

Abstract
CYLD is a deubiquitinating enzyme that plays a crucial role in immunity and inflammation as a negative regulator of NF-κB transcription factor and JNK kinase signaling. Defects in either of these pathways contribute to the progression of numerous inflammatory and autoimmune disorders. Therefore, we set out to unravel molecular mechanisms that control CYLD activity in the context of T cell receptor (TCR) signaling. More specifically, we focused on CYLD phosphorylation at Ser418, which can be detected upon immunoblotting of cell extracts with phospho(Ser418)-CYLD specific antibodies. Jurkat T cells stimulated with either anti-CD3/anti-CD28 or PMA/Ionomycin (to mimic TCR signaling) were used as a model system. The role of specific kinases was analyzed using pharmacological as well as genetic approaches. Our initial data indicated that CYLD is directly phosphorylated by the noncanonical IκB kinases (IKKs) IKKε and TANK Binding Kinase 1 (TBK1) at Ser418 upon TCR stimulation. Treatment with MRT67307, a small compound inhibitor for IKKε and TBK1, inhibited TCR-induced CYLD phosphorylation. However, the phospho(Ser418)-CYLD immunoreactive band was still present in CRISPR/Cas9 generated IKKε/TBK1 double knockout cell lines, where it could still be prevented by MRT67307, indicating that the initially observed inhibitory effect of MRT67307 on TCR-induced CYLD phosphorylation is IKKε/TBK1-independent. Most surprisingly, the phospho(Ser418)-CYLD immunoreactive band was still detectable upon immunoblotting of cell extracts obtained from CYLD deficient cells. These data demonstrate the non-specificity of MRT67307 and phospho(Ser418)-CYLD specific antibodies, implying that previously published results based on these tools may also have led to wrong conclusions. We therefore advise to use genetic knockout studies or alternative approaches for a better validation of antibodies and small compound inhibitors. Interestingly, immunoprecipitation with the phospho(Ser418)-CYLD antibody, followed by immunoblotting with anti-CYLD, revealed that CYLD is phosphorylated by IKKε/TBK1 at Ser418 upon T cell stimulation, but that its direct detection with the phospho(Ser418)-CYLD-specific antibody in a western blot is masked by another inducible protein of the same size that is recognized by the same antibody.
Keywords: CYLD, IKKε, TBK1, T cell receptor, phosphorylation, kinase inhibitor, antibodies
Introduction
Cylindromatosis (CYLD) is a deubiquitinating enzyme that was originally identified as a tumor suppressor in familial cylindromatosis, but has since then also been implicated in other cancer types (Bignell et al., 2000; Massoumi, 2011). The majority of CYLD mutations and truncations found in patients negatively affect its expression or deubiquitinase activity (Massoumi, 2011). Next to its tumor suppressor function CYLD is also a key regulator of immunity and inflammation, which is demonstrated in multiple CYLD genetic mouse models (reviewed in Lork et al., 2017). For example, CYLD knockout mice show defective T cell development and T cells from these mice are hyperresponsive to T cell receptor (TCR)-induced activation and are hyper-proliferative. This hyperresponsive phenotype in CYLD-deficient mice is associated with autoimmune responses and bowel inflammation (Reiley et al., 2006; Zhang et al., 2006). In humans, single nucleotide polymorphisms in the CYLD gene have been associated with inflammatory bowel disease (Cleynen et al., 2014). CYLD is a deubiquitinase capable of cleaving K63-linked as well as M1-linked polyubiquitin chains from target proteins (Komander et al., 2009; Ritorto et al., 2014). CYLD negatively regulates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling by removing polyubiquitin chains from specific target proteins including NF-κB essential modifier (NEMO), TNF receptor associated factor (TRAF) 2 and TRAF6, and Transforming growth factor beta-activated kinase 1 (TAK1) (Brummelkamp et al., 2003; Kovalenko et al., 2003; Trompouki et al., 2003; Yoshida et al., 2005; Reiley et al., 2007). In addition, CYLD was shown to negatively affect c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) signaling pathways, which impacts immune cell function, activation and homeostasis (Yoshida et al., 2005; Zhang et al., 2006; Reiley et al., 2007). Lack of functional CYLD leads to constitutively active downstream NF-κB and MAPK signaling (Reiley et al., 2005; Zhang et al., 2006). Given the importance of CYLD in inflammation and cancer, a better understanding of molecular mechanisms regulating CYLD activity is of considerable interest. CYLD is constitutively expressed in most cell types (Uhlen et al., 2015), suggesting an important role for posttranslational modifications in regulating CYLD activity. Inhibitor of nuclear factor kappa-B kinase (IKK) β- and NEMO-dependent phosphorylation of CYLD on multiple residues within a serine cluster between amino acids 418 and 444 was shown upon stimulation with tumor necrosis factor (TNF), lipopolysaccharide (LPS) and mitogens (Reiley et al., 2005). Other work shows that CYLD can be phosphorylated upon overexpression of the IKK-related kinase IKKε, facilitating IKKε-driven cellular transformation (Hutti et al., 2009).
The serine/threonine kinase IKKε and its homolog TANK binding kinase 1 (TBK1) are referred to as non-canonical IKK kinases as they are closely related to the canonical IKKα and IKKβ, sharing 33% sequence identity within their catalytic kinase domain (Peters et al., 2000; Tojima et al., 2000). IKKε and TBK1 have been intensively studied in the context of type I interferon (IFN) induction in response to viral infection and various pattern recognition receptors, but have also been implicated in the regulation of a number of other processes including autophagy, metabolic regulation and oncogenesis (Shen and Hahn, 2011; Verhelst et al., 2013; Brinkman et al., 2014; Oakes et al., 2017). IKKε/TBK1-mediated type I IFN induction is due to their ability to phosphorylate IFN regulatory factor (IRF) 3 and 7 transcription factors (Fitzgerald et al., 2003; Sharma et al., 2003; Hemmi et al., 2004). Additionally IKKε and TBK1 have been described as NF-κB modulators by phosphorylating IκBα on one of the two critical serines involved in triggering its degradation (Shimada et al., 1999; Bonnard et al., 2000; Peters et al., 2000). Even though TBK1 and IKKε seem to have indistinguishable activities in the activation of IRF3 and IRF7, they do not seem to be fully redundant as they have differential expression patterns and substrate specificities (Fitzgerald et al., 2003; Yu et al., 2012). TBK1 is expressed ubiquitously, while IKKε expression is restricted to particular tissues including the lymphoid tissue, peripheral blood lymphocytes and the pancreas (Shimada et al., 1999; Tojima et al., 2000). TBK1 knockout mice are embryonically lethal and die on embryonic day 14.5 due to massive liver degeneration and apoptosis (Bonnard et al., 2000), while IKKε-deficient mice are viable (Hemmi et al., 2004). TBK1 and IKKε were shown to be activated upon TCR stimulation (Peters et al., 2000; Tojima et al., 2000), but their role in TCR signaling is largely unknown.
We set out to analyze if the noncanonical kinases IKKε and TBK1 regulate TCR-mediated signaling by phosphorylating the deubiquitinase CYLD at Ser418 in Jurkat T cells. Phosphorylation of CYLD at Ser418 was detected upon immunoblotting of cell extracts using phospho(Ser418)-CYLD-specific antibodies. The role of IKKε and TBK1 was assessed using the small compound inhibitor MRT67307 as well as by IKKε/TBK1 double knockout cell lines generated via CRISPR/Cas9. Our data demonstrate that TCR stimulation leads to the phosphorylation of an unknown protein, which is distinct from CYLD but detectable using the phospho(Ser418)-CYLD antibody. Moreover, we show that phosphorylation of this protein can be inhibited by MRT67307 independent of its IKKε and TBK1 inhibitory activity.
Materials and methods
Cells, stimulation, transfection
Jurkat T cells were cultured in Roswell Park Memorial Institute (RPMI) supplemented with 10% FCS, 1 mM sodium pyruvate, 2 mM L-glutamine and HEK 293T cells were maintained under standard culturing conditions in Dulbecco's Modified Eagle Medium (DMEM) from Gibco, supplemented with 10% FCS, 1 mM sodium pyruvate, 2 mM L-glutamine. HEK 293T cells were transfected with the indicated plasmids using calcium phosphate and incubated for 24 h at 37°C/5% CO2 before further use. IKKε and TBK1 plasmids were a gift from Prof. Alain Chariot (University of Liege, Belgium). The CYLD S418A cDNA was a gift from Jessica Hutti, and was recloned into pCAGGS-E-CYLD (BCCM/GeneCorner plasmid collection; LMBP06613).
IKKε knockout mice were a gift from Prof. Alain Chariot. For isolation of primary cells, spleens were collected from wild-type or IKKε deficient mice. Spleens were injected with 5 ml PBS + 0.5% BSA using a 25 g needle. Spleens as well as washed out cells were passed over a 100 μm cell strainer, which was further rinsed with 5 ml PBS + 0.5% BSA. Cells were spun for 8 min at 1300 rpm at 4°C and supernatant was removed. Red blood cells were lysed in 1.7ml ACK buffer per spleen at room temperature for 4 min. 5 ml PBS + 0.5% BSA per spleen were added and cells were passed over a 40 μm cell strainer and the cell strainer was rinsed with 1 ml PBS + 0.5% BSA. Cells were spun for 8 min at 1,300 rpm at 4°C and the supernatant was removed. The cell pellets were resuspended in 2 ml IMDM supplemented with 5% FCS, β-mercaptoethanol, 20 U/ml penicillin and 20 μg/ml streptomycin per spleen. For CD4+ T cells isolation the CD4+ T cell Isolation Kit II protocol (Miltenyi Biotec) was used according to the manufacturer's instructions. Cells were stimulated with different stimuli and inhibitors at the indicated concentrations (Table 1).
Table 1.
Stimuli and inhibitors used.
| Concentration | Cat # | Supplier | |
|---|---|---|---|
| STIMULI | |||
| PMA | 200 ng/ml | P8139 | Sigma |
| Ionomycin | 1 μM | CALB407952 | Merck millipore |
| Anti-CD3 | 10 μg/ml | 553057 | BD pharmingen |
| Anti-CD28 | 10 μg/ml | 553294 | BD pharmingen |
| TNF | 1,000 U/ml | – | VIB protein service facility |
| INHIBITORS | |||
| MRT67307 | 1–10 μM | HY-13018 | MedChemtronica |
| TPCA1 | 5 μM | 2559 | Tocris biosciences |
Cell lysis and immunoprecipitation
Jurkat T cells or primary CD4+ T cells were lysed in Lysis buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton) supplemented with protease and phosphatase inhibitors. Lysates were cleared by centrifugation for 15 min at 14,000 rpm at 4°C. Protein concentration was measured by Bradford protein assay (Bio-Rad). 5x Laemmli buffer (250 mM Tris-HCl pH 8, 10% SDS, 50% glycerol, 0.005% bromophenol blue, 25% β-mercaptoethanol) was added to protein lysates and equal amounts of protein were subjected to immunoblotting. For immunoprecipitation, cell lysates were incubated overnight rotating with specific primary antibodies at 4°C. Then 35 μl of Dynabeads® Protein G (Invitrogen) were added and incubated rotating for 30 min at room temperature (RT). Beads were washed three times with Lysis buffer. Immune complexes were removed from the beads by boiling for 10 min at 90°C in 1x Laemmli buffer and subjected to immunoblotting. For total lysates HEK 293T cells were directly lysed in 1x Laemmli buffer (50 mM Tris-HCl pH 8, 2% SDS, 10% glycerol, 0.001% bromophenol blue, 5% β-mercaptoethanol).
Immunoblotting
Protein samples were resolved by 8 or 10% SDS-PAGE and proteins were then transferred to 0.45 μm protean nitrocellulose membranes (Whatman) or Immobilon-FL transfer membranes (Millipore). The membranes were blocked in 5% milk powder in TBS/0.2% Tween 20 (TBST) for 1 h at room temperature (RT) and probed with specific primary antibodies overnight at 4°C in 5% milk powder in TBST or 5% BSA in TBST. All antibodies used are listed in Table 2. Membranes were washed three times in TBST and then incubated with HRP-conjugated secondary antibodies (GE healthcare) in 5% milk powder in TBST or 5% BSA in TBST for 1h at RT. Membranes were washed 3 times in TBST and proteins were detected using the Western Lightning ECL detection system (Perkin Elmer Life Sciences) according to the manufacturer's instructions.
Table 2.
List of antibodies used.
| MP/BSA | Dilution | Cat # | Supplier | |
|---|---|---|---|---|
| P-CYLD | BSA | 1:1,000 | 4500 | Cell signaling technology |
| CYLD | MP | 1:500 | sc-74435 | Santa Cruz |
| P-IKKε | BSA | 1:1,000 | 8766 | Cell signaling technology |
| IKKε | MP | 1: 1,000 | ab7891 | Abcam |
| P-TBK1 | BSA | 1: 1,000 | 5483 | Cell signaling technology |
| TBK1 | MP | 1: 1,000 | 3013 | Cell signaling technology |
| P-JNK | BSA | 1: 1,000 | 4668 | Cell signaling technology |
| JNK | MP | 1: 1,000 | sc-571 | Santa Cruz |
| P-IkBa | BSA | 1: 1,000 | 9246 | Cell signaling technology |
| Actin | MP | 1:10,000 | MP 6472J | MP biomedicals |
| Tubulin | MP | 1:1,000 | T4026 | Sigma |
| E | MP | 1:2,500 | ab66152 | Abcam |
| Myc | MP | 1:3,000 | IRC PEP core | |
| Flag | MP | 1:1,000 | F-3165 | Sigma |
| HA | MP | 1:1,000 | MMS-101R-B | Babco |
| anti-mouse IgG HRP | MP | 1:3,000 | NA931-1ML | Akta |
| anti-rabbit IgG HRP | MP | 1:3,000 | NA934V | GEAkta |
| anti-goat IgG HRP | MP | 1:3,000 | sc-2020 | Santa Cruz |
MP, Milk Powder.
in vitro kinase assay
250 ng recombinant His-CYLD (#64-0010-050, Ubiquigent) and 35, 70, or 140 ng GST-IKKε (#31177, Active Motif) or GST-TBK1 (#66-0016-050, Ubiquigent) were incubated with 0.6 mM ATP-Mg (#A9187, Sigma) in kinase buffer (20 mM Tris-HCl pH7.5, 10 mM MgCl2, 0.1 mM sodiumorthovanadate, 5 mM β-glycerophosphate, 2 mM DTT) for 30 min at 30°C under constant rotation at 550 rpm. Reactions were stopped by adding 5x Laemmli buffer and analyzed by immunoblotting.
Engineering of knockout cells by CRISPR/Cas9
Knockout cell lines were generated using CRISPR/Cas9 technology as described previously (Ran et al., 2013). Briefly, guide RNAs were designed using the design tool provided by MIT (http://crispr.mit.edu/) and are listed in Table 3. Oligonucleotides (oligos) coding for the guide sequence were extended with CACC in the beginning of the top oligo and CAAA in the end of the bottom oligo. The oligos were phosphorylated and annealed by incubating 1 μl of each oligo (100 μM stock), 1 μl 10x T4 ligation buffer (M1804, Promega) and 1 μl T4 polynucleotide kinase (M4101, Promega) in a total volume of 10 μl in a thermocycler using the following parameters: 37°C for 30 min, 95°C for 5 min, ramp down to 25°C at 5°C/min (=0.1°C/s). The phosphorylated and annealed oligos were diluted 1:200 in ddH2O. Oligos were then cloned into pSpCas9(BB)-2A-GFP (48138, Addgene). The reaction mix contained 100 ng pSpCas9(BB)-2A-GFP, 2 μl oligo, 2 μl 10x Tango buffer (Fermentas), 0.5 mM DTT, 0.5 mM ATP, 1 μl FastDigest BbsI (FD1014, Fermentas) and 0.5 μl T4 ligase (M1804, Promega) in a total volume of 20 μl. The reaction was incubated for 6 cycles: 5 min at 37°C followed by 5 min at 21°C. Plasmids were transformed into DH5α competent E. coli. Plasmid DNA was extracted using the PureLink® Quick Plasmid Miniprep Kit (K210010, Invitrogen) according to the manufacturer's instructions and the sequences were verified by Sequencing from the U6 promoter using the U6-Fwd primer (GAGGGCCTATTTCCCATGATTCC) to check that the 20 nt guide sequence is inserted between the U6 promoter and the remainder of the sgRNA scaffold. The plasmids (2 μg) were transfected into 2 × 106 Jurkat T cells using Amaxa cell line nucleofector kit V (VCA-1003, Lonza) using program X5 according to the manufacturer's instructions. Three days after transfection GFP positive single cells were sorted into 96 well plates using a BD FACS ARIA III (BD Bioscience). Single cell clones were expanded and knockout was confirmed on protein level by immunoblotting and on genomic level by amplification of the region of interest followed by sequencing. For this, genomic DNA was extracted using QuickExtract DNA Extraction Solution (QE09050, Epicenter) according to the manufacturer's instructions and the region of interest was amplified using the primers indicated in Table 3 with the KAPA HiFi HotStart Readymix (KK2602, kapa biosystems) according to the manufacturer's instructions. PCR products were purified using CleanPCR beads (CPCR-0050, Cleanna) according to the manufacturer's instructions and sequenced using the primers indicated in Table 3. The sequences were analyzed using the Tracking of Indels by DEcomposition (TIDE) software (Brinkman et al., 2014).
Table 3.
Oligos and primers used for CRISPR/Cas9 mediated geneome editing.
| Primer | Sequence | |
|---|---|---|
| CYLD | Guide top oligo | CACCAGAGTGGAATCTGTTCTCGG |
| Guide bottom oligo | AAACCCGAGAACAGATTCCACTCT | |
| Forward Primer | ACAAAGACTATCACGTATACGATACCCG | |
| Reverse Primer | TTAACTTCAGCCAATGAGCCCA | |
| Sequencing Primer | AGGAGCGAGAACACTGTT | |
| IKKε | Guide top oligo | CACCGAGAAGTTCGTCTCGGTCTA |
| Guide bottom oligo | AAACTAGACCGAGACGAACTTCTC | |
| Forward Primer | CACCCATCTTGGTTTCCTAGAGAA | |
| Reverse Primer | AGCCTCACCTTTTCCATCTTAGAGAAC | |
| Sequencing Primer | ATGGCTCTGTCAGCCCAT | |
| TBK1 | Guide top oligo | CACCCATAAGCTTCCTTCGTCCAG |
| Guide bottom oligo | AAACCTGGACGAAGGAAGCTTATG | |
| Forward Primer | GATGATTTTGCTTTTGATACTATTATGGC | |
| Reverse Primer | GGTTGTACAAACCCTAATTTTCAACAGT | |
| Sequencing Primer | TATCCATTTCTGAATTCC |
Results
IKKε and TBK1 are activated upon T cell activation and can directly phosphorylate CYLD at Ser418
IKKε and its homolog TBK1 were previously shown to be activated upon crosslinking of CD3 and the co-stimulatory receptor CD28 (Yu et al., 2015). In agreement, we found that treatment of the Jurkat T cell line with a combination of phorbol myristate acetate (PMA) and Ionomycin (I), which mimics TCR signaling by activating PKC and calcium influx, results in the activation of IKKε and TBK1 as assessed by immunoblotting with an antibody that specifically recognizes Ser172 phosphorylation in the IKKε/TBK1 activation loop (Kishore et al., 2002). Similar results were obtained in primary murine CD4+ T cells (Figure 1). We next investigated if CYLD can be phosphorylated by IKKε or TBK1 in the context of TCR signaling. We focused on CYLD phosphorylation at Ser418, which can be easily assessed using a phospho(Ser418)-CYLD-specific antibody. Stimulation of Jurkat T cells with anti-CD3/CD28 or PMA/I led to the rapid phosphorylation of CYLD at Ser418 (Figure 2A). The faster migrating band in the case of immunoblotting for total CYLD corresponds to the previously reported CYLD cleavage product generated by MALT1 (Staal et al., 2011). Although TNF was previously shown to trigger CYLD phosphorylation in Jurkat, HeLa, or HEK 293 cells, leading to the appearance of a slower migrating form of CYLD (Reiley et al., 2005), stimulation of our Jurkat T cells with TNF did not lead to a band shift or detection of a band with the phospho(Ser418)-specific antibody, suggesting either no phosphorylation or phosphorylation at other sites. It should be noted however, that a phospho-CYLD specific signal was detected in several other Jurkat cell clones tested (Figure S1), indicating clonal differences. The TCR- and PMA/I-induced Ser418 phosphorylation of CYLD could be completely prevented by dual inhibition of IKKε and TBK1, using MRT67307, while the IKKβ inhibitor TPCA1 had no effect (Figure 2B). CYLD Ser418 phosphorylation was also observed upon co-expression with IKKε or TBK1, but not upon co-expression of their catalytically inactive counterparts (K38A) in HEK 293T cells (Figure 2C). When the Ser418 residue in CYLD was mutated to alanine, we could no longer detect its IKKε/TBK1-mediated phosphorylation using the phospho(Ser418)-CYLD-specific antibody, confirming phosphorylation at Ser418. However, we still detected a band shift using an antibody against the epitope-tag fused to CYLD, indicating the presence of additional IKKε/TBK1-mediated phosphorylation sites in CYLD upon overexpression of the kinases. Further, in an in vitro kinase assay we could show that recombinant IKKε and TBK1 can directly phosphorylate CYLD at Ser418 (Figure 2D). Together these data suggest that T cell activation is associated with the IKKε/TBK1 mediated phosphorylation of CYLD at Ser418.
Figure 1.
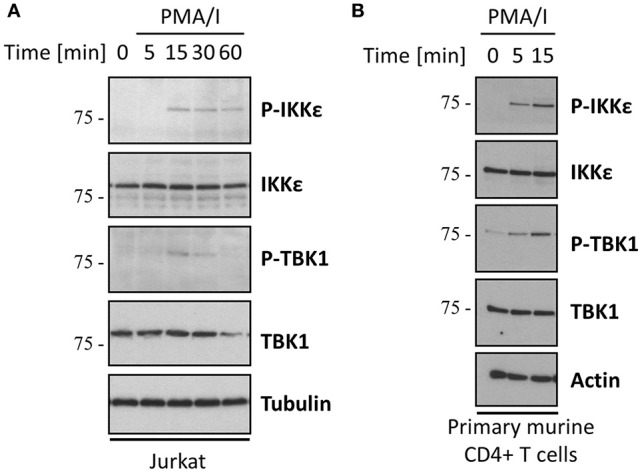
The noncanonical kinases IKKε and TBK1 are activated upon TCR stimulation. Jurkat T cells (A) or primary mouse CD4+ T cells (B) were stimulated with 200 ng/ml PMA and 1 μM Ionomycin for the indicated times. Protein levels were determined by immunoblotting. Activation of IKKε and TBK1 was determined using an antibody specifically recognizing their phosphorylation at Ser172. Results shown are representative for three (A) and two (B) independent experiments.
Figure 2.

The deubiquitinase CYLD is phosphorylated at Ser418 by IKKε and TBK1. (A) CYLD phosphorylation at Ser418 is observed upon TCR- but not TNFR stimulation. Jurkat T cells were stimulated with 200 ng/ml PMA and 1 μM Ionomycin or 20 μg/ml anti-CD3 and anti-CD28 or 1,000 U/ml TNF for the indicated times. (B) CYLD phosphorylation is abolished by IKKε/TBK1, but not IKKβ inhibition Jurkat T cells were stimulated with 200 ng/ml PMA for the indicated times. Cells were pre-incubated in the presence or absence of 10 μM IKKε/TBK1 inhibitor MRT67307 or 5 μM IKKα/β inhibitor TPCA1. (C) IKKε/TBK1 co-expression induced CYLD phosphorylation. HEK 293T cells were transfected with the indicated expression constructs and harvested 24 h post transfection. (D) CYLD is phosphorylated by IKKε and TBK1 in vitro. 250 ng recombinant His-CYLD was incubated with 35 or 70 ng GST-IKKε IKKε or 35, 70, or 140 ng TBK1 in an in vitro kinase assay (A–D) Protein levels were determined by immunoblotting. Results shown are representative at least three independent experiments.
The IKKε/TBK1 inhibitor MRT67307 inhibits CYLD phosphorylation independent of IKKε and TBK1
As MRT67307 blocks both IKKε and TBK1, we wanted to determine which of the two kinases is responsible for CYLD phosphorylation. Therefore, we first tested PMA/I-induced CYLD phosphorylation in primary CD4+ T cells from IKKε deficient mice. As a control, wild-type and IKKε deficient cells were stimulated in the presence of MRT67307. MRT67307 significantly decreased CYLD phosphorylation in wild-type cells (Figure 3A). However, there was no difference in CYLD phosphorylation between wild-type and IKKε knockout cells. Moreover, CYLD phosphorylation was still inhibited by MRT67307 in IKKε deficient cells. Together, these results suggest that TCR-induced CYLD phosphorylation is independent of IKKε. Alternatively, the effect of IKKε deficiency may be masked by the redundant function of TBK1. To further investigate a possible redundancy between IKKε and TBK1, we generated IKKε and TBK1 double deficient Jurkat T cells using CRISPR/Cas9 technology. Several independent clones were derived from single cells. Knockout of IKKε and TBK1 was confirmed by immunoblotting as well as by genomic sequencing of the region of interest. Unexpectedly, a phospho(Ser418)-CYLD signal was still detectable upon PMA/I stimulation in the IKKε/TBK1 double deficient Jurkat cells, indicating that a kinase different from IKKε and TBK1 is responsible for CYLD phosphorylation (Figure 3B). Furthermore, this signal could be inhibited by pre-treatment with MRT67307, suggesting that this inhibitor acts independently of IKKε/TBK1 (Figure 3C). As it was shown previously that MRT67307 can inhibit IKKε and TBK1 at concentrations as low as 1–2 μM (Clark et al., 2011a), the concentration of 10 μM used may have led to off-target effects. Therefore we tested if the phospho(Ser418)-CYLD signal could also be inhibited when using MRT67307 at lower concentrations (Figure 3D). Here we could show that 5 μM MRT67307 completely prevented CYLD phosphorylation, while 1 or 2 μM reduced the phospho(Ser418)-CYLD signal only partially (Figure 3D). Taken together, these results indicate that MRT67307 inhibits the phospho(Ser418)-CYLD signal independent of its IKKε and TBK1 inhibitory properties.
Figure 3.

The TCR-induced phospho(Ser418)-specific band occurs independent of IKKε and TBK1 (A) Primary mouse CD4+ T cells from wild-type (wt) or IKKε knockout (ko) mice were pre-incubated for 30 min in the presence of absence of 10 μM IKKε/TBK1 inhibitor MRT67307 and subsequently stimulated with 200 ng/ml PMA and 1μM Ionomycin for the indicated times. Specific proteins were determined by immunoblotting with the indicated antibodies. (B) Wild-type (wt) or IKKε/TBK1 double deficient Jurkat T cell clones were stimulated with 200 ng/ml PMA and 1 μM Ionomycin for 15 min. Specific proteins were determined by immunoblotting with the indicated antibodies. (C) Wt or IKKε/TBK1 double deficient Jurkat T cell clones were pre-incubated for 30 min in the presence or absence of 10 μM IKKε/TBK1 inhibitor MRT67307 and then stimulated with 200 ng/ml PMA and 1 μM Ionomycin for 15 min. Specific proteins were determined by immunoblotting with the indicated antibodies. (D) Wt Jurkat cells were pre-incubated for 30 min in the presence of 1, 2, 5, or 10 μM IKKε/TBK1 inhibitor MRT67307 and then stimulated with 200 ng/ml PMA and 1 μM Ionomycin for 15 min. Specific proteins were determined by immunoblotting with the indicated antibodies. The data are representative of at least two independent experiments.
The phospho(Ser418)-CYLD-specific antibody detects a protein distinct from CYLD
To further elucidate the importance of CYLD phosphorylation at Ser418 for proximal TCR signal transduction, we generated CYLD knockout cells by CRISPR/Cas9 to subsequently reconstitute with CYLD(S418A) or CYLD(S418E) mutants that prevent or mimic CYLD phosphorylation at Ser418, respectively. Absence of CYLD was confirmed by immunoblotting as well as genomic sequencing. Three different CYLD-deficient Jurkat T cell clones were characterized for signaling in response to PMA/I. While all clones show similar inducible IκBα phosphorylation, they differ strongly in JNK activation (Figure 4A). JNK phosphorylation in clone #10 is comparable to wild-type Jurkat T cells, while clone #8 shows decreased and clone #9 increased JNK phosphorylation. This clonal variation between the different CYLD deficient Jurkat T cell clones illustrates that one should be highly cautious when using specific knockout cell clones generated by CRISPR/Cas9 and that the effect of CYLD reconstitution should be tested in order to make valid conclusions on the impact of CYLD deficiency on JNK activation. The latter was beyond the scope of the current study. Most surprisingly, however, western blotting and detection with the phospho(Ser418)-CYLD-specific antibody still revealed the same PMA/I-inducible phospho(Ser418)-CYLD signal in all CYLD-deficient cell clones (Figure 4A). These data indicate that the phospho(Ser418)-CYLD-specific antibody detects a protein distinct from CYLD, but with the same molecular weight of approximately 110 kDa. The impact of this finding on our current data and previously published results of others using the same antibody will be discussed below. From Figure 2C it is obvious that this antibody specifically recognizes CYLD phosphorylation at Ser418 upon overexpression. At this point, it is not possible to exclude that the unspecific 110 kDa band masks a specific phospho-CYLD band in the Jurkat cell line. We therefore performed an immunoprecipitation using the phospho(Ser418)-CYLD-specific antibody, followed by western blotting with a CYLD antibody (Figures 4B,C). Indeed, CYLD could be readily detected upon immunoprecipitation with the phospho(Ser418)-CYLD-specific antibody from PMA/I-stimulated Jurkat T cells and primary murine CD4+ T cells, showing that CYLD is indeed phosphorylated at Ser418 upon T cell stimulation but that its direct detection with the phospho(Ser418)-CYLD-specific antibody in a western blot is masked by another inducible protein of the same size that is recognized by the same antibody.
Figure 4.

The phospho(Ser418)-CYLD specific antibody still detects a protein of the same size as CYLD in CYLD-deficient cells. (A) Wild-type (wt) or three different CYLD knockout (ko) Jurkat T cell clones were stimulated with 200 ng/ml PMA and 1 μM Ionomycin for the indicated times. (B) Jurkat cells or (C) primary murine CD4+ T cells were stimulated with 200 ng/ml PMA and 1 μM Ionomycin (PMA/I) for the indicated times. Cell extracts were subjected to immunoprecipitation using the phospho(Ser418)-CYLD-specific antibody. Beads without antibody were used as negative control (nc). (A–C) Specific proteins were determined by immunoblotting with the indicated antibodies.
To determine if the specific phosphorylation of CYLD is dependent on IKKε and TBK1 as hypothesized earlier, we performed an immunoprecipitation with the phospho(Ser418)-CYLD-specific antibody in IKKε/TBK1 deficient cells (Figure 5A). While we could detect immunoprecipitated CYLD in PMA/I stimulated wild-type Jurkat T cells, we did not detect any CYLD upon immunoprecipitation with the phospho(Ser418)-CYLD-specific antibody in the IKKε/TBK1 knock-out cells, indicating that CYLD phosphorylation is indeed dependent on these two kinases. Interestingly, we could detect a band with the phospho(Ser418)-CYLD-specific antibody in the total lysate control of the IKKε/TBK1 knock-out clones (Figures 3B,C, 5A), indicating that this antibody specifically detects phosphorylated CYLD under native conditions, while it additionally detects an unknown protein of the same size in denaturing conditions. We further confirmed the importance of IKKε and TBK1 for CYLD phosphorylation by treatment of cells with MRT67307, followed by immunoprecipitation of cell lysates with the phospho(Ser418)-CYLD-specific antibody (Figure 5B). Here we show that specific phosphorylation of CYLD upon immunoprecipitation is already completely inhibited at 2 μM MRT67307 (Figure 5B, left panel), while 10 μM MRT67307 is necessary to completely prevent the phosphorylation band in the total lysate (Figure 5B, right panel). Taken together these data show that CYLD is phosphorylated upon TCR stimulation and that this phosphorylation is dependent on IKKε and TBK1. However, the CYLD phosphorylation band in total cell lysates is masked by the phosphorylation of another protein of the same size which is also detected by the phospho(Ser418)-CYLD-specific antibody. Specific CYLD phosphorylation should thus be studied by immunoprecipitation with this antibody followed by CYLD detection via immunoblotting.
Figure 5.

CYLD is phosphorylated in an IKKε/TBK1 dependent manner, which can be detected by immunoprecipitation of CYLD using the phospho(Ser418)-CYLD-specific antibody. (A) Wild-type (wt) or IKKε/TBK1 double knockout (ko) Jurkat T cell clones were stimulated with 200 ng/ml PMA and 1 μM Ionomycin (PMA/I) for 30 min. (B) Wt Jurkat T cells were pre-incubated for 30 min in the presence of 2 or 10 μM IKKε/TBK1 inhibitor MRT67307 and then stimulated with 200 ng/ml PMA and 1μM Ionomycin for 30 min. (A,B) Cell extracts were subjected to immunoprecipitation using the phospho(Ser418)-CYLD-specific antibody. Beads without antibody were used as negative control (nc). Specific proteins were determined by immunoblotting with the indicated antibodies.
Discussion
In the present study we show that co-expression of CYLD and IKKε/TBK1 in HEK293T cells, as well as incubation with recombinant IKKε/TBK1, results in CYLD phosphorylation that can be detected with a phospho(Ser418)-CYLD-specific antibody upon immunoblotting. A similar signal, sensitive to MRT67307, could be detected in TCR-stimulated Jurkat cells. CYLD is known to be phosphorylated on a serine cluster (amino acids 418-444) in an IKKβ/NEMO-dependent manner in response to TNF, LPS and PMA/I, resulting in a band shift upon immunoblotting (Reiley et al., 2005). It must be noted that we did not observe such a band shift of CYLD upon TCR crosslinking or PMA/I stimulation of Jurkat T cells, while we did detect a clear signal using the phospho(Ser418)-CYLD antibody. At the functional level, CYLD phosphorylation was previously shown to prevent CYLD-mediated TRAF2 deubiquitination and to promote TNF-induced gene expression (Reiley et al., 2005). Similarly, CYLD Ser418 phosphorylation upon co-expression of IKKε was found to decrease its DUB activity (Hutti et al., 2009). IKKε-dependent CYLD Ser418 phosphorylation and inactivation of its DUB activity has also been reported in response to stimulation of the C-type lectin receptor DC-SIGN in dendritic cells (Gringhuis et al., 2014). Remarkably, there is also a contradictory report showing that CYLD Ser418 phosphorylation increases its in vitro DUB activity toward K63-linked polyubiquitin (Thein et al., 2014). Surprisingly, using CYLD deficient cells we found that the phospho(Ser418)-CYLD antibody cross-reacts with another unknown protein of the same size as CYLD, and whose phosphorylation is also inhibited by MRT67307. These findings not only forced us to reevaluate our findings on TCR-induced CYLD phosphorylation, they also imply that one should be very cautious with the interpretation of several published findings using the phospho(Ser418)-CYLD antibody (Table 4). However, by immunoprecipitation with the phospho(Ser418)-CYLD-specific antibody followed by CYLD detection via immunoblotting and detection with anti-CYLD, we were able to detect specific CYLD phosphorylation, and could show that this was indeed dependent on IKKε and TBK1, confirming our initial hypothesis.
Table 4.
Publications related to CYLD phosphorylation on Ser418.
| References | Kinase | Context | Effect |
|---|---|---|---|
| Beli et al., 2012 | nd | TNF, ionizing radiation or etoposide treatment of U2OS cells | nd |
| Gringhuis et al., 2014 | IKKε | DC-SIGN stimulation of dendritic cells | Reduced DUB activity; reduced Bcl3 deubiquitination and nuclear translocation |
| Thein et al., 2014 | IKKα/β | Postsynaptic density | Increased DUB activity |
| Zhu et al., 2014 | IKKε/TBK1 | Co-expression with TBK1 or IKKε in HEK 293 cells, abrogated by MRT67307 treatment | nd |
nd, not determined.
Antibodies are among the most frequently used research tools in basic science and are used in a wide range of applications. Many antibodies are however not properly validated. One of the pitfalls is that detection of a single band of the correct molecular weight on a western blot is often used to demonstrate specificity. However, our results with a commonly used phospho(Ser418)-CYLD antibody demonstrate this is far from true and that there is a clear need for universal standards in antibody validation. The International Working Group for Antibody Validation (IWGAV) proposed a model with standard guidelines for antibody validation based on five conceptual pillars or validation strategies (Uhlen et al., 2015). First, the antibody should be tested on cells in which expression of a protein of interest is reduced or eliminated by RNA interference or knockout using techniques such as CRISPR/Cas9. Second, orthogonal antibody-independent methods such as mass spectrometry or qPCR can validate expression. Third, protein expression can be compared using two distinct antibodies that recognize non-overlapping epitopes. Fourth, the expression of a protein that contains an epitope-tag (e.g., Flag, His) can be analyzed in parallel using an antibody raised against the target protein or an antibody raised against the tag. Fifth, the protein can be affinity-purified with the antibody and analyzed by mass spectrometry. Uhlen and colleagues recommend using at least one of these pillars as a minimal criterion for antibody specificity. However, our own experience taught us that the parallel employment of several strategies (overexpression, use of recombinant proteins and knockout) is necessary for confident proof of antibody specificity. A useful resource to select antibodies is the antibodypedia, a repository of already validated antibodies which reports on primary data, publications and commentaries on commercially available antibodies (https://www.antibodypedia.com/).
Our observation that MRT67307 inhibits CYLD phosphorylation in IKKε and TBK1 deficient cells indicates a role for other MRT67307-sensitive kinases in TCR signaling. While MRT67307 was first described as an inhibitor of TBK1 and IKKε (IC50 value of 19 and 160 nM, respectively) (Clark et al., 2011a), several AMP-activated protein kinase (AMPK)-related kinases were later described as additional targets: MAP/microtubule affinity-regulating kinases (MARK1)-4 (IC50 value of 27–52 nM), NUAK family kinase 1 (NUAK1) (IC50 value of 230 nM), and the salt inducible kinases (SIK) (IC50 value of 250, 67, and 430 nM for SIK1, SIK2, and SIK3, respectively) (Clark et al., 2011a, 2012). Moreover, MRT67307 is also an inhibitor of Unc-51 Like Autophagy Activating Kinase (ULK) 1 and ULK2 with IC50 values of 45 and 38 nM, respectively (Petherick et al., 2015). In addition, the MRC PPU International Centre for Kinase Profiling reported in vitro specificity screens for several inhibitors (http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors), showing that MRT67307 not only inhibits IKKε/TBK1 and the AMPK-related kinases MARK, MELK and NUAK, but also Mixed Lineage Kinase (MLK) 1 and MLK3, Janus kinase (JAK) 2, and Ca(2+)/calmodulin-dependent protein kinase kinase β (CamKKβ). It will be interesting to further investigate if any of the above mentioned kinases is responsible for the inducible phosphoprotein signal detected by the phospho(Ser418)-CYLD-specific antibody. Moreover, the non-specificity of MRT67307 also implies that numerous studies using this compound as an IKKε/TBK1 inhibitor may have to be revisited (summarized in Table 5). The above findings illustrate the importance of validating small compound kinase inhibitors for specificity. Much valuable information is publicly available from selectivity profile data of commonly used kinase inhibitors screened against panels of kinases (Davies et al., 2000; Bain et al., 2003, 2007; Fabian et al., 2005; Fedorov et al., 2007; Bamborough et al., 2008; Karaman et al., 2008). Additional information on kinase screens or kinome scans can be found on http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors or http://lincs.hms.harvard.edu/kinomescan/, respectively. Also specific recommendations on the most optimal inhibitor or combinations of inhibitors to study a number of selected kinases have been published (Davies et al., 2000; Bain et al., 2003, 2007). For example, using at least two structurally different inhibitors or cells expressing a drug resistant mutant of the kinase to validate findings is much desired. We would also recommend using genetic knockout or siRNA mediated knockdown of the specific kinase to complement data obtained from inhibitor studies.
Table 5.
Publications in which MRT67307 was used.
| References | Intended target | Other targets | IC50 [nM] | Effect of inhibitor treatment | Confirmation of inhibitor effects | Concentration |
|---|---|---|---|---|---|---|
| Clark et al., 2011a | TBK1/IKKε | 19/160 | Inhibition of PolyI:C induced IRF3 phosphorylation, but not JNK and p38 phosphorylation and reduced LPS-induced IFNβ production in macrophages; Enhanced NF-κB activation in response to IL-1 or TNF and PolyI:C or LPS in MEF cells and macrophages, respectively | 2μM | ||
| Gleason et al., 2011 | TBK1/IKKε | Reduced OPTN phosphorylation in LPS-treated BMDMs in combination with TAK1 inhibitor 5Z-7-oxozeanol. | ||||
| Smith et al., 2011 | TBK1/IKKε | Inhibition of LPS-induced Pellino-1 induction and IRF3 phosphorylation in BMDMs; Reduced IKKε and TBK1 dependent Pellino 1 phosphorylation in vitro; Reduced LPS- or PolyI:C-induced Pellino 1 activation in macrophages | 2μM | |||
| Clark et al., 2011b | TBK1/IKKε | Increased TBK1 and IKKε activationin Pam3CSK4 stimulated BMDMs | IKKε and TBK1 knockout MEF cells | 2μM | ||
| Dzamko et al., 2012 | TBK1/IKKε | Inhibition of phosphorylation of LRRK2 in Pam3CSK4 stimulated RAW 264.7 cells | 2μM | |||
| Liu et al., 2012 | TBK1 | Abrogation of thapsigargin-dependent IRF3 phosphorylation in MEF cells | 2μM | |||
| Bruni et al., 2013 | TBK1/IKKε | Reduced ERK phosphorylation in response to PolyI:C in BMDMs | IKKε and TBK1 knockout BMDMs | 2μM | ||
| Jiang et al., 2015 | TBK1/IKKε | Decreased IFN-λ1 mRNA expression in short dsRNA-stimulated human monocyte-derived DCs | 0.1–10μM | |||
| Zhu et al., 2014 | TBK1/IKKε | Impaired CYLD (Ser418) phosphorylation upon co-expression with IKKε/TBK1 in HEK cells; decreased LPS-induced expression of IFNβ1, CCL5 and IL-6 in RAW 264.7 cells; impaired viability of KRAS-dependent A549 and HCC44 cells | genetic evidence (Tbk1−/− MEFs), independent inhibitor (CYT387) | 0.1–10μM | ||
| Lopez-Pelaez et al., 2014 | TBK1/IKKε | Inhibition of in vitro phosphorylation of IRF5 by TBK1; no effect on nuclear translocation of IRF5 upon CL097 (TLR7/8) in RAW 264.7 cells | 2μM | |||
| Awuh et al., 2015 | TBK1 | Decreased NF-κB and IRF-1 nuclear translocation in response to M. avium infection in monocyte derived macrophages; Increased M. avium growth | independent inhibitor (BX795) | 5μM | ||
| Pillai et al., 2015 | TBK1 | Inhibition of mitotic progression and formation of multinucleate cells in H460 and HeLa cells | independent inhibitor (BX795) (knockdown in additional experiments) | 2μM | ||
| Yu et al., 2015 | TBK1 | Inhibition of AKT degradation in anti-CD3/CD28 stimulated CD4+ T cells in the presence of cycloheximide | 2μM | |||
| Bruni et al., 2015 | TBK1/IKKε | Increased viral RNA production in pDCs infected with yellow fever life vaccine and reduced type I IFN production | 1μM | |||
| Saric et al., 2016 | TBK1 | No effect on LPS-induced lysosome tubulation in RAW 264.7 macrophages; inhibition of LPS-induced IRF3 phosphorylation | 2μM | |||
| Heo et al., 2015 | TBK1 | Inhibition of recruitment of OPTN, NDP52 and SQSTM1 to depolarized mitochondria in Antimycin A and Oligomycin A treated HeLa cells | TBK1−/− cells | 2μM | ||
| Swamy et al., 2015 | TBK1/IKKε | Reduced Ifnα and Ifnl mRNA induction, but no effect on Ifnγ upregulation in CD3-stimulated intestinal intraepithelial lymphocytes | 1μM | |||
| Schweitzer et al., 2016 | TBK1/IKKε | Marginal decrease in phosphorylation of IκBα and RelA (Ser468) in the cytosol and RelA nuclear translocation in TNF-stimulated HeLa cells | 10μM | |||
| Achard et al., 2017 | TBK1/IKKε | Impaired IFNα production and TRAIL expression in response to measles virus infection or R837 (TLR7 agonist) stimulation in plasmacytoid DCs; Reduced TRAIL expression in response to type I IFN | 8μM | |||
| Clark et al., 2012 | SIK1 | 250 | Increased TLR-induced expression of anti-inflammatory cytokines (IL-10 and IL-1ra) in macrophages | Structurally different inhibitors (MRT199665, HG-9-91-01, KIN112), genetic evidence (LKB1−/− macrophages) | 2μM | |
| SIK2 | 67 | |||||
| SIK3 | 430 | |||||
| MARK1-4 | 27-52 | |||||
| NUAK1 | 230 | |||||
| MacKenzie et al., 2013 | SIK | Increased LPS-induced IL-10 mRNA in BMDMs | Independent inhibitor (KIN112) | 2μM | ||
| Petherick et al., 2015 | ULK1 | 45 | Reduced amino acid starvation-induced ATG13 phosphorylation and autophagy in MEF cells | Structurally different inhibitor (MRT68921) | 10μM | |
| ULK2 | 38 | |||||
| Lazarus and Shokat, 2015 | ULK1/2 | Inhibition of autophagy (LC3 II accumulation) in HeLa cells | Independent inhibitor (BX795) | 4μM | ||
| Draz et al., 2017 | ULK1/2 | Sensitization of LNCaP and C42B cells to cell death in the presence of subtoxic concentrations of 3,3′-diindolylmethane | 10μM |
In conclusion, the present case study clearly illustrates several pitfalls when using antibodies and small compound inhibitors. There is a clear need for better validation of antibodies and small compound inhibitors to make results more reliable and reproducible. More specifically, some previously published data making use of MRT67307 or phospho(Ser418)-CYLD specific antibodies may have led to wrong conclusions or at least need to be validated by independent approaches if not yet done so. Finally, our results also reveal that TCR stimulation results in the IKKε/TBK1-independent phosphorylation of an unknown 110 kDa protein, which can be inhibited by MRT67307, and which masks the detection of phosphorylated CYLD upon western blotting with a phospho(Ser418)-CYLD-specific antibody. The identification of this phosphorylated protein as well as the responsible kinase that is inhibited by MRT67307 will be the topic of future studies.
Author contributions
ML, JS, and RB designed the research. ML and MK performed the experiments. ML analyzed the data. ML, JS, and RB wrote the paper.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer NR and handling Editor declared their shared affiliation.
Footnotes
Funding. This work was supported by grants from the Fund for Scientific Research Flanders (FWO), the Belgian Foundation Against Cancer, Interuniversity Attraction Poles, Concerted Research Actions (GOA) and the Group-ID Multidisciplinary Research Partnership of Ghent University. ML is supported by a predoctoral fellowship from the FWO.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2018.00040/full#supplementary-material
CYLD does not show a band shift indicative of phosphorylation upon stimulation with PMA/I or TNF. Different Jurkat T cell clones were stimulated with 200 ng/ml PMA and 1μM Ionomycin or 1,000 U/ml TNF for the indicated times. Protein levels were determined by immunoblotting. Jurkat-E (from Shigekazu Nagata, Osaka), Jurkat E6.1 (ATCC), Jurkat TNFR2 (gift from Nicholas Harper, Leicester).
References
- Achard C., Guillerme J. B., Bruni D., Boisgerault N., Combredet C., Tangy F., et al. (2017). Oncolytic measles virus induces tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated cytotoxicity by human myeloid and plasmacytoid dendritic cells. Oncoimmunology 6:e1261240. 10.1080/2162402X.2016.1261240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awuh J. A., Haug M., Mildenberger J., Marstad A., Do C. P., Louet C., et al. (2015). Keap1 regulates inflammatory signaling in Mycobacterium avium-infected human macrophages. Proc. Natl. Acad. Sci. U.S.A. 112, E4272–E4280. 10.1073/pnas.1423449112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J., McLauchlan H., Elliott M., Cohen P. (2003). The specificities of protein kinase inhibitors: an update. Biochem. J. 371(Pt 1), 199–204. 10.1042/BJ20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., et al. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. 10.1042/BJ20070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P., Drewry D., Harper G., Smith G. K., Schneider K. (2008). Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J. Med. Chem. 51, 7898–7914. 10.1021/jm8011036 [DOI] [PubMed] [Google Scholar]
- Beli P., Lukashchuk N., Wagner S. A., Weinert B. T., Olsen J. V., Baskcomb L., et al. (2012). Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225. 10.1016/j.molcel.2012.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell G. R., Warren W., Seal S., Takahashi M., Rapley E., Barfoot R., et al. (2000). Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25, 160–165. 10.1038/76006 [DOI] [PubMed] [Google Scholar]
- Bonnard M., Mirtsos C., Suzuki S., Graham K., Huang J., Ng M., et al. (2000). Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19, 4976–4985. 10.1093/emboj/19.18.4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman E. K., Chen T., Amendola M., van Steensel B. (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42:e168. 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp T. R., Nijman S. M., Dirac A. M., Bernards R. (2003). Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424, 797–801. 10.1038/nature01811 [DOI] [PubMed] [Google Scholar]
- Bruni D., Chazal M., Sinigaglia L., Chauveau L., Schwartz O., Despres P., et al. (2015). Viral entry route determines how human plasmacytoid dendritic cells produce type I interferons. Sci. Signal. 8:ra25. 10.1126/scisignal.aaa1552 [DOI] [PubMed] [Google Scholar]
- Bruni D., Sebastia J., Dunne S., Schroder M., Butler M. P. (2013). A novel IRAK1-IKKepsilon signaling axis limits the activation of TAK1-IKKbeta downstream of TLR3. J. Immunol. 190, 2844–2856. 10.4049/jimmunol.1202042 [DOI] [PubMed] [Google Scholar]
- Clark K., MacKenzie K. F., Petkevicius K., Kristariyanto Y., Zhang J., Choi H. G., et al. (2012). Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U.S.A. 109, 16986–16991. 10.1073/pnas.1215450109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Peggie M., Plater L., Sorcek R. J., Young E. R., Madwed J. B., et al. (2011a). Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 434, 93–104. 10.1042/BJ20101701 [DOI] [PubMed] [Google Scholar]
- Clark K., Takeuchi O., Akira S., Cohen P. (2011b). The TRAF-associated protein TANK facilitates cross-talk within the IkappaB kinase family during Toll-like receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 17093–17098. 10.1073/pnas.1114194108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleynen I., Vazeille E., Artieda M., Verspaget H. W., Szczypiorska M., Bringer M. A., et al. (2014). Genetic and microbial factors modulating the ubiquitin proteasome system in inflammatory bowel disease. Gut 63, 1265–1274. 10.1136/gutjnl-2012-303205 [DOI] [PubMed] [Google Scholar]
- Davies S. P., Reddy H., Caivano M., Cohen P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351(Pt 1), 95–105. 10.1042/bj3510095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draz H., Goldberg A. A., Titorenko V. I., Tomlinson Guns E. S., Safe S. H., Sanderson J. T. (2017). Diindolylmethane and its halogenated derivatives induce protective autophagy in human prostate cancer cells via induction of the oncogenic protein AEG-1 and activation of AMP-activated protein kinase (AMPK). Cell. Signal. 40, 172–182. 10.1016/j.cellsig.2017.09.006 [DOI] [PubMed] [Google Scholar]
- Dzamko N., Inesta-Vaquera F., Zhang J., Xie C., Cai H., Arthur S., et al. (2012). The IkappaB kinase family phosphorylates the Parkinson's disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS ONE 7:e39132. 10.1371/journal.pone.0039132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. A., Biggs W. H., III., Treiber D. K., Atteridge C. E., Azimioara M. D., Benedetti M. G., et al. (2005). A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336. 10.1038/nbt1068 [DOI] [PubMed] [Google Scholar]
- Fedorov O., Marsden B., Pogacic V., Rellos P., Muller S., Bullock A. N., et al. (2007). A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. U.S.A. 104, 20523–20528. 10.1073/pnas.0708800104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., et al. (2003). LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055. 10.1084/jem.20031023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason C. E., Ordureau A., Gourlay R., Arthur J. S., Cohen P. (2011). Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J. Biol. Chem. 286, 35663–35674. 10.1074/jbc.M111.267567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringhuis S. I., Kaptein T. M., Wevers B. A., Mesman A. W., Geijtenbeek T. B. (2014). Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKepsilon- and CYLD-dependent Bcl3 activation. Nat. Commun. 5:3898. 10.1038/ncomms4898 [DOI] [PubMed] [Google Scholar]
- Hemmi H., Takeuchi O., Sato S., Yamamoto M., Kaisho T., Sanjo H., et al. (2004). The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650. 10.1084/jem.20040520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J. M., Ordureau A., Paulo J. A., Rinehart J., Harper J. W. (2015). The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 60, 7–20. 10.1016/j.molcel.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti J. E., Shen R. R., Abbott D. W., Zhou A. Y., Sprott K. M., Asara J. M., et al. (2009). Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol. Cell 34, 461–472. 10.1016/j.molcel.2009.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M., Osterlund P., Fagerlund R., Rios D. N., Hoffmann A., Poranen M. M., et al. (2015). MAP kinase p38alpha regulates type III interferon (IFN-lambda1) gene expression in human monocyte-derived dendritic cells in response to RNA stimulation. J. Leukoc. Biol. 97, 307–320. 10.1189/jlb.2A0114-059RR [DOI] [PubMed] [Google Scholar]
- Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., et al. (2008). A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132. 10.1038/nbt1358 [DOI] [PubMed] [Google Scholar]
- Kishore N., Huynh Q. K., Mathialagan S., Hall T., Rouw S., Creely D., et al. (2002). IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 277, 13840–13847. 10.1074/jbc.M110474200 [DOI] [PubMed] [Google Scholar]
- Komander D., Reyes-Turcu F., Licchesi J. D., Odenwaelder P., Wilkinson K. D., Barford D. (2009). Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 10, 466–473. 10.1038/embor.2009.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A., Chable-Bessia C., Cantarella G., Israel A., Wallach D., Courtois G. (2003). The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424, 801–805. 10.1038/nature01802 [DOI] [PubMed] [Google Scholar]
- Lazarus M. B., Shokat K. M. (2015). Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg. Med. Chem. 23, 5483–5488. 10.1016/j.bmc.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. P., Zeng L., Tian A., Bomkamp A., Rivera D., Gutman D., et al. (2012). Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J. Immunol. 189, 4630–4639. 10.4049/jimmunol.1102737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pelaez M., Lamont D. J., Peggie M., Shpiro N., Gray N. S., Cohen P. (2014). Protein kinase IKKbeta-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc. Natl. Acad. Sci. U.S.A. 111, 17432–17437. 10.1073/pnas.1418399111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lork M., Verhelst K., Beyaert R. (2017). CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell Death Differ. 24, 1172–1183. 10.1038/cdd.2017.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie K. F., Clark K., Naqvi S., McGuire V. A., Noehren G., Kristariyanto Y., et al. (2013). PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J. Immunol. 190, 565–577. 10.4049/jimmunol.1202462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoumi R. (2011). CYLD: a deubiquitination enzyme with multiple roles in cancer. Future Oncol. 7, 285–297. 10.2217/fon.10.187 [DOI] [PubMed] [Google Scholar]
- Oakes J. A., Davies M. C., Collins M. O. (2017). TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10:5 10.1186/s13041-017-0287-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters R. T., Liao S. M., Maniatis T. (2000). IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 5, 513–522. 10.1016/S1097-2765(00)80445-1 [DOI] [PubMed] [Google Scholar]
- Petherick K. J., Conway O. J., Mpamhanga C., Osborne S. A., Kamal A., Saxty B., et al. (2015). Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 290, 11376–11383. 10.1074/jbc.A114.627778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S., Nguyen J., Johnson J., Haura E., Coppola D., Chellappan S. (2015). Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat. Commun. 6:10072. 10.1038/ncomms10072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley W. W., Jin W., Lee A. J., Wright A., Wu X., Tewalt E. F., et al. (2007). Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 204, 1475–1485. 10.1084/jem.20062694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley W. W., Zhang M., Jin W., Losiewicz M., Donohue K. B., Norbury C. C., et al. (2006). Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 7, 411–417. 10.1038/ni1315 [DOI] [PubMed] [Google Scholar]
- Reiley W., Zhang M., Wu X., Granger E., Sun S. C. (2005). Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol. Cell. Biol. 25, 3886–3895. 10.1128/MCB.25.10.3886-3895.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritorto M. S., Ewan R., Perez-Oliva A. B., Knebel A., Buhrlage S. J., Wightman M., et al. (2014). Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 5:4763. 10.1038/ncomms5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saric A., Hipolito V. E., Kay J. G., Canton J., Antonescu C. N., Botelho R. J. (2016). mTOR controls lysosome tubulation and antigen presentation in macrophages and dendritic cells. Mol. Biol. Cell 27, 321–333. 10.1091/mbc.E15-05-0272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer K., Pralow A., Naumann M. (2016). p97/VCP promotes Cullin-RING-ubiquitin-ligase/proteasome-dependent degradation of IkappaBalpha and the preceding liberation of RelA from ubiquitinated IkappaBalpha. J. Cell. Mol. Med. 20, 58–70. 10.1111/jcmm.12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., tenOever B. R., Grandvaux N., Zhou G. P., Lin R., Hiscott J. (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. 10.1126/science.1081315 [DOI] [PubMed] [Google Scholar]
- Shen R. R., Hahn W. C. (2011). Emerging roles for the non-canonical IKKs in cancer. Oncogene 30, 631–641. 10.1038/onc.2010.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Kawai T., Takeda K., Matsumoto M., Inoue J., Tatsumi Y., et al. (1999). IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 11, 1357–1362. 10.1093/intimm/11.8.1357 [DOI] [PubMed] [Google Scholar]
- Smith H., Liu X. Y., Dai L., Goh E. T., Chan A. T., Xi J., et al. (2011). The role of TBK1 and IKKepsilon in the expression and activation of Pellino 1. Biochem. J. 434, 537–548. 10.1042/BJ20101421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal J., Driege Y., Bekaert T., Demeyer A., Muyllaert D., Van Damme P., et al. (2011). T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 30, 1742–1752. 10.1038/emboj.2011.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy M., Abeler-Dorner L., Chettle J., Mahlakoiv T., Goubau D., Chakravarty P., et al. (2015). Intestinal intraepithelial lymphocyte activation promotes innate antiviral resistance. Nat. Commun. 6:7090. 10.1038/ncomms8090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein S., Pham A., Bayer K. U., Tao-Cheng J. H., Dosemeci A. (2014). IKK regulates the deubiquitinase CYLD at the postsynaptic density. Biochem. Biophys. Res. Commun. 450, 550–554. 10.1016/j.bbrc.2014.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojima Y., Fujimoto A., Delhase M., Chen Y., Hatakeyama S., Nakayama K., et al. (2000). NAK is an IkappaB kinase-activating kinase. Nature 404, 778–782. 10.1038/35008109 [DOI] [PubMed] [Google Scholar]
- Trompouki E., Hatzivassiliou E., Tsichritzis T., Farmer H., Ashworth A., Mosialos G. (2003). CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 424, 793–796. 10.1038/nature01803 [DOI] [PubMed] [Google Scholar]
- Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347:1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Verhelst K., Verstrepen L., Carpentier I., Beyaert R. (2013). IkappaB kinase epsilon (IKKepsilon): a therapeutic target in inflammation and cancer. Biochem. Pharmacol. 85, 873–880. 10.1016/j.bcp.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Jono H., Kai H., Li J. D. (2005). The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J. Biol. Chem. 280, 41111–41121. 10.1074/jbc.M509526200 [DOI] [PubMed] [Google Scholar]
- Yu J., Zhou X., Chang M., Nakaya M., Chang J. H., Xiao Y., et al. (2015). Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat. Commun. 6:6074. 10.1038/ncomms7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Yi Y. S., Yang Y., Oh J., Jeong D., Cho J. Y. (2012). The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm. 2012:979105 10.1155/2012/979105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Stirling B., Temmerman S. T., Ma C. A., Fuss I. J., Derry J. M., et al. (2006). Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin. Invest. 116, 3042–3049. 10.1172/JCI28746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Aref A. R., Cohoon T. J., Barbie T. U., Imamura Y., Yang S., et al. (2014). Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 4, 452–465. 10.1158/2159-8290.CD-13-0646 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CYLD does not show a band shift indicative of phosphorylation upon stimulation with PMA/I or TNF. Different Jurkat T cell clones were stimulated with 200 ng/ml PMA and 1μM Ionomycin or 1,000 U/ml TNF for the indicated times. Protein levels were determined by immunoblotting. Jurkat-E (from Shigekazu Nagata, Osaka), Jurkat E6.1 (ATCC), Jurkat TNFR2 (gift from Nicholas Harper, Leicester).