

Abstract
Causal genes of chronic obstructive pulmonary disease (COPD) remain elusive. The current study aims at integrating genome-wide association studies (GWAS) and lung expression quantitative trait loci (eQTL) data to map COPD candidate causal genes and gain biological insights into the recently discovered COPD susceptibility loci. Two complementary genomic datasets on COPD were studied. First, the lung eQTL dataset which included whole-genome gene expression and genotyping data from 1038 individuals. Second, the largest COPD GWAS to date from the International COPD Genetics Consortium (ICGC) with 13 710 cases and 38 062 controls. Methods that integrated GWAS with eQTL signals including transcriptome-wide association study (TWAS), colocalization and Mendelian randomization-based (SMR) approaches were used to map causality genes, i.e. genes with the strongest evidence of being the functional effector at specific loci. These methods were applied at the genome-wide level and at COPD risk loci derived from the GWAS literature. Replication was performed using lung data from GTEx. We collated 129 non-overlapping risk loci for COPD from the GWAS literature. At the genome-wide scale, 12 new COPD candidate genes/loci were revealed and six replicated in GTEx including CAMK2A, DMPK, MYO15A, TNFRSF10A, BTN3A2 and TRBV30. In addition, we mapped candidate causal genes for 60 out of the 129 GWAS-nominated loci and 23 of them were replicated in GTEx. Mapping candidate causal genes in lung tissue represents an important contribution to the genetics of COPD, enriches our biological interpretation of GWAS findings, and brings us closer to clinical translation of genetic associations.
Introduction
Chronic obstructive pulmonary disease (COPD) is among the leading causes of hospitalization in industrialized countries and is the third leading cause of death worldwide (1). It was recently estimated that the absolute number of COPD cases in developed countries will increase by more than 150% from 2010 to 2030 (2). The lack of understanding of the molecular mechanisms underlying the pathogenesis of COPD has hampered efforts to develop new biomarkers and effective therapies.
Cigarette smoking is the main modifiable environmental risk factor for COPD. However, only 20–25% of smokers develop clinically significant airflow obstruction (3). There is strong evidence for genetic contribution to COPD. Candidate gene and genome-wide association studies (GWAS) have identified many genetic variants associated with COPD and its related phenotypes (4–9). The latest GWAS was performed by the International COPD Genetics Consortium (ICGC) which included 15 256 cases and 47 936 controls, and replication of significant signals in additional 9498 cases and 9748 controls (10). The combined meta-analysis identified 22 COPD susceptibility loci at genome-wide significance. However, it is likely that many additional loci contributing to COPD pathogenesis were missed because of the stringent statistical threshold typically used in GWAS.
Biological interpretation of genetic association results remains a major challenge. Most GWAS-associated variants have regulatory function and are associated with changes in gene expression (11,12). The mapping of expression quantitative trait loci (eQTL) in disease-relevant tissues has been successfully used to identify the candidate causal genes underpinning GWAS-nominated loci (13). Using lung eQTL derived from 1038 subjects, (14) we have previously identified the likely causal genes within COPD susceptibility loci (15–18). Recently developed methods allow more advanced integration of GWAS and eQTL results to colocalize GWAS and eQTL signals (19,20) as well as to perform transcriptome-wide association study (TWAS) (21) to identify candidate causal genes and functional single nucleotide polymorphisms (SNPs) underlying biological traits and diseases. In this study, we used three complementary methods to integrate the ICGC COPD GWAS results (10) and previously published COPD loci with lung tissue eQTLs (14) to identify the most likely causal COPD genes.
Results
Overall study design
The study design is shown in Figure 1. The ICGC COPD GWAS and lung eQTL datasets were integrated using three methods: TWAS, colocalization and Mendelian randomization-based (SMR) approach. These methods were first applied at the genome-wide level to identify new COPD candidate genes/loci. Results were then further evaluated in risk loci derived from published literature of GWAS on COPD and related phenotypes. Direct eQTL evaluation of GWAS SNPs (eSNP) was also assessed for literature-based COPD loci. The same integrative methods were then repeated using the GTEx lung eQTL dataset in order to replicate the results. Finally, a fourth genomics integrative method, S-PrediXcan, was evaluated to compare with other methods.
Figure 1.

Overview of the study design. Candidate causal genes enumerated at the bottom are those discovered using the lung eQTL dataset. The asterisk indicates those that replicated in the GTEx lung eQTL dataset. COLOC, colocalization; ICGC, International COPD Genetics Consortium; SMR, Mendelian randomization-based (SMR) approach; TWAS, transcriptome-wide association study.
Genome-wide integrative approaches
TWAS
A Manhattan plot showing transcriptome-wide associations in lung tissue with COPD is shown in Figure 2A. The 11 gene-COPD associations (corresponding to 16 probe sets) that reached genome-wide significance (PTWAS < 0.0001, Supplementary Material, Table S1) are shown. Of these, 12 probe sets resided in literature-based GWAS loci including four on 6p24.3, two on 5q32, two on 16p11.2 and two on 19q13.2. In contrast, MROH1 on 8q24.3 and SYCE1 on 10q26.3 represented novel candidate causal genes for COPD (Table 1).
Figure 2.
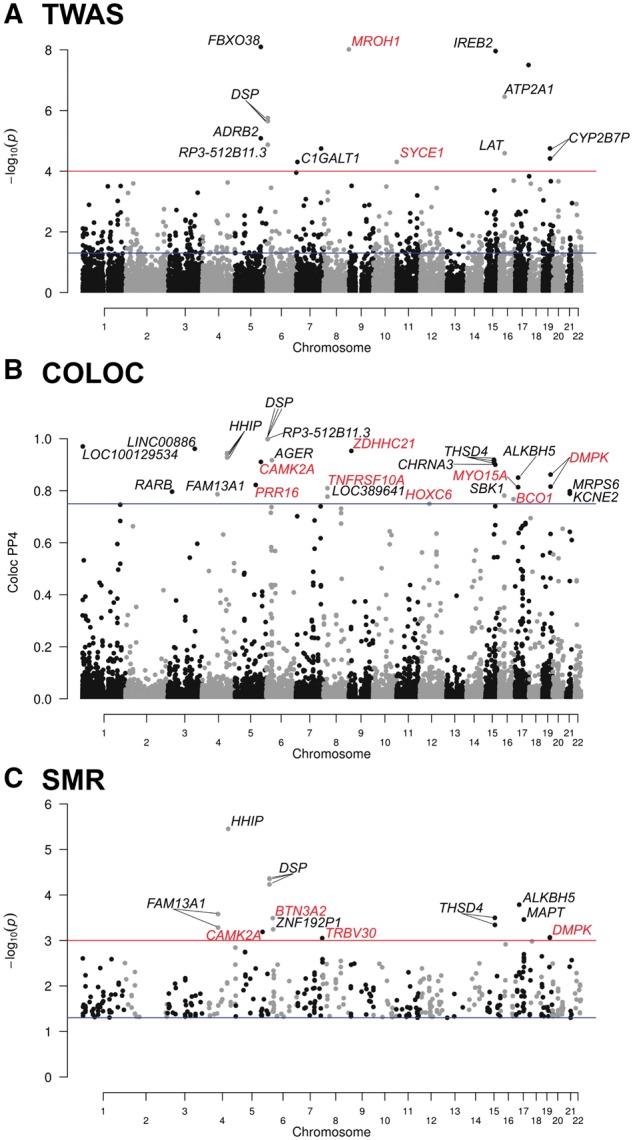
Manhattan plots of TWAS, colocalization and SMR. (A) TWAS results showing P-values on the −log10 scale on the y-axis for gene expression–COPD associations. The blue horizontal line represents PTWAS of 0.05. The red horizontal line represents the genome-wide significant threshold used in this study (PTWAS < 0.0001). Annotations for significant probe sets are indicated. (B) Colocalization results showing posterior probability of shared signals (PP4) on the y-axis. The blue horizontal line represents a standard colocalization threshold of 75%. Probe sets above this threshold are annotated. (C) SMR results showing P-values on the −log10 scale on the y-axis. By default, SMR only considered probe sets with at least one cis-eQTL < 5.0 × 10−8. The blue horizontal line represents PSMR of 0.05. The red horizontal line represents the significant threshold used in this study (PSMR < 0.001). Probe sets showing heterogeneity (PHEIDI < 0.05) were excluded. Probe sets with PSMR > 0.05 were also removed as no heterogeneity (HEIDI) tests were performed. Gene annotations for probe sets with PSMR < 0.001 are indicated. In the three Manhattan plots, gene names in red are new candidate gene/loci discovered in this study.
Table 1.
The 12 novel COPD candidate genes/loci
| Gene | Locus | Top SNP (PGWAS) | PTWAS | PP4COLOC | PSMR | PeQTL (SNP)a | Gene functionb | GTExc |
|---|---|---|---|---|---|---|---|---|
| MROH1 | 8q24.3 | rs11782029 | 9.65E-09 | 0.03 | – | 2.868E-60 | Lysosomal regulator | NA |
| (4.25E-04) | (rs113954825) | |||||||
| SYCE1 | 10q26.3 | rs9629930 | 4.93E-05 | 0.05 | – | 1.286E-08 | Meiosis, cancer | NA |
| (5.85E-04) | (rs146332556) | |||||||
| ZDHHC21 | 9p22.3 | rs10756585 | 0.0003 | 0.95 | 0.0034 | 2.988E-25 | Endothelial barrier integrity | No |
| (9.26E-05) | (rs10756585) | |||||||
| CAMK2A | 5q32 | rs6885505 | 0.206 | 0.91 | 0.0006 | 6.611E-14 | Calcium signalling | Yes |
| (2.03E-05) | (rs930212) | |||||||
| DMPK | 19q13.32 | rs116959973 | 0.028 | 0.86 | 0.0009 | 2.055E-23 | Anti-oxidant, development | Yes |
| (4.44E-05) | (rs7253302) | |||||||
| PRR16 | 5q23.1 | rs6869780 | 0.843 | 0.82 | – | 7.466E-07 | Regulator of cell size | No |
| (1.71E-05) | (rs10053752) | |||||||
| MYO15A | 17p11.2 | rs55918833 | 0.002 | 0.81 | 0.0050 | 1.98E-12 | Actin-organization in inner ear hair cells | Yes |
| (3.14E-05) | (rs9916193) | |||||||
| TNFRSF10A | 8p21.3 | rs13278062 | 0.0005 | 0.81 | 0.0033 | 8.191E-32 | Apoptosis | Yes |
| (4.30E-04) | (rs13278062) | |||||||
| BCO1 | 16q23.2 | rs11642391 | 0.0002 | 0.77 | – | 5.72E-07 | Alveolar development and repair | No |
| (1.12E-04) | (rs72823838) | |||||||
| HOXC6 | 12q13.13 | rs746423 | – | 0.75 | – | 7.881E-07 | Development and cancer | No |
| (1.67E-04) | (rs746423) | |||||||
| BTN3A2 | 6p22.2 | rs16891727 | 0.0004 | 0.72 | 0.0003 | 6.10E-240 | Immune system, anti-tumour | Yes |
| (6.98E-05) | (rs9366653) | |||||||
| TRBV30 | 7q34 | rs386718767 | – | 0.74 | 0.0009 | 3.211e-39 | Immune response | Yes |
| (2.45E-06) | (rs17267) |
PTWAS < 0.0001, PP4 > 0.75, PSMR < 0.001 and PeQTL < 1 × 10−8 are in bold.
Top eQTL for the probe set.
See Supplementary Material, Table S13 for more details.
Full results for replication in GTEx lung are provided in Supplementary Material, Table S8.
Colocalization (COLOC)
A Manhattan plot showing colocalization results is shown in Figure 2B. Posterior probability of shared signals (PP4) >75% are observed at 18 loci (Supplementary Material, Table S2). Nine of them colocalized in literature-based COPD risk loci. The others represent novel candidate causal genes for COPD including ZDHHC21 on 9p22.3, CAMK2A on 5q32, DMPK on 19q13.32, PRR16 on 5q23.1, MYO15A on 17p11.2, TNFRSF10A on 8p21.3, BCO1 on 16q23.2 and HOXC6 on 12q13.13 (Table 1).
SMR
Figure 2C and Supplementary Material, Table S3 show significant candidate genes identified using the SMR method. Excluding the known COPD risk loci, the four genome-wide significant SMR genes (PSMR = 0.001) were BTN3A2 on 6p22.2, CAMK2A on 5q32, DMPK on 19q13.32 and TRBV30 on 7q34. The four genes were also supported by colocalization (PP4 > 0.72) (Table 1).
Literature-based COPD risk loci
Lung eQTL
The results of 36 GWAS on COPD and related phenotypes are summarized in Supplementary Material, Table S4. Risk loci were extracted from publications as well as key genes and SNPs reported/discussed by the authors. For the top GWAS SNP at each reported locus, we tested its effect on lung gene expression in cis (1 Mb distance on either side of the SNP). All eSNP-regulated genes with PeQTL < 1 × 10−8 are reported in Supplementary Material, Table S5. Top eSNP-regulated genes (ones with the lowest lung eQTL P-value) included KANSL1-AS1 and MAPT on chromosome 17q21.31, DSP on 6p24.3, HSD17B12 on 11p11.2, PSORS1C3 on 6p21.33, ARNT on 1q21.3, HLA-DQB2 on 6p21.32 and IREB2 on 15q25 (Supplementary Material, Fig. S1). All eSNP-regulated genes with a nominal PeQTL < 0.05 by loci and studies are reported in Supplementary Material, Table S4.
Combining results from different approaches
GWAS results were arranged in 129 non-overlapping COPD risk loci (Supplementary Material, Table S6). For each locus, GWAS summary statistics from ICGC and the lung eQTL study were integrated to find the most likely causal gene(s) using TWAS, colocalization, SMR and eSNP-regulated gene approaches. For all loci, Supplementary Material, Table S6 provides the boundaries of each locus, lead GWAS SNP in ICGC, top lung eQTL SNP, TWAS genes (PTWAS < 0.05), colocalizing genes (PP4 > 60%) and SMR genes (PSMR < 0.05). Table 2 summarizes loci for which insightful results were obtained about the candidate causal gene and that were consistent for at least two integrative approaches. Supplementary Material, Table S7 presents results for the 129 loci. The most consistent candidate causal genes identified in this study were DSP on 6p24.3, C1GALT1 on 7p22.1 and THSD4 on 15q23. For these three loci, TWAS, colocalization, SMR and direct eSNP assessment consistently converged on these three potential causal genes (Fig. 3 and Supplementary Material, Fig. S2). For 13 additional loci, the same candidate causal gene was identified by three approaches (Supplementary Material, Fig. S3). Direct lung eQTL assessment of GWAS SNP, TWAS and SMR supported PADI2 on 1p36 (Supplementary Material, Fig. S3a), OXCT2 on 1p34.3 (Supplementary Material, Fig. S3b), MLF1 on 3q25.32 (Supplementary Material, Fig. S3c), TRIM4 on 7q22.1 (Supplementary Material, Fig. S3d), GSTO2 on 10q25 (Supplementary Material, Fig. S3e), ATXN3 on 14q32.12 (Supplementary Material, Fig. S3f) and MAPKBP1 on 15q15.1 (Supplementary Material, Fig. S3g). Direct lung eQTL assessment of GWAS SNP, TWAS and colocalization supported TGFB2 on 1q41 (Supplementary Material, Fig. S3h) and IREB2 on 15q25 (Supplementary Material, Fig. S3i). Direct lung eQTL assessment of GWAS SNP, colocalization and SMR supported FAM13A1 on 4q22.1 (Supplementary Material, Fig. S3j), TUFM on 16q11.2 (Supplementary Material, Fig. S3k), MAPT on 17q21.31 (Supplementary Material, Fig. S3l) and MRPS6 on 21q22.11 (Supplementary Material, Fig. S3m). Candidate causal genes supported by at least two approaches were found at nine other loci: ARNT on 1q21.3, HHIP on 4q31.21, ITGA2 on 5q11.2, CAMSAP1 on 9q33.1, DYDC2 on 10q22.3, FGD6 on 12q23.1-q22, CISD3 on 17q12, NT5C3B on 17q21.2 and MTCL1 on 18p11.22 (Table 2). Possible candidate causal genes supported by a single approach were found at 35 loci, including 8 lung eSNP-regulated genes, 17 TWAS genes, 1 colocalization gene and 9 SMR genes (Supplementary Material, Table S7). Overall, insightful results about the candidate causal genes were provided for 60 loci. For 23 of these (38%), the target gene was different from that reported in the GWAS (Supplementary Material, Fig. S4). For 18 loci (30%), the suspected gene from the GWAS was confirmed. Finally, for 19 loci (32%), the investigation refined the search to a single gene among the list of genes suspected by the GWAS.
Table 2.
The most likely causal genes in literature-based COPD-risk loci
| Approaches |
||||||
|---|---|---|---|---|---|---|
| Loci | Genes reported in GWAS | eSNP-regulated gene | TWAS | COLOC | SMR | GTExa |
| All approaches | ||||||
| 6p24.3 | DSP, BMP6 | DSP> RP3-512B11.3 | DSP> RP3-512B11.3 | DSP> RP3-512B11.3 | DSP | Yes |
| 7p22.1 | C1GALT1 | C1GALT1 | C1GALT1 | C1GALT1 | C1GALT1 | Yes |
| 15q23 | THSD4 | THSD4 | THSD4 | THSD4> U79293 | THSD4 | Yes |
| Three approaches | ||||||
| 1p36 | MFAP2 | BI715270 > PADI2> MFAP2 | BC044863 > PADI2 | MFAP2> CROCC > PADI2 | Yes | |
| 1p34.3 | LOC101929516, PABPC4 | OXCT2 | OXCT2/OXCT2P1 > PPIE | OXCT2 | No | |
| 1q41 | LYPLAL1, RNU5F-1, SLC30A10, TGFB2 | TGFB2 | TGFB2 | TGFB2 | No | |
| 3q25.32 | AK097794, MLF1, RSRC1 | LOC100996447 > RSRC1 > MLF1 | MLF1 | AY070437 > MLF1 | No | |
| 4q22.1 | FAM13A, TIGD2 | FAM13A> AK023526 | FAM13A-AS1 | FAM13A1 | FAM13A1 | Yes |
| 7q22.1 | ZKSCAN1 | PILRB/STAG3L5P-PVRIG2P-PILRB > PILRB > TRIM4 | TRIM4> ZKSCAN1 > ZNF3 > PVRIG | GATS > TRIM4> MBLAC1 | Yes | |
| 10q25 | GSTO2 | GSTO2 | GSTO2 | AK024150 | GSTO2 | Yes |
| 14q32.12 | ATXN3, FBLN5, RIN3, TRIP11 | AX721199 > BC033643 > ATXN3 | ATXN3> CATSPERB | ATXN3> BC033643 > AX721199 | Yes | |
| 15q15.1 | MGA | MAPKBP1 | MAPKBP1> LTK | JMJD7 > MAPKBP1 | No | |
| 15q25 | ADAMTS7, AGPHD1, CHRNA3, CHRNA5, CHRNB4, HYKK, IREB2, PSMA4 | AF147302 > IREB2> CHRNA5 > PSMA4 | IREB2> AF147302 > RASGRF1 > CTSH > AL109708 | CHRNA3 > IREB2 | No | |
| 16p11.2 | CCDC101, IL27, TUFM | TUFM | ATP2A1 > LAT > NUPR1 | SBK1 > TUFM | TUFM | Yes |
| 17q21.31 | ARHGAP27, ARL17A, ARL17B, CRHR1, FMNL1, KANSL1, LRRC37A, LRRC37A2, LRRC37A4, MAPT, NSF, NUDT1, PLEKHM1, SPPL2C, WNT3 | KANSL1-AS1 > LRRC37A4P > AW749333 > MAPT> PLEKHM1 > WNT3 > KANSL1 | CRHR1-IT1 | MAPT | MAPT> AW749333 > KANSL1-AS1 > KANSL1 | Yes |
| 21q22.11 | KCNE2, LINC00310 | MRPS6 | KCNE2 | MRPS6> KCNE2 | MRPS6 | Yes |
| Two approaches | ||||||
| 1q21.3 | ARNT, ENSA, GOLPH3L, LASS2, MCL1 | ARNT> GOLPH3L > CERS2 > HORMAD1 > RP11-54A4.2 | C1orf54 | ARNT> GOLPH3L | Yes | |
| 4q31.21 | HHIP | AK024689 > HHIP | HHIP> AK024689 | AK024689 | Yes | |
| 5q11.2 | ITGA1 | ITGA2 | ITGA2 | Yes | ||
| 9q33.1 | CARD9, DNLZ, INPP5E, LHX3, QSOX2 | CAMSAP1 | CAMSAP1 | Yes | ||
| 10q22.3 | ANXA11, SFTPD | DYDC2 | DYDC2> FAM213A | Yes | ||
| 12q23.1/12q22 | CCDC38, FGD6, SNRPF | NTN4> SNRPF | FGD6> VEZT | FGD6 | NTN4> VEZT | No |
| 17q12 | CISD3 | PCGF2 | CISD3 | CISD3 | Yes | |
| 17q21.2 | NT5C3B | NT5C3B | NT5C3B | Yes | ||
| 18p11.22 | MTCL1 | RAB12 | MTCL1 | MTCL1 | Yes | |
Candidate causal genes are illustrated in bold. For some loci, evidences for a second candidate causal gene (underline) were nearly as supportive.
Full results of replication in GTEx lung are provided in Supplementary Material, Table S9.
Figure 3.

The most consistent candidate causal genes identified in this study: (A) DSP on chromosome 6p24.3, (B) C1GALT1 on chromosome 7p22.1 and (C) THSD4 on 15q23. The upper left panel shows the genetic associations with COPD in ICGC. The bottom left panel shows the lung eQTL statistics for the target genes (DSP, C1GALT1 or THSD4). The boundaries of the loci are defined in Supplementary Material, Table S6 and the location of genes in this locus is illustrated at the bottom. The upper right panel shows boxplots of gene expression levels in the lung according to genotype groups for Laval, University of British Columbia (UBC) and Groningen samples. The y-axis shows the mRNA expression levels. The x-axis represents the three genotype groups for the SNP most strongly associated with mRNA expression of the target genes with the number of individuals in parenthesis. The risk allele identified in the ICGC GWAS is shown in red. Box boundaries, whiskers and centre mark in boxplots represent the first and third quartiles, the most extreme data point which is no more than 1.5 times the interquartile range, and the median, respectively. The table shows the top GWAS SNPs in ICGC and then the most likely causal gene(s) based on TWAS, colocalization and SMR combining summary statistics at these loci from the ICGC GWAS and the lung eQTL study. The linkage disequilibrium plots of selected SNPs on 6p24.3, 7p22.1 and 15q23 loci are provided in Supplementary Material, Figure S2.
The 12 novel COPD candidate genes/loci identified in this study are mapped in Figure 4. In addition, Figure 4 illustrates the 129 non-overlapping COPD risk loci derived from GWAS as well as the corresponding candidate causal genes for 60 of them revealed in this study.
Figure 4.

The COPD gene map. The map is an ideogram of the 22 autosomal human chromosomes and shows the location of candidate causal genes identified in this study. The map includes 12 genes in red located in new COPD candidate loci identified with the lung eQTL dataset. Those replicated in the GTEx lung eQTL dataset are indicated by an asterisk. The 129 non-overlapping COPD-risk loci derived from published GWAS on COPD, lung function and related phenotypes are also illustrated and are colour-coded based on phenotypes (see colour key). For 60 of these loci, the candidate causal genes identified using the lung eQTL dataset are indicated. Twenty-three of these candidate genes replicated in GTEx and are illustrated by an asterisk. The boundaries of each locus are provided in Supplementary Material, Table S6. The alternating grey and white colours on the chromosomes have been used to distinguish cytogenic bands from the adjacent ones and do not correspond to the band colours observed on giamsa-stained chromosomes. Information to construct the ideogram was obtained from the UCSC Genome Browser (hg19).
Replication in GTEx
To replicate our findings, we used lung eQTL data from 278 individuals available in GTEx (version 6). Significant cis-heritability is required to evaluate genes using the TWAS approach (21), i.e. a significant part of expression variability must be explained by SNPs. In GTEx, 2880 genes had significant cis-heritability and thus expression weights to be evaluated with TWAS. This is in contrast to 12 474 probe sets (correspond to 7126 unique genes) out of 40 359 with significant cis-heritability in our lung discovery eQTL dataset. Accordingly, applying the TWAS approach on lung eQTL from GTEx, we were only able to attempt replication for a fraction of the genes (1687 genes in common, or 24%). Similarly, for SMR tests only probe sets with at least one cis-eQTL at PeQTL < 5 × 10−8 were evaluated, and at this threshold, replication in GTEx lung was not feasible for some loci. Table 1 summarizes replication results in lung data from GTEx for the 12 novel COPD candidate genes/loci. Overall, 10 of these loci were tested and six (representing 60% of total gene tested) were replicated. TWAS, COLOC and SMR results for these 12 loci are provided in Supplementary Material, Table S8. Lung data from GTEx were also used for replication of the 60 candidate causal genes mapped in GWAS-nominated loci. Supplementary Material, Table S9 shows the results from our lung eQTL and GTEx lung data. For TWAS, expression weights were available for 20 genes, and for SMR, 47 out of the 60 genes had at least one significant eQTL for testing. Overall, for 39 out of the 60 genes that could be evaluated in GTEx, 23 were replicated (59%). The genes that replicated in GTEx lung data are presented in Table 2 and Figure 4.
S-PrediXcan results
To further validate our results and pinpoint the most consistent candidate causal genes, the ICGC GWAS on COPD and the lung eQTL set were analysed using S-PrediXcan, which is a recently developed integrative method (22). A Manhattan plot showing the S-PrediXcan results is provided in Supplementary Material, Figure S5. The 13 gene–COPD associations that reached genome-wide significance are provided in Supplementary Material, Table S10. Of these, four were on chromosome 15q25 pointing in the order of significance to IREB2, CHRNA3, CHRNA5 and HYKK as the candidate causal genes. This suggests again multiple candidate causal genes at this locus and consistent with other methods highlights IREB2 as the top significant gene. Six additional candidate causal genes in GWAS-nominated loci were consistent with other integrative methods described above including HHIP, FBXO38, FAM13A, DSP, THSD4 and TUFM. S-PrediXcan also identified ZDHHC21 on 9p22.3 as a new COPD candidate locus. On 7q22.1, GATS was the top candidate gene using S-PrediXcan, while the other integrative methods have identified TRIM4 at that locus. Finally, SNRPD2 on 19q13.32 was not identified by other integrative methods and is located outside known COPD GWAS-nominated loci. Similar to other integrative methods, replication of S-PrediXcan results in GTEx lung data was only feasible for a fraction of genes. For 5 out of the 13 genes that could be evaluated, four of them (80%) were replicated including DSP, CHRNA5, SNRPD2 and TUFM (Supplementary Material, Table S10).
S-PrediXcan results for both our lung eQTL dataset and GTEx lung for the 12 novel COPD candidate genes/loci as well as the 60 candidate causal genes in GWAS-nominated loci were also evaluated. In our lung eQTL set, 11 out of the 12 novel COPD genes could be evaluated using S-PrediXcan and 10 of them (91%) were replicated (Supplementary Material, Table S11). In GTEx lung, 7 out of the 12 novel COPD candidate genes could be evaluated and six of them (86%) were replicated (Supplementary Material, Table S11). For the 60 candidate causal genes in GWAS-nominated loci, 57 out of the 60 candidate causal genes were evaluated in our lung eQTL set. Among them, 39 (68%) were replicated (Supplementary Material, Table S12). In GTEx lung, 32 out of the 60 candidate causal genes in GWAS-nominated loci were evaluated and 13 (41%) were replicated (Supplementary Material, Table S12). This is a relatively high percentage of replicated genes considering that these 60 genes were identified using different integrative approaches.
Discussion
This is a comprehensive study to investigate the regulatory mechanisms in lung tissue underlying GWAS loci for COPD and its related phenotypes. Genome-wide integration of the largest GWAS on COPD with the largest lung eQTL study revealed 12 novel COPD candidate genes/loci and six of them were replicated in an independent lung eQTL dataset. This study also summarized 129 susceptibility loci from published GWAS on COPD and related phenotypes. Insightful results about the most likely causal genes were provided for 60 (47%) of them including 23 that were replicated in GTEx lung. Finally, while the results from each method were slightly different, TWAS, SMR, COLOC, S-PrediXcan and direct eSNP assessment converged on three genes: DSP on 6p24.3, C1GALT1 on 7p22.1 and THSD4 on 15q23.
At the genome-wide level, novel candidate loci (after excluding literature-based COPD risk loci, Supplementary Material, Table S6) identified through TWAS approach included MROH1 on 8q24.3 and SYCE1 on 10q26.3. Significant colocalization was also observed at eight novel candidate loci: ZDHHC21 on 9p22.3, CAMK2A on 5q32, DMPK on 19q13.32, PRR16 on 5q23.1, MYO15A on 17p11.2, TNFRSF10A on 8p21.3, BCO1 on 16q23.2 and HOXC6 on 12q13.13. Finally, four SMR genes were identified including two overlapping with those discovered by colocalization, namely CAMK2A and DMPK as well as BTN3A2 on 6p22.2 and TRBV30 on 7q34. The biology of these genes and their potential link to COPD pathobiology is provided in Table 1, with more details in Supplementary Material, Table S13. Interestingly, the top colocalization gene (ZDHHC21) is implicated in lung vascular endothelial barrier integrity (23). The P-values of the top GWAS SNPs in ICGC located 500 kb up and downstream of genome-wide discovered genes varied from 1.71 × 10−5 to 5.85 × 10−4, suggesting that the largest GWAS on COPD alone was underpowered to identify these genes at genome-wide significance. The findings support the utility of leveraging transcriptome data to uncover biologically relevant genes.
Complementary and as a functional follow-up of GWAS, methods used herein represent an important step to uncover genes whose expression changes in lung tissue are causally related to COPD. In this study, we provided insightful results about candidate causal genes for 60 out of the 129 COPD-risk loci derived from the literature. For 18 (30%) of these loci, we confirmed the target gene suspected by the GWAS. These include C1GALT1 on 7p22.1 and THSD4 on 15q23. The 7p22.1-C1GALT1 locus was recently reported as one of the 43 new signals for lung function (24). The 15q23-THSD4 locus was repeatedly associated with lung function (24,25) and COPD (10). In this study, the different integrative methods consistently pointed to these genes as being causally linked to COPD. With the wealth of susceptibility loci being reported (>100 loci), our study provides much needed information to prioritize follow-up functional studies. In addition, we identified the most likely causal gene for 19 loci (32%) where more than one gene was reported by GWAS. This includes 6p24.3 with two genes suspected from GWAS, namely BMP6 (24,26) and DSP (10). Integrative analyses consistently support DSP as the causal gene and demonstrate again how our study is narrowing down the investigational space underneath GWAS loci. Finally, for 23 loci (38%) the gene showing the most convincing evidence of causality was not reported by GWAS. As illustrated in Supplementary Material, Figure S4, the causal genes supported in this study were not necessarily the nearest annotated gene to the lead GWAS SNP. For the 60 candidate genes in GWAS-nominated loci identified using our lung eQTL dataset, 23 of them were replicated in GTEx (Fig. 4).
To the best of our knowledge, we have the largest lung eQTL dataset available (n = 1038). Replication of our results was attempted in a smaller lung eQTL set from GTEx (n = 278). It should also be mentioned that concerns were raised about the lung transcriptome data in GTEx. Indeed, extensive heterogeneity in gene expression variation was observed in this dataset, mostly due to sampling location in the lung and treatment-related changes (e.g. mechanical ventilation) (27). Considering the differences in sample size and lung tissue processing between our lung eQTL study and the GTEx lung eQTL, we were not expecting to be able to fully validate our results. Despite these differences, we were able to replicate approximately 60% of candidate causal genes highlighted with our lung eQTL dataset.
For many COPD risk loci, the most likely causal genes were not identified with the current data and will require further investigation. This is consistent with the lower than expected number of variants that colocalized between GWAS on glucose- and insulin-related traits and eQTL in human pancreatic islets and 44 different tissues in the GTEx portal (28). In many cases, the biological mechanisms underlying GWAS loci will not be mediated by eQTL. In this study, we used the most disease-relevant tissue to study COPD, but unavoidably we may have missed gene regulation processes that are specific to other tissues. There is a scope in future studies to investigate other tissues. Studying the whole lung transcriptome of patients undergoing surgery, does not allow to find eQTL specific to a disease-relevant cell type or eQTL that are context dependent, i.e. observed at certain stages of life or disease. It must be emphasized that the results from integrative approaches will require experimental work to confirm the role of identified genes in COPD. In addition, for some COPD-risk loci, our results implicate more than one candidate causal gene and further research is needed to explore multiple causal genes in a single locus.
In this study, we leveraged the largest lung eQTL dataset and GWAS on COPD available to provide insights about causality genes. We found 12 new COPD candidate genes outside GWAS loci including six that replicated in a second lung eQTL dataset. By synthesizing the GWAS literature on COPD, we collated 129 non-overlapping risk loci and provided insightful results about the candidate causal gene(s) for 60 of them including 23 that replicated in GTEx. Many of these genes were not the closest to, or harbouring the lead GWAS variant. Overall, by identifying plausible causal COPD genes, this study translates genetic associations into knowledge that is one step closer to clinical applications.
Materials and Methods
Published GWAS-risk loci for COPD
Supplementary Material, Table S4 shows the COPD susceptibility loci identified by review of the literature. This table is an extension of our previous review on the genetics of COPD (4) and was manually curated by reviewing the published GWAS on COPD, lung function, lung function decline, emphysema, chronic bronchitis and other related phenotypes published before 1 March 2017. For each study, we provided the reference, sample size, specific phenotype, suspected susceptibility genes and key SNPs as reported in the publications. Susceptibility loci were further validated and complemented using the GWAS catalogue (29). Results of GWAS in Supplementary Material, Table S4 were then arranged by locus in Supplementary Material, Table S6. These loci are considered literature-based COPD-risk loci and their boundaries were defined as follows: key SNPs derived from scientific publications were tabulated for each locus and the locations of the most 5′ and 3′ SNPs were identified. The boundaries of each locus were then defined by adding 500 kb downstream of the most 5′ SNP and 500 kb upstream of the most 3′ SNP. When windows overlapped, the intervals were amalgamated into a single interval with 500 kb on either side of each hit. The final boundaries are provided in Supplementary Material, Table S6.
ICGC GWAS on COPD
The ICGC has recently reported the world’s largest GWAS on COPD risk (10). For the current study, only GWAS results for individuals of European ancestry were considered consisting of 20 studies with 13 710 COPD cases and 38 062 controls. Cases were defined by moderate-to-severe airflow limitation based on pre-bronchodilator spirometry measurements (% predicted FEV1 < 80% and FEV1/FVC < 0.7) and GOLD recommendations (1) indicative of COPD GOLD stage 2 or worse. Genome-wide genotyping data were obtained for cases and controls, and additional SNPs were imputed using the 1000 genomes reference set. Quality control details have been published previously (10) and SNPs were considered in the GWAS analysis if they were included in at least 13 of the studies. The GWAS was performed using logistic regression of genotype dosage on COPD case–control status in each cohort separately adjusting for age, sex, smoking status (ever smoking and current smoking), pack-years smoking and ancestry-based principal components as needed. Results were then meta-analyzed in METAL (30) using fixed-effect model with inverse variance weighting. Summary statistics were available for 6 948 071 SNPs including chromosome position, alleles, allele frequencies, P-values, effect estimates and standard errors for all studies evaluated. For ICGC, each cohort obtained approval from appropriate ethical/regulatory bodies; informed consent was obtained for all individuals. The genome-wide association summary statistics from the ICGC are available at the database of Genotypes and Phenotypes (dbGaP) under accession phs000179.v5.p2.
Lung eQTL mapping study
The lung eQTL study has been described previously (14–16). Human lung tissues from subjects who underwent lung surgery were obtained at three academic sites: Laval University, University of British Columbia (UBC) and University of Groningen. All patients provided written informed consent and the study was approved by the ethics committees of the Institut universitaire de cardiologie et de pneumologie de Québec (IUCPQ) and the UBC-Providence Health Care Research Institute Ethics Board for Laval and UBC, respectively. The study protocol was consistent with the Research Code of the University Medical Center Groningen and Dutch national ethical and professional guidelines (‘Code of conduct; Dutch federation of biomedical scientific societies’; http://www.federa.org). Genotyping, gene expression and lung cis-eQTL analyses are described in the online supplementary materials. Gene expression data for the lung eQTL dataset are available in the Gene Expression Omnibus repository through accession number GSE23546.
Methods of GWAS-eQTL integration
Transcriptome-wide association study (TWAS)
The TWAS was performed using FUSION (21). The 1038 individuals for whom both gene expression and genetic variants were measured (i.e. the lung eQTL dataset) were combined with summary-level GWAS data from ICGC to estimate association statistics between gene expression and COPD. Briefly, this method can be conceptualized as having imputed expression data for all cases and controls in ICGC (using the part of expression that can be explained by SNPs in the lung eQTL dataset) and then test for association between imputed gene expression and COPD. To do so, normalized gene expression from Laval, UBC and Groningen were first combined using ComBat adjustment method (31) to correct for study site. Second, the genetic values of expression were computed one probe set at a time using SNP genotyping data located 500 kb on both sides of the probe sets using prediction models implemented in FUSION including (1) the single most significant lung eQTL-SNP as the predictor (top1), (2) LASSO regression and (3) elastic net regression (enet). All probe sets that passed QC in the lung eQTL were evaluated (n = 40 359) and 12 474 of them showed significant cis-heritability (i.e. part of expression variability that can be explained by SNPs). The expression weights of these cis-heritable probe sets were then combined with summary statistics from ICGC to obtain Z-score for each probe set. Genome-wide significant TWAS genes were considered at PTWAS < 0.0001. A higher cut-off threshold of PTWAS < 0.05 was used for literature-based COPD-risk loci as we aim to identify the most likely causal gene in these previously established COPD-risk loci.
Bayesian colocalization
Summary statistics, more specifically regression coefficients and their variance, from the ICGC GWAS and lung eQTL results were combined using COLOC package version 2.3-6 in R (19). Briefly, this method assesses whether two association signals, in this case GWAS and eQTL, are consistent with shared causal variant(s). Default prior probabilities of the software were used, i.e. P1= 1 × 10−04, P2= 1 × 10−04, P12= 1 × 10−05. Genes that demonstrated a high posterior probability (PP4 >75%) indicating that the COPD GWAS and lung eQTL signals colocalized were reported. PP4 > 60% was also considered within literature-based COPD risk loci.
Summary data-based Mendelian randomization SMR
GWAS and lung eQTL data were also integrated using the SMR method (20). Conceptually this approach is similar to standard Mendelian randomization analysis, where measured variations in genes are used as instrumental variables to test for causative effect of an exposure on disease. Here, the SNPs (instrumental variables) are used to test for the causative effect of gene expression (exposure) on COPD (disease). By default, SMR only considered probe sets with at least one cis-eQTL PeQTL < 5 × 10−8. In this analysis, 8679 probe sets were evaluated. A cut-off threshold of PSMR < 0.001 with no evidence of heterogeneity (PHEIDI > 0.05) was used for genome-wide analysis. For literature-based COPD-risk loci, SMR genes were those with a PSMR < 0.05 with no evidence of heterogeneity (PHEIDI > 0.05).
eQTL analysis with GWAS SNPs (eSNP)
This method was performed at the individual SNP level and tested whether GWAS SNPs from the literature act as lung eQTL. Lung eQTL-regulated genes by GWAS SNPs were considered eSNP-regulated genes. eSNP-regulated genes with PeQTL < 1 × 10−8 were considered statistically significant. However, eSNP-regulated genes with PeQTL < 0.05 were also explored as these analyses were performed at previously established COPD loci.
S-PrediXcan
The summary GWAS data from ICGC was also integrated with the lung eQTL dataset using S-PrediXcan (22) (bioRxiv, http://www.biorxiv.org/content/early/2017/10/03/045260). Gene expression traits were first trained with elastic net linear models (alpha = 0.5, n_k_folds = 10, window = 500 kb) using the lung eQTL set (n = 1038). Models with FDR < 0.05 were obtained for 19 546 probe sets. Predicted expression levels from the lung in the ICGC study set were then tested for association with COPD within the MetaXcan framework. Genome-wide significantly associated genes were considered at PPrediXcan < 0.0001.
Replication in GTEx
Lung data from GTEx (V6) were used to replicate our results. The lung eQTL dataset in GTEx comprises 278 individuals. TWAS, COLOC, SMR and S-PrediXcan were performed similar to our lung eQTL dataset. Analyses were restricted to the 12 novel COPD candidate genes/loci as well as the 60 candidate causal genes in GWAS-nominated loci identified in our lung eQTL dataset. Thresholds of significance for replication were set at PTWAS < 0.05, PP4 > 0.6, PSMR < 0.05 and PPrediXcan < 0.05.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank the staff at the Quebec Respiratory Health Network Tissue Bank for their valuable assistance with the lung eQTL dataset at the Laval University.
Conflict of Interest Statement. None declared.
Funding
M.L. was the recipient of a doctoral studentship from the Fonds de recherche Québec—Santé (FRQS). J.C.B. is the recipient of doctoral scholarships from the Canadian Respiratory Research Network (CRRN) and FRQS. M.O. is a Fellow of the Parker B. Francis Foundation. M.O. is also a recipient of British Columbia Lung Association Research Grant. K.H. is partially supported by the National Natural Science Foundation of China (Grant No. 21477087, 91643201) and by the Ministry of Science and Technology of China (Grant No. 2016YFC0206507). Y.B. holds a Canada Research Chair in Genomics of Heart and Lung Diseases. This study was supported by grants from the Chaire de pneumologie de la Fondation JD Bégin de l’Université Laval, the Fondation de l’Institut universitaire de cardiologie et de pneumologie de Québec, the Quebec Respiratory Health Network and the Canadian Institutes of Health Research [MOP – 123369] to Y.B. Research reported in this publication was supported by the National Heart, Lung, and Blood Institute [R01 HL113264 to M.H.C]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1. Vestbo J., Hurd S.S., Agusti A.G., Jones P.W., Vogelmeier C., Anzueto A., Barnes P.J., Fabbri L.M., Martinez F.J., Nishimura M.. et al. (2013) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med., 187, 347–365. [DOI] [PubMed] [Google Scholar]
- 2. Khakban A., Sin D.D., FitzGerald J.M., McManus B.M., Ng R., Hollander Z., Sadatsafavi M. (2017) The projected epidemic of chronic obstructive pulmonary disease hospitalizations over the next 15 years. A population-based perspective. Am. J. Respir. Crit. Care Med., 195, 287–291. [DOI] [PubMed] [Google Scholar]
- 3. Lokke A., Lange P., Scharling H., Fabricius P., Vestbo J. (2006) Developing COPD: a 25 year follow up study of the general population. Thorax, 61, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bossé Y. (2012) Updates on the COPD gene list. Int. J. Chron. Obstruct. Pulmon. Dis., 7, 607–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cho M.H., Boutaoui N., Klanderman B.J., Sylvia J.S., Ziniti J.P., Hersh C.P., DeMeo D.L., Hunninghake G.M., Litonjua A.A., Sparrow D.. et al. (2010) Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet., 42, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cho M.H., Castaldi P.J., Wan E.S., Siedlinski M., Hersh C.P., Demeo D.L., Himes B.E., Sylvia J.S., Klanderman B.J., Ziniti J.P.. et al. (2012) A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum. Mol. Genet., 21, 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pillai S.G., Ge D., Zhu G., Kong X., Shianna K.V., Need A.C., Feng S., Hersh C.P., Bakke P., Gulsvik A.. et al. (2009) A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet., 5, e1000421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cho M.H., McDonald M.L., Zhou X., Mattheisen M., Castaldi P.J., Hersh C.P., Demeo D.L., Sylvia J.S., Ziniti J., Laird N.M.. et al. (2014) Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir. Med., 2, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bossé Y. (2009) Genetics of chronic obstructive pulmonary disease: a succinct review, future avenues and prospective clinical applications. Pharmacogenomics, 10, 655–667. [DOI] [PubMed] [Google Scholar]
- 10. Hobbs B.D., de Jong K., Lamontagne M., Bossé Y., Shrine N., Artigas M.S., Wain L.V., Hall I.P., Jackson V.E., Wyss A.B.. et al. (2017) Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet., 49, 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Torres J.M., Gamazon E.R., Parra E.J., Below J.E., Valladares-Salgado A., Wacher N., Cruz M., Hanis C.L., Cox N.J. (2014) Cross-tissue and tissue-specific eQTLs: partitioning the heritability of a complex trait. Am. J. Hum. Genet., 95, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet., 6, e1000888.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bossé Y. (2013) Genome-wide expression quantitative trait loci analysis in asthma. Curr. Opin. Allergy Clin. Immunol., 13, 487–494. [DOI] [PubMed] [Google Scholar]
- 14. Hao K., Bossé Y., Nickle D.C., Pare P.D., Postma D.S., Laviolette M., Sandford A., Hackett T.L., Daley D., Hogg J.C.. et al. (2012) Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet., 8, e1003029.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamontagne M., Couture C., Postma D.S., Timens W., Sin D.D., Pare P.D., Hogg J.C., Nickle D., Laviolette M., Bossé Y. (2013) Refining susceptibility loci of chronic obstructive pulmonary disease with lung eQTLS. PLoS One, 8, e70220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamontagne M., Timens W., Hao K., Bossé Y., Laviolette M., Steiling K., Campbell J.D., Couture C., Conti M., Sherwood K.. et al. (2014) Genetic regulation of gene expression in the lung identifies CST3 and CD22 as potential causal genes for airflow obstruction. Thorax, 69, 997–1004. [DOI] [PubMed] [Google Scholar]
- 17. Obeidat M., Hao K., Bossé Y., Nickle D.C., Nie Y., Postma D.S., Laviolette M., Sandford A.J., Daley D.D., Hogg J.C.. et al. (2015) Molecular mechanisms underlying variations in lung function: a systems genetics analysis. Lancet Respir. Med., 3, 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lamontagne M., Joubert P., Timens W., Postma D.S., Hao K., Nickle D., Sin D.D., Pare P.D., Laviolette M., Bossé Y. (2016) Susceptibility genes for lung diseases in the major histocompatibility complex revealed by lung expression quantitative trait loci analysis. Eur. Respir. J., 48, 573–576. [DOI] [PubMed] [Google Scholar]
- 19. Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M.. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
- 21. Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W., Jansen R., de Geus E.J., Boomsma D.I., Wright F.A.. et al. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet., 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barbeira A.N., Dickinson S.P., Torres J.M., Bonazzola R., Zheng J., Torstenson E.S., Wheeler H.E., Shah K.P., Edwards T., Garcia T.. et al. (2017) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. bioRxiv, http://www.biorxiv.org/content/early/2017/10/03/045260. [DOI] [PMC free article] [PubMed]
- 23. Beard R.S. Jr, Yang X., Meegan J.E., Overstreet J.W., Yang C.G., Elliott J.A., Reynolds J.J., Cha B.J., Pivetti C.D., Mitchell D.A.. et al. (2016) Palmitoyl acyltransferase DHHC21 mediates endothelial dysfunction in systemic inflammatory response syndrome. Nat. Commun., 7, 12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wain L.V., Shrine N., Artigas M.S., Erzurumluoglu A.M., Noyvert B., Bossini-Castillo L., Obeidat M., Henry A.P., Portelli M.A., Hall R.J.. et al. (2017) Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet., 49, 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Repapi E., Sayers I., Wain L.V., Burton P.R., Johnson T., Obeidat M., Zhao J.H., Ramasamy A., Zhai G., Vitart V.. et al. (2010) Genome-wide association study identifies five loci associated with lung function. Nat. Genet., 42, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loth D.W., Soler Artigas M., Gharib S.A., Wain L.V., Franceschini N., Koch B., Pottinger T.D., Smith A.V., Duan Q., Oldmeadow C.. et al. (2014) Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nat. Genet., 46, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCall M.N., Illei P.B., Halushka M.K. (2016) Complex sources of variation in tissue expression data: analysis of the GTEx lung transcriptome. Am. J. Hum. Genet., 99, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hormozdiari F., van de Bunt M., Segre A.V., Li X., Joo J.W., Bilow M., Sul J.H., Sankararaman S., Pasaniuc B., Eskin E. (2016) Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet., 99, 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L.. et al. (2014) The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willer C.J., Li Y., Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson W.E., Li C., Rabinovic A. (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.