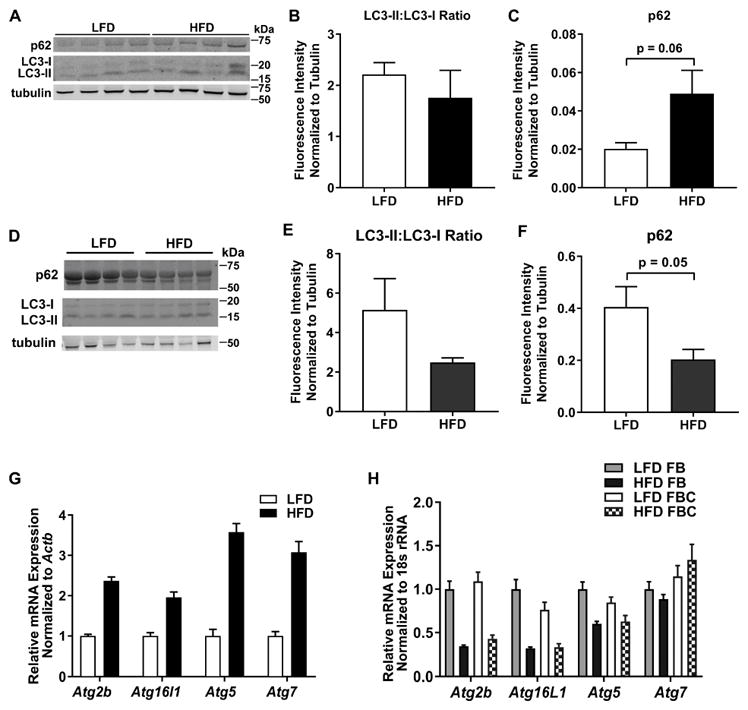
Male WT mice were fed a LFD or HFD for 3 months, beginning at 8 weeks of age. A. Protein expression of p62 and LC3 in whole eWAT was determined by Western blotting. B. Quantification of A; The ratio of LC3-II:LC3-I normalized to tubulin is reported in n=4 mice/group. C. Quantification of A; The ratio of p62 normalized to tubulin is reported in n=4 mice/group. D. Protein expression of LC3 and p62 in eWAT floating adipocyte fraction. E. Quantification of D; The ratio of LC3-II:LC3-I normalized to tubulin is reported in n=4 mice/group. F. Quantification of D; The ratio of p62 normalized to tubulin is reported in n=4 mice/group. G. mRNA expression of the indicated autophagy genes in eWAT floating adipocyte fraction, n=5 mice/group. H. mRNA expression of the indicated autophagy genes in FB and FBC eWAT ATMs, n=4 LFD FB, n=4 HFD FB, n=3 LFD FBC, n=4 HFD FBC. FBC = F4/80+ CD11b+ CD11c+, FB = F4/80+ CD11b+ CD11c−
3.2 Increases in macrophage lipid content do not alter expression of autophagy proteins
It was previously established that ATMs in mice with obesity have increased lipid content as compared to WT controls [5]. In mice fed a 60% HFD, we observed a significant increase in neutral lipid content in ATMs of HFD-fed mice as compared to LFD-fed mice; p<0.05 (Fig. 2A – 2B). In primary BMDMs treated with a mixture of saturated (palmitate) and unsaturated (oleate) fatty acids to mimic the components of a HFD, we observed a significant increase in neutral lipid content as compared to control bovine serum albumin (BSA)-treated BMDMs (Fig. 2C – 2D). However, there was no significant difference in LC3-II:LC3-I nor p62 protein between fatty acid- and control-treated BMDMs, treated with either 100 μM palmitate (Fig. 2E – 2G) or a 500 μM palmitate/500 μM oleate mixture (Fig. 2H – 2J). Treatment with bafilomycin, a lysosome inhibitor, showed an increase in LC3-II:LC3-I protein ratio, ruling out effects due to lysosome dysfunction (Fig. 2E – 2G). In Fig. 2F, ANOVA analyses show a significant interaction effect (p < 0.05) between bafilomycin and fatty acid treatment. Tukey HSD post-hoc testing was used for multiple group testing, which showed a significant increase in bafilomycin treatment only (p < 0.0001). There was no significant difference between BSA and Palmitate treatment. ANOVA analyses did not show significant interactions for 2G. Taken together, these data suggest that lipid loading in ATMs and BMDMs does not result in changes in autophagy, at least under the conditions employed in these experiments.
Figure 2. Lipid loading does not affect macrophage autophagy: ex vivo and in vitro studies.

A. Neutral lipid species in total eWAT ATMs of LFD and HFD mice were detected by LipidTox™ staining. A representative image is shown. B. Quantification of neutral lipid species represented in A, n=15 mice/group. C. Neutral lipid species in BMDMs treated for 24 h with 500 μM palmitate and 500 μM oleate were detected by LipidTox™ staining. A representative image is shown. D. Quantification of neutral lipid species represented in C, BMDMs from n=4 mice/group. E – J. The ratio of LC3-II:LC3-I or levels of p62 normalized to tubulin is reported by Western blotting in BMDMs treated with the indicated fatty acid for 24 h from n=4 mice/group. Where indicated, BMDMs were treated with 10 μm bafilomycin for the last 3 h in the 24 h fatty acid treatment time period. F and G are quantifications of E. I and J and quantifications of H. FMO = fluorescence minus one, Palm = palmitate, Baf = bafilomycin * = p<0.05, ** = p<0.01, **** = p<0.0001
3.3 Deletion of autophagy gene, Atg16l1, in the myeloid compartment does not affect obesity, IR, or ATM lipid content in vivo
To definitively test if myeloid autophagy affects the response to high fat feeding, we fed littermate male mice deficient in myeloid Atg16l1, LysMcre(+/−).Atg16l1flox/flox, vs. control LysMcre(−/−).Atg16l1flox/flox mice [19, 20] a LFD or HFD for 3 months beginning at 8 weeks of age. After 3 months on diet, we assessed a number of metabolic responses in these animals, including body mass, fat mass vs lean mass, food consumption, fasting blood glucose, and glucose- and insulin sensitivity. There was no signficiant difference nor interaction effect between HFD- and LFD-fed myeloid-deficient Atg16l1 mice vs control mice for any of these metabolic endpoints (Fig. 3A – 3I), despite equivalent energy intake (Fig. 3D). Furthermore, we observed no differences in total neutral lipid content of the eWAT ATMs between the two genotypes, although the expected increases in eWAT ATM lipid content by HFD diet were observed (Fig. 3J – 3K). Taken together, these data indicate that deletion of myeloid Atg16l1 does not impact body mass, lean or fat mass, IR, or ATM lipid content compared to littermate control mice fed HFD.
Figure 3. Deletion of autophagy gene, Atg16l1, in the myeloid compartment does not affect body mass or the metabolic response to HFD in mice.
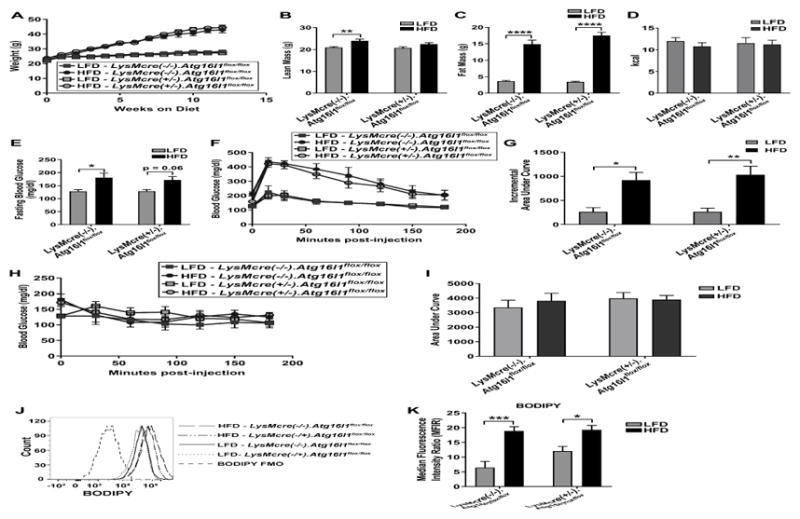
Male LysMcre(+/−).Atg16l1flox/flox mice vs littermate LysMcre(−/−).Atg16l1flox/flox mice were fed a LFD or HFD for 3 months, beginning at 8 weeks of age. All data shown reflect analyses at 3 months on diet, unless otherwise indicated. A. Body mass (g), n=16 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=15 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=12 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, n=13 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. B. DEXA scan for determination of lean mass. C. DEXA scan for determination of fat mass. B – C. n=10 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=12 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=12 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and n=11 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. D. Energy intake over 24 h, n=9 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=8 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=7 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and n=7 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. E. Fasting blood glucose. F. Glucose tolerance test. G. Incremental area under curve of F. H. Insulin tolerance test. I. Area under curve of H. J. eWAT ATM neutral lipids, representative image. K. eWAT ATM neutral lipid quantification. E – K. n=7 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=7 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=5 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and n=6 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. * = adj p<0.05, ** = adj p<0.01, *** adj p<0.001, **** = adj p<0.0001
3.4 Myeloid Atg16l1 deletion does not affect adipose tissue inflammation in HFD feeding
It was previously established that in HFD-fed mice, the total number of macrophages in VAT increases from 10% to 50%, and peak infiltration of these macrophages occurs at 12 weeks of feeding [3]. We therefore assessed the ATM content in the eWAT of these myeloid-Atg16l1 deleted vs control mice by flow cytometry after 3 months of HFD feeding, and found no difference nor interaction effect in total macrophage content (Fig. 4A) or in the ratio of FBC to FB macrophage content (Fig. 4B). We also assessed ATM content in whole eWAT by quantifying the mRNA expression of Adgre1, which is the gene that encodes the macrophage marker, F4/80. We observed no significant differences nor significant interaction effects in Adgre1 expression (Fig. 4C). To specifically address adipose tissue inflammation, we measured the mRNA expression levels of 4 pro-inflammatory (Itgax, Ccl2, Tnfα, Nos2) and 4 anti-inflammatory (Il10, Arg1, Mrc1, CD163) genes in whole eWAT from these mice. CT values of all genes in eWAT from LFD fed mice were too low to be detected (data not shown). In eWAT from HFD fed mice, there were no significant changes in expression in any of the inflammatory genes (Fig. 4D), indicating that myeloid Atg16l1 likely does not affect adipose tissue inflammation in HFD feeding.
Figure 4. Myeloid Atg16l1 deletion does not affect adipose tissue inflammation.

Male LysMcre(+/−).Atg16l1flox/flox mice vs littermate LysMcre(−/−).Atg16l1flox/flox mice were fed a LFD or HFD for 3 months, beginning at 8 weeks of age. All data shown reflect analyses at 3 months on diet. A. eWAT ATM content. B. FBC:FB ratio of ATM content. A – B. n=7 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=7 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=5 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and n=6 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice. C. eWAT Adgre1 mRNA expression, n=3 LFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=4 HFD-fed LysMcre(+/−).Atg16l1flox/flox mice, n=4 LFD-fed LysMcre(−/−).Atg16l1flox/flox mice, and n=3 HFD-fed LysMcre(−/−).Atg16l1flox/flox mice.. D. Gene expression of the indicated pro- or anti-inflammatory genes in whole eWAT from HFD fed mice, n=4 LysMcre(+/−).Atg16l1flox/flox mice, n=3 LysMcre(−/−).Atg16l1flox/flox mice. FBC = F4/80+ CD11b+ CD11c+, FB = F4/80+ CD11b+ CD11c−
4. Discussion
The role of autophagy in obesity, IR, and VAT inflammation appears to be context dependent. Different studies yield distinct results depending on the specific diet, age and sex of mice, the duration of high fat feeding, the mediators of obesity in the different models, and the deletion of distinct genes linked to autophagy. Two common obesity models used to study the putative role of autophagy are a genetically obese Lepob model or LPS stimulation in a HFD-fed mouse model. However, both of these models have limitations for studying the mechanistic progression of obesity to T2D. Lepob mice are deficient in the production of leptin, a hormone that induces satiety. Although obesity adversely affects leptin function, mutations in the human leptin gene are rare [23]. A HFD fed model is another mechanistic approach, but the addition of LPS may complicate results pertaining to VAT inflammation. HFD feeding alone increases circulating LPS, but in a more pathophysiologically relevant manner compared to direct injection [24]. LPS induces inflammation, as it is an endotoxin present on the outer coat of gram negative bacteria [25]. Chronic LPS infusion in LFD fed mice increases adiposity and IR, independent of HFD feeding [26, 27]. Therefore, adding excess LPS to a HFD-fed model may amplify inflammation to a degree not likely to be observed in vivo. In the present study, we utilized a HFD model of obesity without additional stimulation to address this issue.
The two main autophagy proteins previously studied, ATG7 and ATG5, are both involved in LC3 lipidation, a process involved in expansion of the autophagosomal membrane [16]. However, there are other crucial components of the autophagy pathway including autophagolysosome fusion and autophagosomal membrane biogenesis [16]. WIPI-2B, an autophagosomal biogenesis protein, binds directly to ATG16L1 to recruit the ATG16L1-ATG5-ATG12 lipidation complex to the autophagosomal membrane [15]. By focusing on ATG16L1, our study examines the effect of inhibiting the connection between autophagosome biogenesis and autophagosomal membrane lipidation on the lipid-inflammation-obesity axis.
Our results add to the body of literature demonstrating that autophagy is not likely to be a contributing mechanism by which ATM lipid metabolism is connected to whole body IR. We report that HFD alone does not change autophagy protein or gene expression in whole eWAT, eWAT floating adipocyte fractions, or eWAT ATMs, despite the increase in neutral lipid species in ATMs. In vitro treatment of macrophages with fatty acids to mimic a HFD yields analogous results. Furthermore, myeloid-specific deletion of Atg16l1 does not change metabolic endpoints on a LFD or HFD, nor does it affect ATM number, the amount of neutral lipids within these cells, or adipose tissue pro- vs. anti-inflammatory gene expression. These data suggest that autophagy is not an important mechanism in obesity induced VAT inflammation, despite inhibition of autophagy at an earlier point in the pathway.
Surprisingly, we did not observe a significant HFD induced increase in macrophage infiltration into eWAT by flow cytometry or Adgre1 expression. However, there is a trending increase in the FBC subset of macrophages in the HFD fed mice (Fig 4B) as would be expected [28]. Furthermore, there is an increase in inflammatory gene expression in HFD vs LFD fed mice, as the CT values in the LFD mice were too low to be detected compared to the HFD mice where CT values were detected for all genes. There was no effect, however, of myeloid deletion of Atg16l1 on expression of adipose tissue inflammatory genes in mice fed HFD.
Myeloid ATG16L1 is important in pathogen induced inflammation and lipid metabolism [18, 19], although our results show no role in VAT macrophage lipid content or inflammation in HFD feeding. Our results are similar to other studies of ATG16L1-related inflammation. In VAT from humans with inflammatory Crohn’s disease, there is no change in Atg16l1 mRNA expression as compared to anti-inflammatory controls [29]. In a Salmonella infection model in mice, only epithelial deletion of Atg16l1 increased ileal and cecal inflammation as compared to no inflammatory changes in mice bearing macrophage and dendritic cell deletion of Atg16l1 [13]. However, in a murine colitis model, myeloid deletion of Atg16l1 exacerbated gut inflammation, and increased the percentage of pro-inflammatory macrophages specifically in the gut [17]. Therefore, it is possible that ATG16L1 is a gut-specific inflammatory regulator, although the mechanistic details remain to be clarified.
Other genes and proteins in the autophagy pathway may be more important than ATG16L1 in autophagy’s role in metabolic disease. ATG2 is suggested to be important in the biogenesis of lipid droplets and autophagosomes [30, 31], and as such may be more important in the autophagy-lipid metabolism connection. Although we did not see changes in Atg2b gene expression in our studies, it may play a larger role in adipocytes or in other tissues. WIPI-2B, ATG16L1’s binding partner, could also be more important in studying the effects of obesity on autophagosomal biogenesis.
Autophagy may be more important in other cell types and tissues in the progression of obesity to T2D. Deletion of Atg7 in skeletal muscle protects mice from DIO and IR by increasing fatty acid oxidation and WAT browning [32]. However, liver specific deletion of Atg7 results in an increase in lipid accumulation [33]. We show that in the eWAT floating adipocyte fraction, HFD does not affect autophagy protein expression, but may cause lysosomal dysfunction, as assessed by trends to decreased p62 protein in the floating adipocytes of HFD- vs. LFD-fed mice (Figure 1F). Other studies show signficiant HFD induced autophagy changes in adipocytes. In vitro, palmitate treatment of 3T3L1 adipocytes increases autophagic flux [34], and adipocyte-specific deletion of Atg7 in mice fed a HFD yields leaner mice with smaller adipocytes and increased insulin sensitivity [35]. These divergent findings may underscore the importance of differences in time point of assessment (early vs later time point exposures to high fat feedng), the fat content of the specific diets, genetic background of the mice, and sex of the mice. They may also highlight differences in difficult to control facility specific conditions, such as vivarium temperature, water source, and pathogen status. All of these differences might influence the composition of the gut microbiome, a factor that is known to affect obesity and metabolic syndrome [36]. Taken together, such considerations suggest distinct organ-specific roles for autophagy in obesity and HFD feeding.
Our finding that HFD may cause changes in lysosomal function is similar to other studies that suggest roles for HFD inducing lysosomal dysfunction, albeit using different experimental conditions and stratgies. For example, adipocytes from mice fed a HFD and 3T3L1 adipocyte-like cells treated in vitro with palmitate both displayed lysosome permeabilization, although levels of p62 were not monitored [37]. Also, HFD-fed mice deficient in cathepsin B, a lysosomal protease, were protected from lysosome permeabilization and reduced ATM inflammation [37]. HFD was shown to induce lysosomal hypertrophy in kidney proximal tubular cells of mice fed HFD [38]. Furthermore, cathepsin K, another lysosomal protease, was found to be increased in the hearts of HFD-fed mice, and palmitate treatment of cardiomyocytes induced release of cathepsin K into the cytoplasm [39]. Taken together, the results of these published studies suggest that the association of lysosomal dysfunction with HFD feeding or fatty acid treatment in cultured adipocyte-like cells is highly condition-dependent.
In summary, our results add to the evidence suggesting that myeloid autophagy is not critically involved in obesity induced VAT inflammation or systemic metabolic dysfunction.
Supplementary Material
Highlights.
Myeloid Atg16l1 deletion does not affect visceral adipose tissue inflammation.
Myeloid Atg16l1 deletion does not block diet-induced obesity or insulin resistance.
Macrophage lipid content does not correlate with expression of autophagy proteins.
LC3 expression in visceral adipose tissue does not change in high fat feeding.
Acknowledgments
We acknowledge Latoya Woods, New York University Langone Health, for her help in assembling the manuscript. Cell sorting/flow cytometry services were provided by Michael Gregory, Kamilah Ryan, and the NYU School of Medicine’s Cytometry Core, which is supported in part by grant P30CA016087 from the National Institutes of Health/National Cancer Institute. This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, grant number 1R01DK109675.
Abbreviations
- T2D
type 2 diabetes
- VAT
visceral adipose tissue
- IR
insulin resistance
- LFD
low fat diet
- HFD
high fat diet
- ATM
adipose tissue macrophage
- DIO
diet induced obesity
- BMDM
bone marrow derived macrophage
- WT
wildtype
- eWAT
epididymal white adipose tissue
- FB
F4/80+, CD11b+, CD11c−
- FBC
F4/80+, CD11b+, CD11c+
Footnotes
Author Contributions: EMSL designed the studies, performed the experiments, analyzed the data, and wrote and edited the manuscript. MYG and KS performed the experiments and edited the manuscript. JH and HL reviewed all the statistical analyses and edited the manuscript. KC provided mice, gave guidance on experimental design, and edited the manuscript. AMS designed and supervised the studies and study design, discussed data interpretation, and revised and edited the manuscript. All authors approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–80. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 3.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prieur X, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. 2011;60(3):797–809. doi: 10.2337/db10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu X, et al. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18(6):816–30. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang YH, et al. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget. 2016;7(24):35577–35591. doi: 10.18632/oncotarget.9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee HY, et al. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy. 2016;12(8):1390–403. doi: 10.1080/15548627.2016.1184799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grijalva A, Xu X, Ferrante AW., Jr Autophagy Is Dispensable for Macrophage-Mediated Lipid Homeostasis in Adipose Tissue. Diabetes. 2016;65(4):967–80. doi: 10.2337/db15-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu K, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11(2):271–84. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, et al. Inhibition of mTOR reduces lipotoxic cell death in primary macrophages through an autophagy-independent mechanism. J Leukoc Biol. 2016;100(5):1113–1124. doi: 10.1189/jlb.3A1015-463R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He L, Weber KJ, Schilling JD. Glutamine Modulates Macrophage Lipotoxicity. Nutrients. 2016;8(4):215. doi: 10.3390/nu8040215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conway KL, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145(6):1347–57. doi: 10.1053/j.gastro.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lassen KG, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111(21):7741–6. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dooley HC, Wilson MI, Tooze SA. WIPI2B links PtdIns3P to LC3 lipidation through binding ATG16L1. Autophagy. 2015;11(1):190–1. doi: 10.1080/15548627.2014.996029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavel M, Rubinsztein DC. Mammalian autophagy and the plasma membrane. FEBS J. 2016 doi: 10.1111/febs.13931. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, et al. Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis. J Immunol. 2017 doi: 10.4049/jimmunol.1601293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchiando AM, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe. 2013;14(2):216–24. doi: 10.1016/j.chom.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang S, et al. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012;11(4):397–409. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carstensen M, Thomsen C, Hermansen K. Incremental area under response curve more accurately describes the triglyceride response to an oral fat load in both healthy and type 2 diabetic subjects. Metabolism. 2003;52(8):1034–7. doi: 10.1016/s0026-0495(03)00155-0. [DOI] [PubMed] [Google Scholar]
- 22.Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008 doi: 10.1101/pdb.prot5080. pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 23.Rehman Khan A, Awan FR. Leptin Resistance: A Possible Interface Between Obesity and Pulmonary-Related Disorders. Int J Endocrinol Metab. 2016;14(1):e32586. doi: 10.5812/ijem.32586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boutagy NE, et al. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie. 2016;124:11–20. doi: 10.1016/j.biochi.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seeley JJ, Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol. 2017;101(1):107–119. doi: 10.1189/jlb.3MR0316-118RR. [DOI] [PubMed] [Google Scholar]
- 26.Neves AL, et al. Metabolic endotoxemia: a molecular link between obesity and cardiovascular risk. J Mol Endocrinol. 2013;51(2):R51–64. doi: 10.1530/JME-13-0079. [DOI] [PubMed] [Google Scholar]
- 27.Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 28.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leal RF, et al. Autophagy is decreased in mesenteric fat tissue but not in intestinal mucosae of patients with Crohn’s disease. Cell Tissue Res. 2012;350(3):549–52. doi: 10.1007/s00441-012-1491-8. [DOI] [PubMed] [Google Scholar]
- 30.Pfisterer SG, et al. WIPI beta-propellers at the crossroads of autophagosome and lipid droplet dynamics. Biochem Soc Trans. 2014;42(5):1414–7. doi: 10.1042/BST20140152. [DOI] [PubMed] [Google Scholar]
- 31.Velikkakath AK, et al. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23(5):896–909. doi: 10.1091/mbc.E11-09-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim KH, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19(1):83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Lopez N, Singh R. Autophagy and Lipid Droplets in the Liver. Annu Rev Nutr. 2015;35:215–37. doi: 10.1146/annurev-nutr-071813-105336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin J, et al. Palmitate induces endoplasmic reticulum stress and autophagy in mature adipocytes: implications for apoptosis and inflammation. Int J Mol Med. 2015;35(4):932–40. doi: 10.3892/ijmm.2015.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh R, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119(11):3329–39. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouter KE, et al. Role of the Gut Microbiome in the Pathogenesis of Obesity and Obesity-Related Metabolic Dysfunction. Gastroenterology. 2017 doi: 10.1053/j.gastro.2016.12.048. [DOI] [PubMed] [Google Scholar]
- 37.Gornicka A, et al. Adipocyte hypertrophy is associated with lysosomal permeability both in vivo and in vitro: role in adipose tissue inflammation. Am J Physiol Endocrinol Metab. 2012;303(5):E597–606. doi: 10.1152/ajpendo.00022.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto T, et al. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J Am Soc Nephrol. 2017;28(5):1534–1551. doi: 10.1681/ASN.2016070731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hua Y, et al. Cathepsin K knockout mitigates high-fat diet-induced cardiac hypertrophy and contractile dysfunction. Diabetes. 2013;62(2):498–509. doi: 10.2337/db12-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.