

Abstract
Synchronous colorectal cancers (syCRCs), which present two or more lesions at diagnosis, are rare and pose a great challenge for clinical management. Although some predisposing factors associated with syCRCs have been studied with limited accession, the full repertoire of genomic events among the lesions within an individual and the causes of syCRCs remain unclear. We performed whole-exome sequencing of 40 surgical tumour samples of paired lesions from 20 patients to characterize the genetic alterations. Lesions from same patient showed distinct landscapes of somatic aberrations and shared few mutations, which suggests that they originate and develop independently, although they shared the similar genetic background. Canonical genes, such as APC, KRAS, TP53 and PIK3CA, were frequently mutated in the syCRCs, and most of them show different mutation profile compared with solitary colorectal cancer. We identified a recurrent somatic alteration (K15fs) in RPL22 in 25% of the syCRCs. Functional analysis indicated that mutated RPL22 may suppress cell apoptosis and promote the epithelial–mesenchymal transition (EMT). Potential drug targets were identified in several signalling pathways, and they present great discrepancy between lesions from the same patient. Our data show that the syCRCs within the same patient present great genetic heterogeneity, and they may be driven by distinct molecular events and develop independently. The discrepancy of potential drug targets and mutation burden in lesions from one patient provides valuable information in clinical management for patients with syCRCs.
The syCRCs within the same patient present great genetic heterogeneity and may be driven by distinct molecular events and develop independently. The discrepancy of potential drug targets in lesions from one patient provides valuable information in clinical management.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in men and the second in women, with an estimated 1.4 million cases and 693 900 deaths occurring in 2012 (1). Approximately, 3.5% of all patients with CRC have synchronous colorectal cancer (syCRC)—the presence of two or more primary colorectal carcinomas at initial presentation (2). Compared with patients with solitary CRC, syCRC patients were significantly associated with poor prognosis (3).
The causes and subsequent mechanisms leading to this cancer type remain poorly understood. Patients with inflammatory bowel diseases, hyperplastic polyposis and Lynch syndrome have been reported to be predisposed to syCRCs (4), but they account for less than 10% of cases. Most studies investigating syCRCs have focused on clinical characteristics and epidemiology (5), and only a few have provided genome-wide characterization of syCRCs (6). A prospective cohort study showed that syCRCs have more frequent mutations in BRAF and have the CpG island methylator phenotype-high and microsatellite instability-high status more frequently than do solitary CRCs (3). Mutations in NTHL1 also have been associated with the onset of syCRCs (7). However, these studies merely focused on candidate genes and failed to provide the molecular landscape of this cancer type. Furthermore, whether the syCRCs originate independently and the mechanisms underlying the evolution of the molecular aberrations remain to be revealed. Precision medicine, especially target therapy for patients with syCRCs, also has never been guided.
To determine the genetic profile of syCRCs and critical genomic events leading to the development of syCRCs, as well as potential targets for therapy, we performed whole-exome sequencing on 40 tumour samples as well as adjacent normal controls from 20 patients with syCRCs (Supplementary Table 1, available at Carcinogenesis Online). Comprehensive genomic analysis revealed that paired syCRCs within one patient have distinct landscapes of somatic aberrations and share few mutations. These data, which represent the largest known set of syCRCs, provide us with new insights into the syCRC genomic profile and advance our knowledge of disease molecular mechanisms, biological pathways and potentially actionable targets.
Materials and methods
Patients and sample collection
All fresh samples were collected from patients who had undergone surgical resection of synchronous colorectal carcinomas (syCRCs). The paired syCRCs from the same patient were separated by at least 50 mm of pathological normal bowel wall. Samples were obtained from eight patients from the First Affiliated Hospital of Chongqing Medical University (Chongqing, China) and 12 patients from the Research Institute of Surgery, Third Military Medical University (Chongqing, China). Individuals were excluded from entry into the study if they had a known history of inflammatory bowel disease or familial adenomatous polyposis. All patients provided written informed consent, and the study was approved by the ethics committee of our institution.
Whole-exome capture and sequencing
The qualified genomic DNA was randomly fragmented by using Covaris technology (Woburn, MA). The prepared DNA fragments were amplified by ligation-mediated PCR, purified and hybridized to the Agilent SureSelect 51M Capture kit (Agilent Technologies, Santa Clara, CA) for enrichment. Each qualified captured library was then loaded on Illumina HiSeq 2000 (Illumina, San Diego, CA) platforms and subjected to high-throughput sequencing.
Read mapping and variation calling
Reads from the Illumina machine containing sequencing adapters and low-quality reads with more than five unknown bases were removed to obtain clean data. The clean data were mapped to the human reference genome (GRCh37/hg19) using the Burrows–Wheeler Aligner (8). Picard (v1.54; http://picard.sourceforge.net/) was used to identify duplicates, followed by the Genome Analysis Toolkit (v1.0.6076, GATK IndelRealigner) (9). Potential single-nucleotide variants (SNVs) were called by Varscan2.2.5 (with parameters as mpileup-Q0 and Varscan2.2.5-min-coverage 10-min-coverage-normal 10-min-coverage-tumour 10-min-var-freq 0.1-min-freq-for-hom 0.75-min-avg-qual 0). Then, we used our in-house pipeline to do filtration, with major criteria as the following: adjacent somatic mutation distance, mapping quality, base quality, allele frequency change between tumour and adjacent normal, mutation should not be in gap-aligned reads, mutations should not be significantly enriched within 5 bp of 5′ or 3′ ends of the reads and mutations should not be in simple repeat region. Somatic indels were predicted by GATK SomaticIndelDetector with default parameters. Then, we used our own pipeline to obtain high-confidence somatic indels, as following: combined normal and tumour bam were reused to perform local realignment, and germline indels were filtered for high confidently indels; normal coverage and tumour coverage should be ≥10. Highly confident somatic SNVs and indels were annotated using ANNOVAR (10).
Somatic copy number variation analysis
Somatic copy number variation analysis was performed using GATK4 Alpha (http://gatkforums.broadinstitute.org/gatk/discussion/5640/recapseg-overview). Specifically, sample reads, targets and reference were employed to create proportional coverage. The panel of normal was created by collecting proportional coverage from each normal sample and storing the median proportional coverage, which was used to encapsulate sequencing noise. Then, the proportional coverage of each tumour and panel of normal was used to normalize the coverage profile, followed by tangent normalization. Segmented coverage was obtained by normalized coverage segmentation. The copy-ratio profiles were segmented with circular binary segmentation. Variations in the X and Y chromosome were excluded during this analysis.
Mutational signature analysis
All variants used for signature analysis were stringently filtered, which has been addressed in variants calling section. Non-negative matrix factorization was used to decipher signatures of mutational processes from mutational catalogues of cancer genomes. The profile of each signature was displayed using the six substitution subtypes: C>A, C>G, C>T, T>A, T>C and T>G. Each substitution was examined by incorporating information on the bases immediately 5′ and 3′ to each mutated base to generate 96 possible mutation types. The mutational signatures were displayed and reported based on the observed trinucleotide frequency of the human genome. The number of signatures was determined by considering the extracted processes stability and reconstruction error as described previously by Alexandrov (11). The computational framework for deciphering mutational signatures was downloaded from http://www.mathworks.com/matlabcentral/fileexchange/38724, which is supported by the Wellcome Trust Sanger Institute.
Cancer cell fraction evaluation of candidate gene mutations
For each mutation, cancer cell fraction (CCF) was calculated by ABSOLUTE, which integrated information from copy number change, purity of sequenced samples and variant allele counts. It computes a probability distribution of the CCF for each mutation with 95% confidence intervals and includes a probability that each mutation is subclonal (12).
Neoantigen prediction
Human leucocyte antigen (HLA) alleles of patients were identified from autologous normal tissue using HLAVBseq (13). Neoantigens were predicted by NetMHC, NetMHCpan, PickPocket, PSSMHCpan and SMM (14). We calculated the average IC50 value of normal peptides and mutant peptides based on these tools. We retained the mutant peptides as neoantigens if they meet the two criteria (1): IC50 < 500 at least in two tools (2); MT score < WT score.
Validation of RPL22 mutation by PCR and Sanger sequencing
Genomic region of 268 bp long encompassing the mutations were sequenced by 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA). Forward and reverse sequences were inspected respectively. Following primers were used:
Forward primer ACCACCCGAGTGGCAATAAG
Reverse primer CAAAGGGAGCACACTTCCGT
Histology
The Ki-67 protein detection was evaluated by immunohistochemistry using rabbit polyclonal antibody (NO.19972-1-AP, Proteintech, Wuhan, China) in formalin-fixed, paraffin-embedded tissue.
Immunofluorescent staining
According to the protocol recommended by the manufacturer, TUNEL staining was performed with the In Situ Cell Death Detection Kit, POD(NO.11684817910, Roche, Mannheim, Germany) for immunofluorescent detection and quantification of apoptosis (programmed cell death) in formalin-fixed, paraffin-embedded tissues. Sections were incubated with a 1:50 dilution of rabbit anti-RPL22 Polyclonal antibody (NO.25002-1-AP, Proteintech). A 1:100 dilution of Cy3-labeled Goat Anti-Rabbit IgG (H+L; NO. A0516, Beyotime, Shanghai, China) was used as the secondary antibody. The localization of RPL22 in the syCRCs was examined under confocal laser scanning microscopy (FV1200 IX83, Olympus Corporation, Tokyo, Japan).
Lentivirus transductions
Genomic regions of 386 bp long encompassing the human RPL22 mutation p.K15fs were amplified by PCR and subcloned into the lentiviral vector pLenti-EF1a-EGFP-P2A-Puro-CMV-3Flag-RPL22(mutation). The 387 bp long DNA corresponding to the human RPL22 wild-type (WT) was subcloned into the lentiviral vector pLenti-EF1a-EGFP-P2A-Puro-CMV-3Flag-RPL22. Recombinant lentiviruses were purchased from NeuronBiotech (Shanghai, China). To obtain RPL22 mutated or WT stable cell lines, HT-29 cells were transfected with 4.75 × 108 and 5.26 × 108 transducing units per millilitre of lentiviruses, respectively, and selected with 2.0μg/ml puromycin for 2 weeks. The stably expressing cell lines were identified using real-time PCR and western blot. CRC cells HT-29 used in this study were authenticated by Short Tandem Repeat profiling and obtained from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai, China in January 2016.
RT-PCR and quantitative real-time PCR
One microgram of total RNA extract was reverse transcribed using the PrimeScript™RT reagent Kit (NO.#RR037A, Takara, Dalian, China). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using the CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA) and SYBR Premix Ex Taq II (NO.#RR820A); GAPDH was used for normalization. The primers used for qRT-PCR of Rpl22 and other genes are listed in Supplementary Table 2, available at Carcinogenesis Online.
Western blotting
Lysates of Cells and frozen pulverized tumours were separated on an 10 or 15% SDS-PAGE gel, followed by incubation with the following antibodies: RPL22 (NO.SC-136413, SantaCruz Biotechnology, SantaCruz), E-cadherin(NO.60335-1-Ig, Proteintech), vimentin (NO.60330-1-Ig, Proteintech), Bcl-2 (NO.12789-1-AP, Proteintech) and Bax (NO.50599-2-Ig, Proteintech). The secondary antibodies IRDye® 800CW goat Anti-Rabbit IgG (P/N925-32211, LI-COR Biosciences, Lincoln, NE) and IRDye® 800CW goat anti-mouse IgG (P/N925-32210, LI-COR Biosciences) were used, followed by visualization using the LI-COR ODYSSEY system (Li-COR Biosciences) and Quantity One software (Bio-Rad).
Results
The molecular landscape of syCRCs
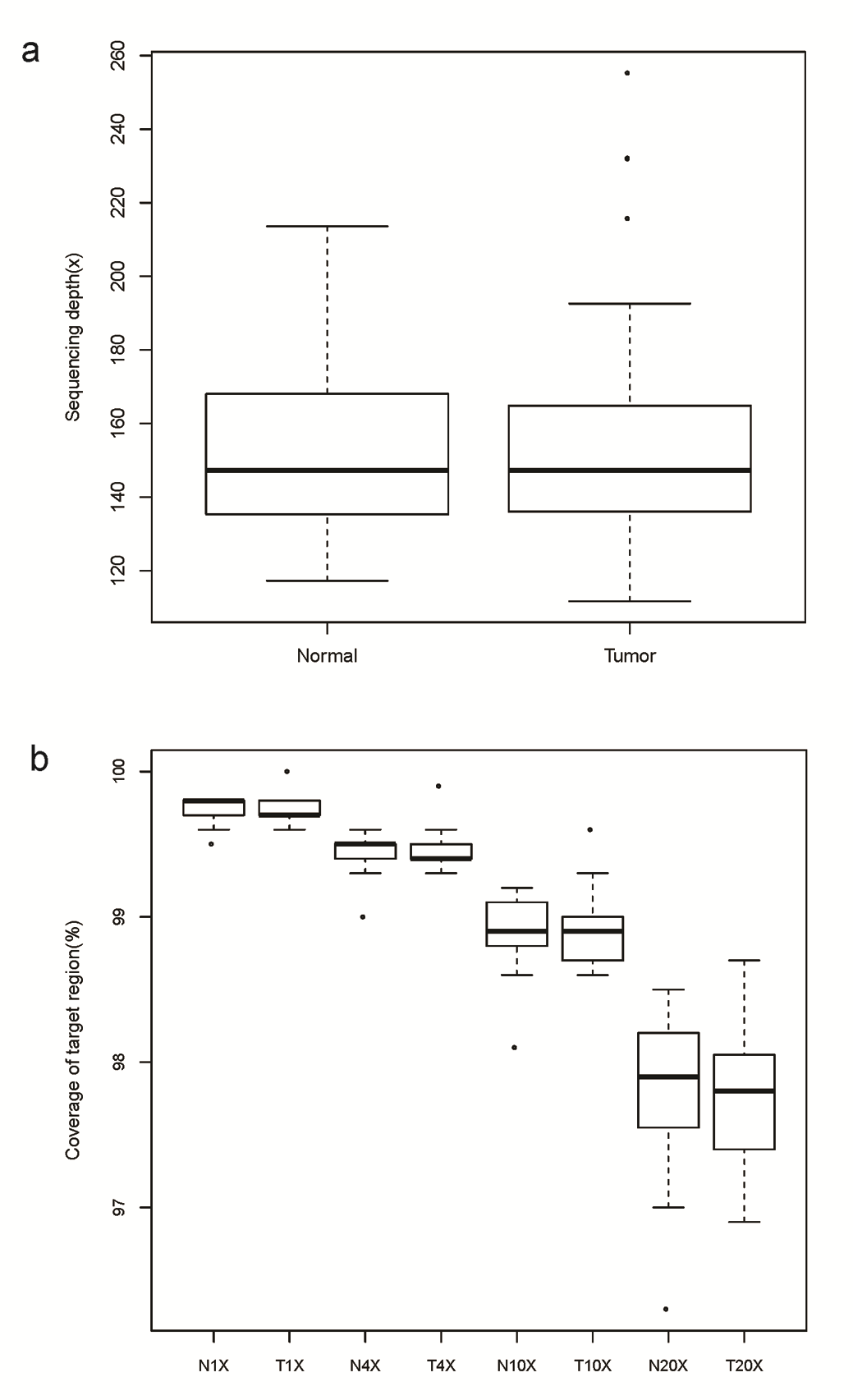
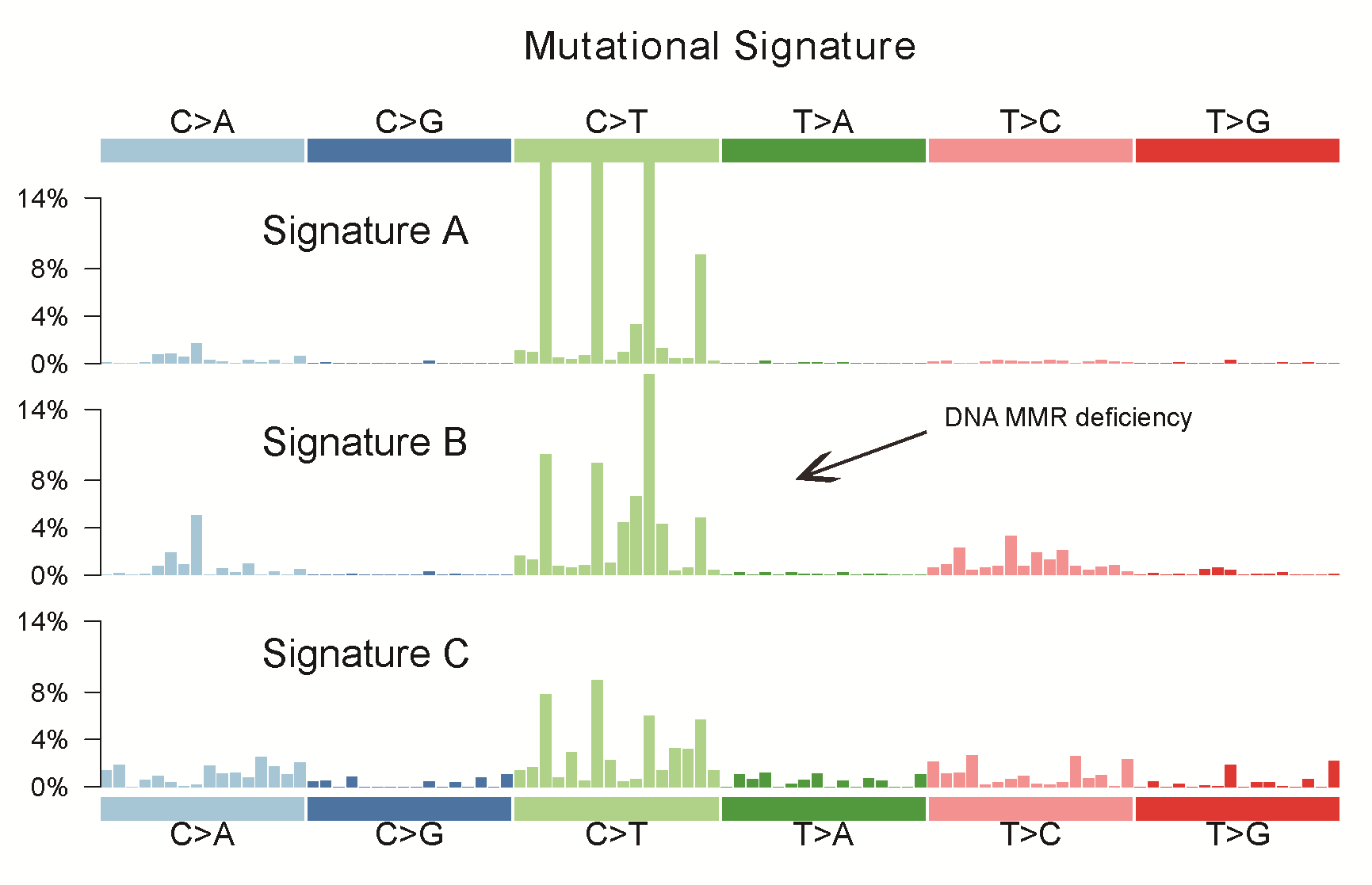
We performed whole-exome capture and sequencing of syCRCs and matched adjacent non-tumour intestinal tissue from 20 patients to identify somatic mutations. The average sequencing depth of the target was 156× for tumours and 153× for normal controls (Supplementary Figure 1, available at Carcinogenesis Online). In total, 25 133 somatic SNVs were identified in the tumour samples, of which 17 843 were non-silent mutations predicted to affect coding proteins. We also detected 4435 somatic small insertions and deletions (indels) among the tumour samples (Supplementary Table 3, available at Carcinogenesis Online). The somatic mutation rate varied widely among the tumours; some of them had a mutation rate <1/million bases, whereas others reached 70/million bases (Figure 1). We separated tumours with a mutation rate <6/million bases as non-hypermutated and those with mutation rate >15/million bases as hypermutated. The number of mutations in tumours was not significantly associated with the tumour stage (Fisher’s exact test, P = 0.23). To investigate whether this variability could be attributed to mismatch repair gene (MMR) deficiency, we integrated the context of the mutation spectrum and extracted signature B (cosine similarities of 0.97 with COSMIC signature 6) which is related to the MMR deficiency (Supplementary Figure 2, available at Carcinogenesis Online). Analysis of copy number aberrations revealed 9071 segments in total, ranging from 64 to 505 for each tumour.
Figure 1.

Landscape of genomic aberrations in syCRCs. Each column represents a tumour; contiguous tumours are syCRCs from one patient. The top panel shows the number of somatic mutations. Group 1 comprised patients in whom both syCRCs are hypermutated. Group 2 comprised patients in whom one tumour is hypermutated, but the other is non-hypermutated. Group 3 comprised patients in whom both syCRCs are non-hypermutated. The next panel presents patients’ information including tumour location, stage, gender and mutation status (Mut status). The middle panel presents the frequently mutated genes in our cohort study, and the frequency is shown on the right. Germline mutations of common CRC-related hereditary genes are also shown (specific mutations are on the right). The bottom panel indicates the somatic copy number aberrations.
To clearly illustrate the landscape of somatic mutations in syCRCs, we divided the patients into three groups based on mutation burdens (Figure 1). For five patients in Group 1, both syCRCs were hypermutated, and we speculated that germline mutations in MMRs predisposed the patients to these phenotypes. The results showed that P6 and P10 had damaging germline mutations in MLH1 (p.L73P and p.R226P, respectively), and the remaining three patients showed no trace. Group 2 consisted of three patients with one tumour being hypermutated and the other tumour non-hypermutated. No damaging germline mutations in MMRs were observed in these patients. Nevertheless, somatic mutations in MSH6 and POLE were found in tumour P15T02, which is hypermutated. Somatic POLE mutations affecting the exonuclease domain have been predicted to cause a defect in the correction of mispaired bases inserted during DNA replication leading to hypermutated (15). Group 3 comprised 12 patients, in which all tumours were non-hypermutated. As expected (16), tumours with hyper-mutations had less somatic copy number alterations than did those with non-hyper-mutations (Figure 1).
Alterations between paired tumours reveal heterogeneity
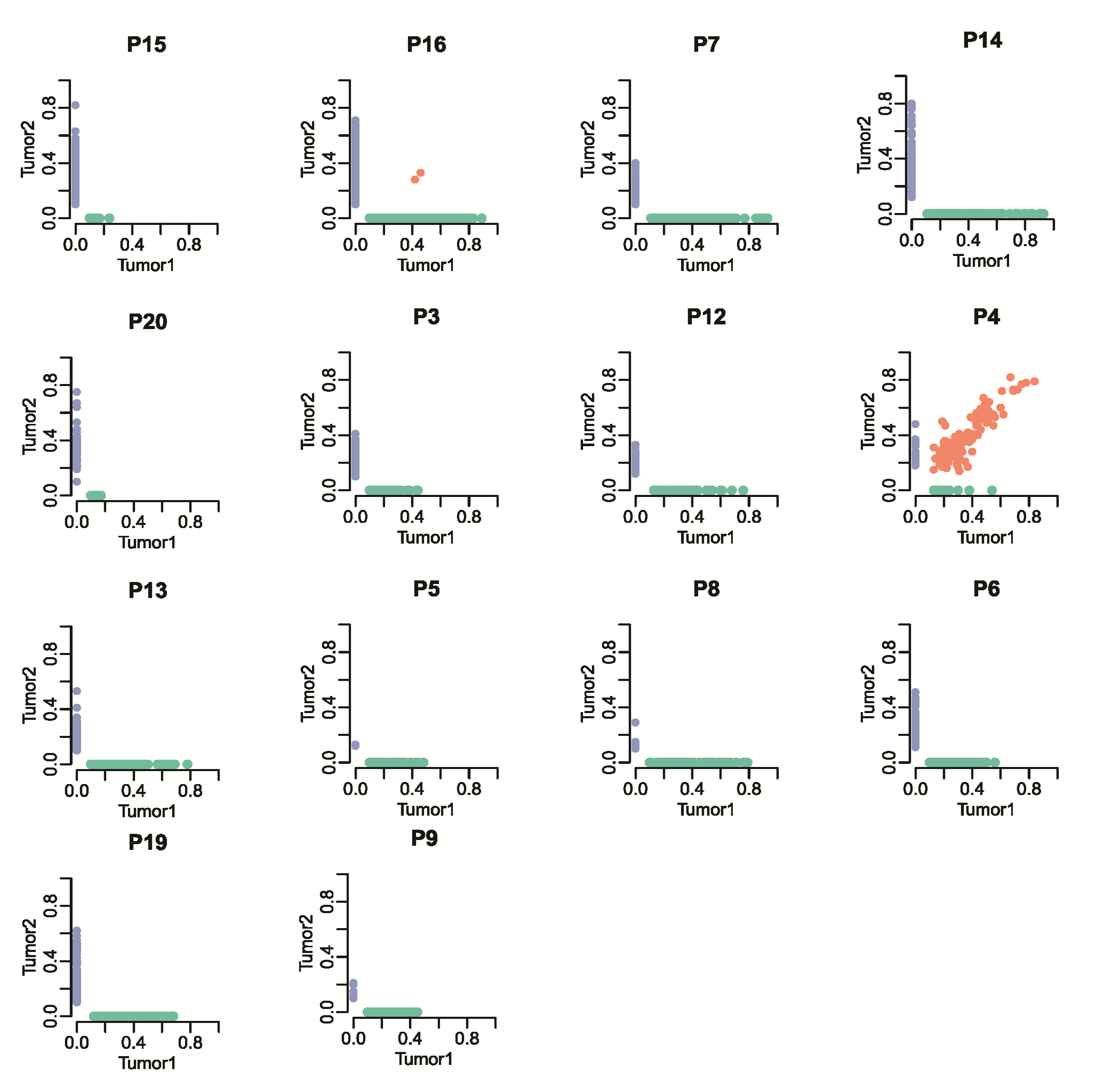
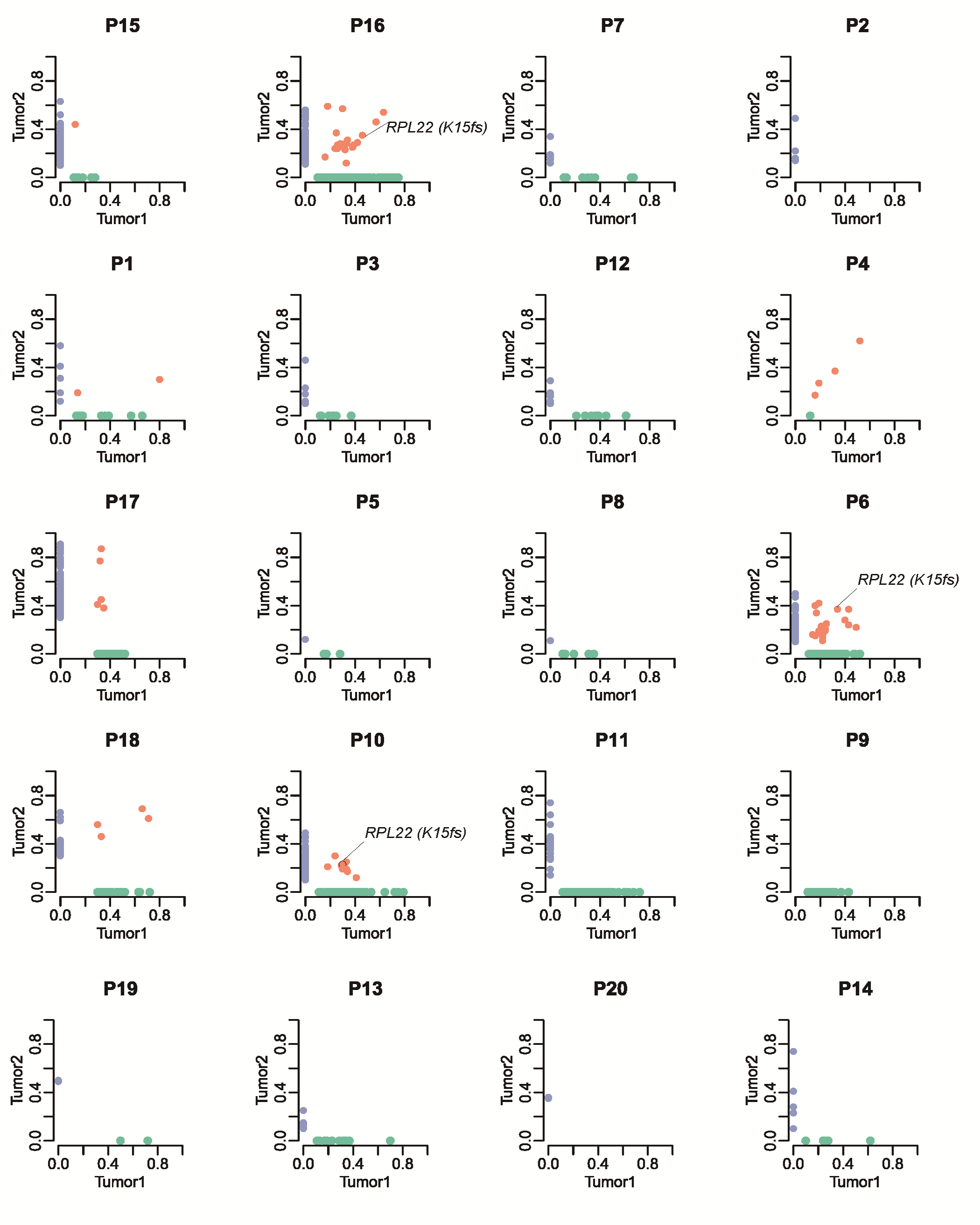
We compared the profile of somatic mutations between paired syCRCs from each individual patient. Comparing the mutations within tumour pairs showed a high level of discrepancy; for most of the patients, there were very few mutations shared in the paired lesions, except for those in patient P4, which presented 89.4 and 86.3% overlap in each tumour, respectively (Figure 2a; Supplementary Table 4, available at Carcinogenesis Online). Apart from patient P4, we identified 16 shared SNVs in 8 patients, and 7 of them (in four patients) were non-synonymous mutations (Figure 2b; Supplementary Figure 3, available at Carcinogenesis Online). Notably, in patients P1 and P18, both synchronous tumours shared the mutation p.G13D in KRAS. This mutation is a well-known pathogenic allele associated with non-small cell lung cancer (17). The pathogenic mutation p.R43H in TP53 was found in both tumours of patient P11; this mutation has been reported previously to be a germline mutation predisposing to human cancer (18). In patient P10, one mutation in NPHP1 and one mutation in DNAH3 were observed, although they were not predicted to affect protein function (p.T3285M: SIFT score = 0.09, p.A97T: SIFT score = 0.09). The effects of the p.E361D mutation in APOBR identified in patient P2 remain unknown. The remaining shared mutation in patient P17, p.R387C in FBXW7, was predicted to be harmful (SIFT score =0); FBXW7 has been shown to have an important role in the pathogenesis of human cancers (19) and is a highly significantly mutated gene in The Cancer Genome Atlas (TCGA) CRC cohort (21%). The details of somatic indels were showed in Supplementary Figure 4, available at Carcinogenesis Online.
Figure 2.

Genomic heterogeneity of syCRCs. (a) Proportion of SNVs and indels in exonic region shared by paired syCRCs or specific to one of them. (b) Variant frequency distribution of identified SNVs between the paired syCRCs from each patient. Each dot represents a variation. The values shown on the axes denote the variant frequency in each tumour. The light green dots are specific to tumour1, the light blue dots are specific to tumour2, and the light red dots represent variations that are present in both of the tumours. (c) Proportion of all mutated genes and well-known cancer genes with non-silent mutations shared by paired syCRCs or specific to one of them. (d) The number of mutations shared or private was distinguished by the indicated branches. APC, KRAS and TP53 mutations were labelled.
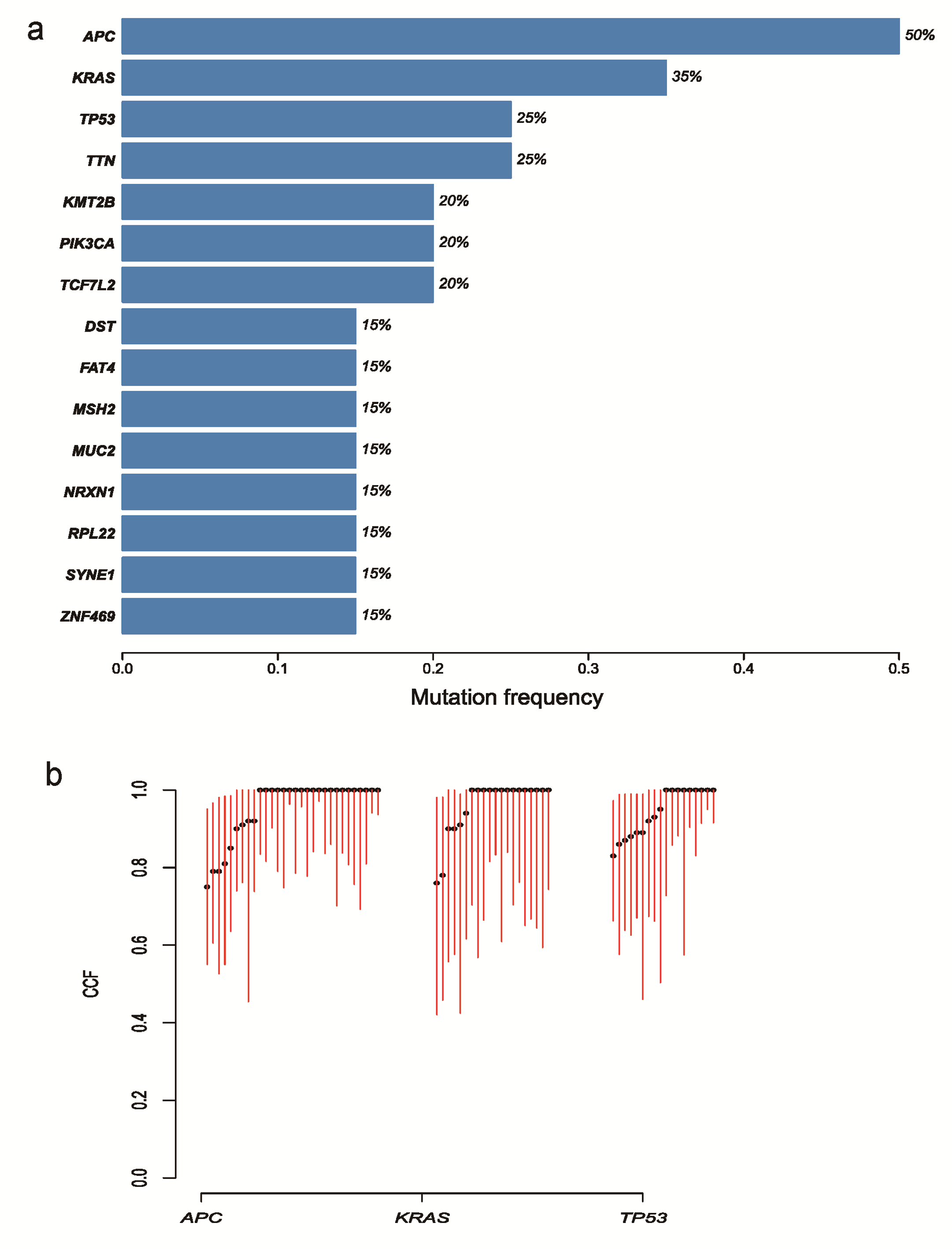
In addition to the specific mutation sites, we explored mutated genes between paired samples (Figure 2c). Most of the mutated genes were specific to only one of each syCRC pair, excluding the hypermutated tumours and patient P4. The mean number of genes that were mutated in both of a pair of syCRCs in non-hypermutated tumours was 2.23. Furthermore, we screened the well-known cancer genes (20) shared by syCRCs (Supplementary Figure 5a, available at Carcinogenesis Online); the top three were APC (50%), KRAS (35%) and TP53 (25%), which all have been shown to have a crucial role in the development of CRC (21). Loss of function of APC was associated with the initial step in tumorigenesis, as it promotes the degradation of β-catenin and limits the transcription of Wnt target genes involved in regulating the cell cycle (22). With the tumour progression, acquired mutations in KRAS and TP53 may be the key events among the genetic abnormalities (22). We evaluated the likely fractions of sampled cancer cells (CCF) that carried each mutation in these three genes; they present various CCF among the tumours (Supplementary Figure 5b, available at Carcinogenesis Online). Most of mutations in these three genes show different sites within syCRCs (Figure 2d). As mentioned above, although P1 and P18 share the p.G13D in KRAS, the paired lesions in the two patients present different APC and TP53 mutations. In patient P10, both syCRCs have distinct mutations in APC and KRAS. Thus, although syCRCs showed great heterogeneity, they might share the mutated genes that are important in tumour initiation and progression.
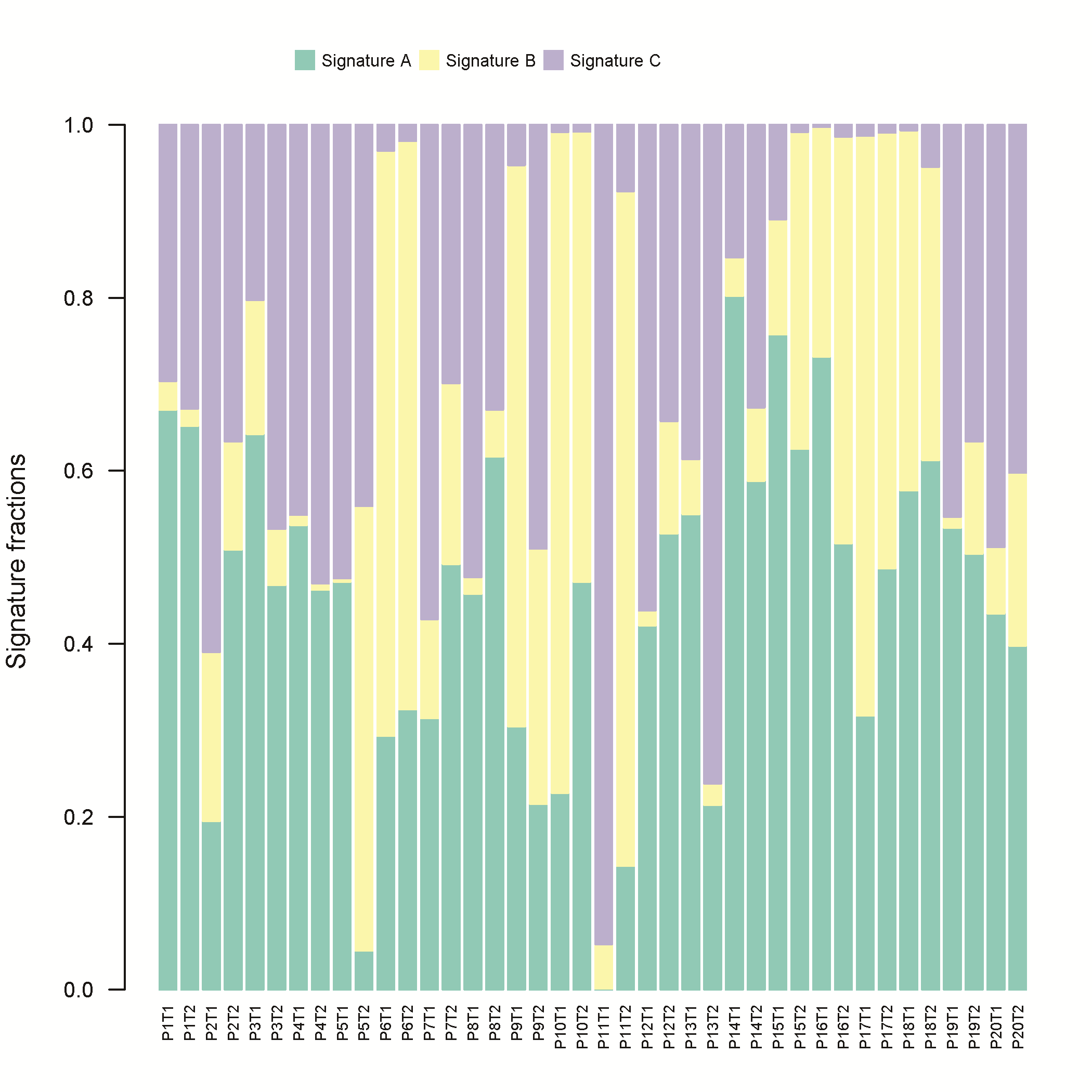
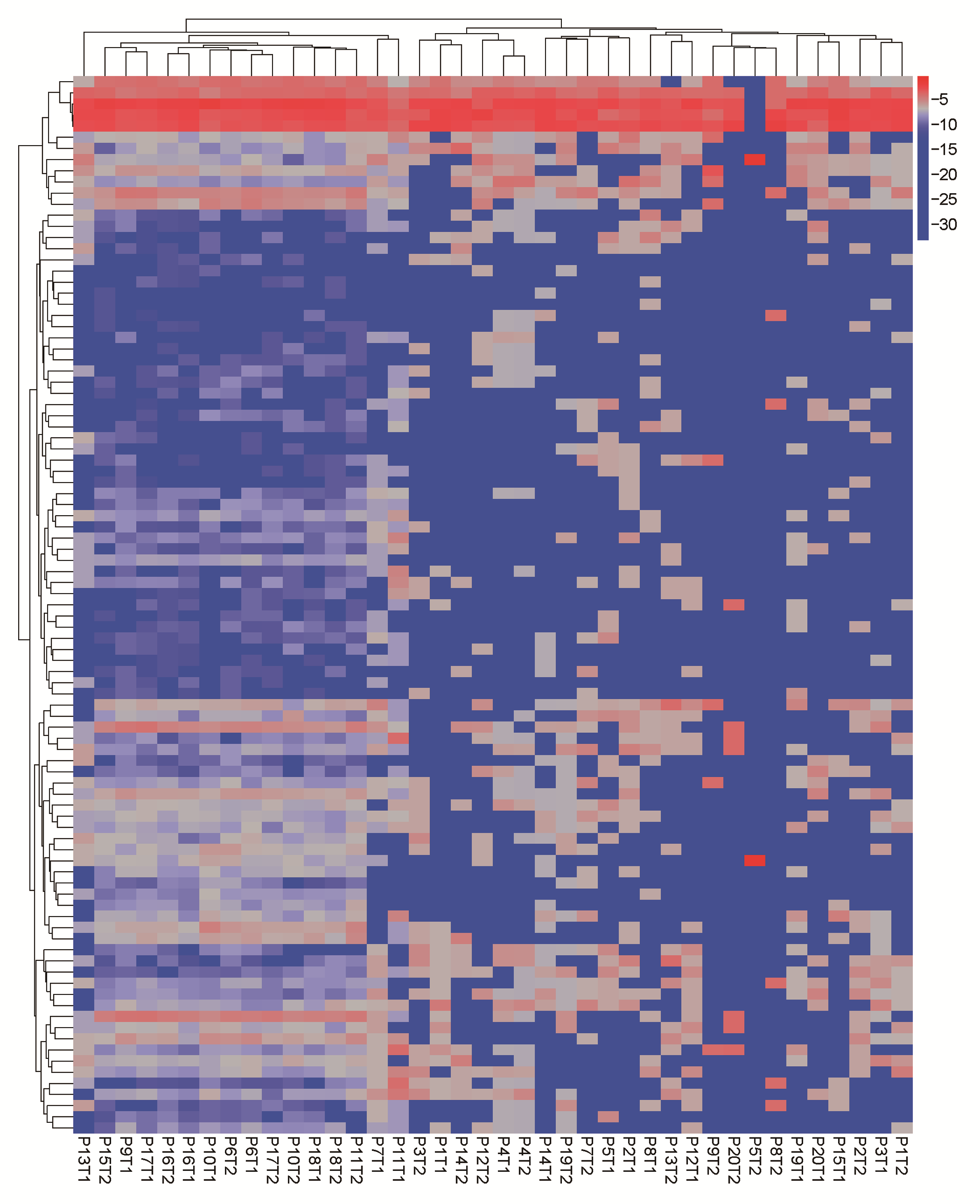
Next, we investigated the syCRC mutational signature for each tumour to assess whether the paired tumours evolved from the same mutational process (Supplementary Figure 6, available at Carcinogenesis Online). The contribution of the three extracted signatures was calculated for each tumour, and two synchronous tumours from one individual showed great discrepancy. The signature B makes great contributions to samples with high hypermutated status, which is associated with defective DNA mismatch repair and consistent with previous report (23). For patient P6 and P10 with MLH1 germline mutation, both lesions of the two patients have higher signature B contribution (Supplementary Table 5, available at Carcinogenesis Online). Furthermore, we conducted hierarchical clustering of all tumours according to their nucleotide context-specific exonic mutation rates and found that most of the paired syCRCs were not clustered together, thus indicating the distinct context-specific spectrum and mutation rate among the syCRCs (Supplementary Figure 7, available at Carcinogenesis Online).
We also considered whether any germline mutations predisposed to syCRCs. To assess this, we screened the well-known genes (Supplementary Table 6, available at Carcinogenesis Online) related to hereditary CRC (24) and found four potential candidate gene mutations in three patients. Among them, MLH1 (p.L73P) and TCF7L2 (p.G208S) were found in patient P6; none of these mutations has been reported in the Exome Aggregation Consortium (ExAC) (25). The other two mutations, GALNT12 (p.P240L) and MLH1 (p.R226P), were found in patients P13 and P10, respectively. GALNT12 (p.P240L) has been reported in ExAC at a frequency of 0.05% and has unknown clinical significance. Patient P6 has been diagnosed as Lynch syndrome, and his father and brother are CRC patients. However, the other patients with any of these potentially damaging mutations showed no family history of cancer.
Frequently mutated genes in patients with syCRCs
To advance our knowledge about genomic events underlying the development of syCRCs, we integrated all of the tumour mutations and determined the most frequent and significantly mutated genes in syCRCs by MusigCV. We found that four genes, KRAS, TP53, APC and RPL22 were significantly mutated in the cohort (Supplementary Table 7, available at Carcinogenesis Online).
Our study identified the KRAS mutation at a frequency of 55% (22/40), among which 10 samples contained the hotspot mutations p.G13D and p.G12V (Figure 3a). These results are consistent with the TCGA data, which report a 37% frequency of KRAS mutation. Moreover, the in-frame deletion p.154_155del was found in one tumour from P10 but not in the paired-synchronous tumour. This mutation has been reported in the lymphoid neoplasm (26) (COSM1360823).
Figure 3.

Profile of frequently mutated genes in syCRCs. (a–d) Profile of frequently mutated genes, including KRAS, TP53, APC and PIK3CA. For each gene, the upper panel indicates the mutations in syCRCs, and the lower panel depicts the mutations in TCGA CRC data.
TP53, a well-known tumour suppressor gene, was mutated in 42.5% of the tumours (51% in TCGA; Figure 3b). The mutations include two in-frame insertions, 1 splicing mutation, 14 non-synonymous substitutions and 1 stop-gain substitution; most of these mutations have been reported (27).
APC was identified as the most frequently mutated gene with non-silent mutations in our cohort, with 67% of the tumours containing protein-altering mutations (Figure 3c). These mutations seem to be randomly distributed among the APC domains. Over half (55.3%) of the mutations were stop-gain substitutions, and the remainder included 10 frameshift deletions, 4 frameshift insertions and 3 missense mutations. Although the hotspot mutation p.R1450* occurred in 6% (38/629) of the TCGA CRC data, we did not find this mutation in our cohort. Only four mutations in APC were mutated in more than one patient, p.K1345*, p.R1432*, p.R216* and p.R223*, all of which were stop-gain substitutions. Except for the p.R216* mutation that was present in seven tumours in the TCGA data, all of the other mutations have not been reported. The p.K1345* mutation has been reported in a previous study (28).
PIK3CA was identified in 32.5% of the cohort (Figure 3d), which plays a key role by recruiting PH domain-containing proteins to the membrane, including AKT1 and PDPK1, activating signalling cascades involved in cell growth, survival, proliferation, motility and morphology. It has been reported as an important driver gene in CRC (16).
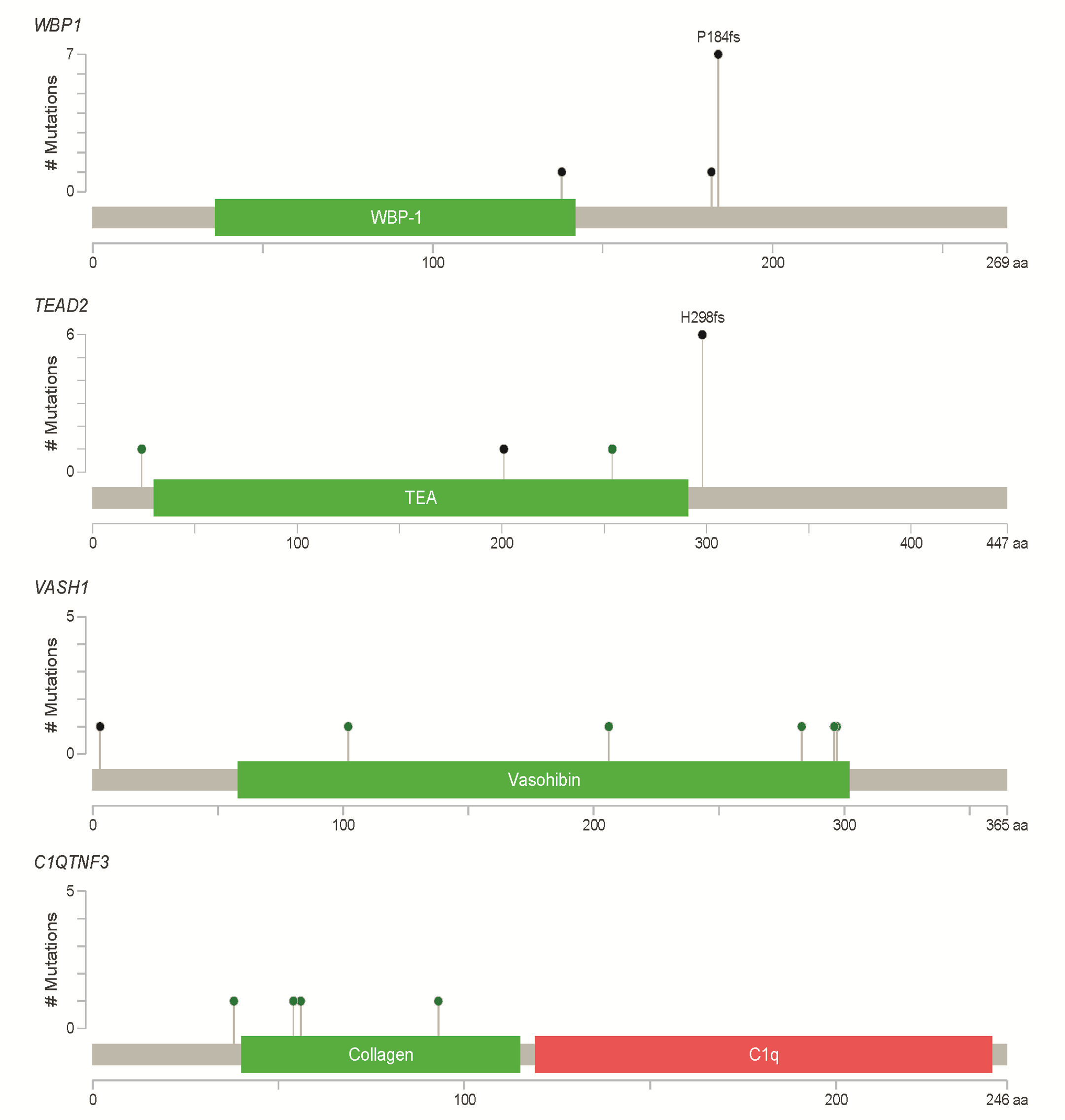
In addition to the well-known previously reported CRC genes, we identified several additional genes that were also frequently mutated in our cohort, including WBP1 (22.5%), TEAD2 (20%), VASH1 (15%) and C1QTNF3 (10%; Supplementary Figure 8, available at Carcinogenesis Online). WBP1, which encodes a ligand of the WW domain of the yes kinase-associated protein, is known to play an important role in mediating protein–protein interactions (29). Nine tumours were determined to contain WBP1 frameshift deletions, and six of them were p.P184fs. The TEAD2 transcription factor regulates the Hippo signalling pathway, which is involved in organ size control and tumour suppression by restricting proliferation and promoting apoptosis (30). This gene was mutated in nine tumours with three distinct mutation types, one splice site mutation, two missense substitutions and six hotspot frameshift deletion p.H298fs. VASH1 has been reported to function as an inhibitor of cell migration, proliferation and network formation by endothelial cells and of angiogenesis (31). VASH1 might have an important role in lung cancer (32); however, its role in CRC remains to be determined. Concerning VASH1, we detected five different missense mutations in the vasohibin (PF14822) domain. Four distinct missense mutations in C1QTNF3 were observed in our data, which has been reported to have be implicated in prostate cancer (33).
Taken together, the majority of the frequently mutated genes found in our cohort have been reported in TCGA cohort, and most of them show similar mutation frequency and pattern. These data also highlight several novel genes that are frequently mutated in syCRC; extensive functional characterization of these mutations is required to further refine their role in syCRC pathogenesis and evolution.
Functional characterization of RPL22 mutation
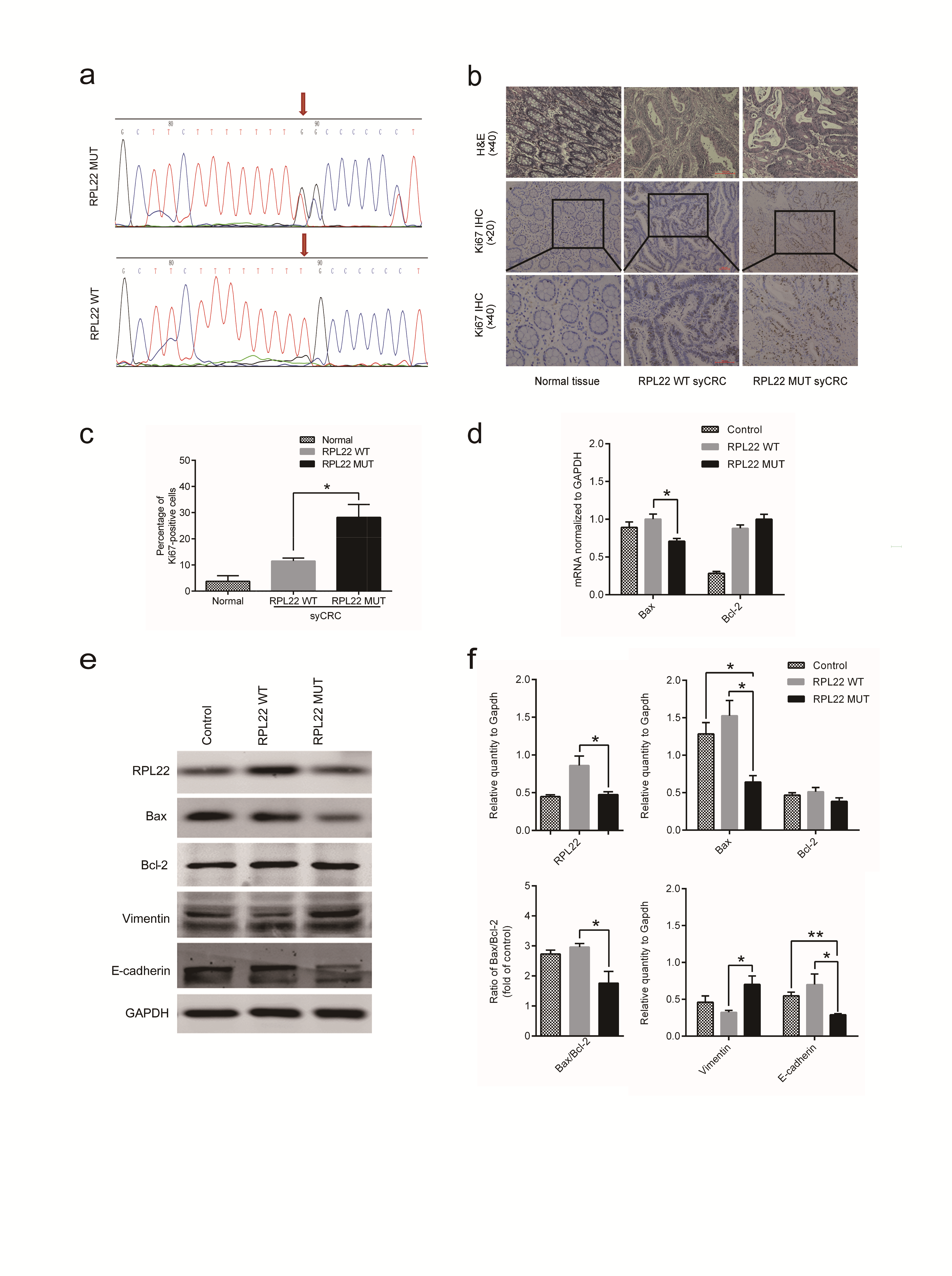
As mentioned above, RPL22 as a frequently mutated gene in our cohort has never been widely characterized in syCRCs. RPL22 encodes for a cytoplasmic ribosomal protein, which is a key component of ribosome subunit 60S that belongs to the L22E family of ribosomal proteins. We observed that RPL22 with the hotspot mutation p.K15fs occurred in 25% (10/40) of the syCRC tumours (Figure 4a), whereas the frequency in TCGA CRC was 4% (25/629). Hypermutated syCRC tumours were more likely to harbour mutations in RPL22 (P < 0.05). However, RPL22 with this mutation was independent of the syCRC tumour size (P = 0.35) and stage (P = 0.75) and shows no significant relationship to the tumour location (P = 0.25). We also confirmed the mutation by Sanger sequencing (Supplementary Figure 9a, available at Carcinogenesis Online), and the residue K15 was highly conserved in closely related species (Figure 4b).
Figure 4.

Characterization of the RPL22 gene in syCRCs. (a) The RPL22 hotspot (p.K15fs) mutation. (b) Conservation of residue K15 in the coding region among near species. (c) Immunofluorescent staining with the RPL22 antibody (red) in normal adjacent tissues and syCRCs with RPL22 WT and RPL22 mutation (MUT; ×40 magnification). DAPI (blue) was used to locate the nuclei of the cells. Scale bars, 100 μm. (d, e) Immunoblotting was used to analyze expression levels of RPL22 protein in adjacent normal tissue and syCRCs with RPL22 WT and RPL22 MUT. GAPDH was used as loading control. (f) Quantitative reverse transcription (qRT)-PCR analysis of RPL22 WT and MUT mRNA expression in syCRCs. (g, h) Representative images of TUNEL positive cells(green) and DAPI (blue) were captured in normal adjacent tissues and syCRCs with RPL22 WT and RPL22 MUT by a fluorescence microscope(×40 magnification). Scale bars, 100 μm. Percentage of TUNEL positive cells in each sample was calculated based in total number of DAPI positive cells for that sample. (i–k) Western blotting analysis of Bax and Bcl-2 protein expression in normal adjacent tissues and syCRCs with RPL22 WT and RPL22 MUT. GAPDH was used as loading control. All experiments were performed at least three independent experiments; Data are mean ± SEM. The P values (determined by the unpaired t-test, two-tailed) relative to normal adjacent tissues or syCRCs with RPL22 WT are shown. *P < 0.05, **P < 0.01, ***P < 0.001. WT, wild-type; MUT, mutation.
Mutation p.K15fs in RPL22 may result in disrupt protein products, which are expected to promote syCRCs oncogenesis. The immunofluorescence staining results suggested that RPL22 protein is mainly localized in the cytoplasm of colonic epithelium cells and expressed lower in syCRCs with mutated (MUT) RPL22 (Figure 4c). In support of this possibility, we evaluated expression level of WT and mutated (MUT) RPL22 in syCRCs. Immunoblotting analysis detected lower RPL22 protein expression in syCRCs with mutated RPL22 than with WT (P < 0.05) or adjacent normal tissue (Figure 4d and e), consistent with mRNA expression in quantitative PCR data (Figure 4f). TUNEL assay exhibited fewer apoptotic cells in syCRC tissues with RPL22 p.K15fs mutation (P < 0.05; Figure 4g and h). We then evaluated the expression of the apoptotic marker Bax and Bcl-2 protein in syCRCs. The ratio of Bax/Bcl-2 protein and the expression of Bax protein were significantly decreased in RPL22 mutated tumours (P < 0.05; Figure 4i, j and k). Moreover, positive Ki67 staining in colonic epithelium of syCRC tumours bearing the RPL22 mutation might be related to more proliferative capacity by immunohistochemistry (P < 0.05; Supplementary Figure 9b and c, available at Carcinogenesis Online).
Furthermore, to explore the functional role of RPL22 mutation in colon cancer cells, lentiviral vectors encoding WT and p.K15fs-mutated RPL22 were transfected into colon cancer cell HT29. A significantly reduced expression of RPL22 protein was observed in HT29 cells transfected with the RPL22 mutation (P < 0.05; Supplementary Figure S9e and f, available at Carcinogenesis Online). We then detected that mRNA expression of Bax was significantly decreased in RPL22 mutated samples (P < 0.05; Supplementary Figure 9d, available at Carcinogenesis Online), and protein level showed the consistent result (P < 0.05; Supplementary Figure 9e and f, available at Carcinogenesis Online). These findings suggested the potential role of RPL22 in regulating apoptosis.
The epithelial–mesenchymal transition (EMT) has been associated with tumour invasion and metastasis (34). Therefore, we evaluated EMT markers (E-cadherin and vimentin) in HT29 cells transfected with the RPL22 p.K15fs mutation versus RPL22 WT by immunoblotting analysis. The result showed that vimentin protein was significantly upregulated (P < 0.05) and E-cadherin protein decreased (P < 0.05; Supplementary Figure S9e and f, available at Carcinogenesis Online). These findings suggest that the RPL22 mutation might be related to EMT process in colon cancer cells HT-29.
Discussion
In this study, we employed whole-exome capture and next-generation sequencing to obtain complete information in the protein coding sequence of synchronous CRCs from 20 patients. To our knowledge, this is the largest sample cohort to date in which syCRC genomic profiling has been performed on fresh tissue. Our findings indicate that the paired syCRCs showed great heterogeneity and might have independent genetic origins, although they share a comparable genetic background and exposure history.
We found that there are very few mutations shared in the paired lesions, and they also present different copy number profile. Three main signatures were extracted from the cohort, and the contribution of each signature for the synchronous tumours from one individual also varied. These results indicated that the two tumours originated independently and experienced different mutational process. APC, KRAS and TP53 ranked the top three of the shared mutated cancer genes in synchronous tumours, and they are also frequently mutated in solitary CRCs and demonstrated to drive tumorigenesis by modulating driver pathways that are involved in proliferation, differentiation and apoptosis (22). We comprehensively characterized mutations in these genes between synchronous tumours, and most of the mutations were clonal mutations that confer the selective growth advantages although they might be at different loci, indicating that the timing at which they accumulated were different.
The hotspot mutation (K15fs) in RPL22 was observed in 25% of our cohort, whereas the frequency in TCGA CRC was 4%. Most CRCs in TCGA are solitary tumours, which suggested that RPL22 mutations might be more aggregated in syCRCs. RPL22 has been identified as a driver gene in adrenocortical carcinoma (35) and gastric cancer (36). Our preliminary data indicated that the inactivation of RPL22 may suppress tumour apoptosis and promote cell proliferation. A recent study has shown that ribosomal proteins play a crucial role in the ribosomal protein-MDM2-p53 signaling pathway, thus providing a molecular switch to the response to nucleolar stress. Ribosomal proteins such as RPL22 could bind to MDM2 and block MDM2-mediated p53 ubiquitination and degradation, resulting in p53-dependent cell-cycle arrest to inhibit cell division (37). Another study reported that RPL22 could control morphogenesis by modulating the splicing of SMAD2 pre-mRNA, which is an essential transcriptional effector of Nodal/TGF-β signalling (38). The molecular mechanism and role in the related pathways of RPL22 in syCRCs and solitary CRC require further investigation. Nevertheless, the high incidence of the mutation in syCRCs suggests a novel potential biomarker for therapy and prognosis.
At present, no common therapy strategies have been established for syCRCs, and their clinical management is mostly similar to that of solitary CRC. Because of the heterogeneity of syCRCs, the potentially actionable targets may be diverse between the paired tumours; therefore, the therapy decision, especially the usage of targeted reagents, should be made carefully, and every lesion in a patient should be treated independently. We analyzed potential drug targets in these syCRCs and found that most aberrations occurred in only one of paired tumours (Figure 5). BRAF (p.V600E) is the most prevalent mutation in many cancer types and has been determined to be an activating mutation. Patients with this mutation have been reported to be sensitive to RAF inhibitors such as vemurafenib and dabrafenib; as an alternative, the MEK inhibitor trametinib has been appraised successfully in BRAF mutant melanomas (39). One of paired tumours from patient P3 and P13 harboured BRAF (p.V600E) mutations, whereas the other of the same patient did not. Because of the heterogeneity of the mutation in paired syCRCs from one patient, more targets may be necessary for the patient when making therapeutic decision.
Figure 5.

Alterations in actionable genes in syCRCs. Each row represents a tumour, and two lesions from one patent were listed together. The mutation type of each actionable gene is marked. Genes were grouped according to pathways. The number of predicted neoantigens for each tumour was shown on the right.
The amplification of EGFR has been proven to respond to tyrosine kinase inhibitors, most notably gefitinib and erlotinib (40), and it is a clinically relevant target in CRC (41). We identified focal amplification of EGFR in P10T2 and P19T1; however, we also observed the KRAS mutations which may predict resistance to anti-EGFR therapy in these tumours. For these patients, not only variant group in one tumour but also the combination of different targets from both tumours in the same patient should be considered.
Neoantigens have been proved as promising markers for cancer immunotherapy, and tumours with high mutation burden may be more susceptible to immune checkpoint blockade (42). Paired tumours in P9, P11 and P15 showed great discrepancy in mutation burden, and the number of predicted neoantigens is also various (Figure 5). When making immunotherapeutic strategies, this kind of heterogeneity in mutation burden should be taken into account.
Overall, the present work provides several lines of evidence that syCRCs may originate independently and present high heterogeneity. Because of the heterogeneity within paired syCRCs, clinical approaches towards patients with syCRC should be standardized and redefined for precision medical. The genes that we identified as being frequently mutated may lead to promising clinical approaches, such as new drugs development and biomarker identification. This study also provides novel insights into the genetic mechanisms of syCRC initiation; in addition to the genetic factors, it would be of interest to investigate the role of the gastrointestinal microbiome in determining its potential role in the initiation of syCRCs in future study.
Supplementary material
Supplementary material are available at Carcinogenesis online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Conflicts of Interest Statement:
The authors declared that there is no conflict of interest.
Funding
This work was supported by grants from the National High Technology Research and Development Program of China (863 Program)(No. 2012AA02A204), National Natural Science Foundation of China (81270461 and 81570483), Youth Science Development of Third Military Medical University(2016XPY19), National Natural Science Foundation of China (81672818), Guangzhou science and technology program key projects 201604020005. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Abbreviations
- CRC
colorectal cancer
- EMT
epithelial–mesenchymal transition
- MMR
mismatch repair gene
- SNVs
single-nucleotide variants
- syCRCs
synchronous colorectal cancers
- WT
wild-type
References
- 1. Torre L.A., et al. (2015)Global cancer statistics, 2012. CA. Cancer J. Clin., 65, 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Lam A.K., et al. (2014)Synchronous colorectal cancer: clinical, pathological and molecular implications. World J. Gastroenterol., 20, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nosho K., et al. (2009)A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology, 137, 1609–20.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenstein A.J., et al. (1986)A comparison of multiple synchronous colorectal cancer in ulcerative colitis, familial polyposis coli, and de novo cancer. Ann. Surg., 203, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kato T., et al. (2016)Clinical characteristics of synchronous colorectal cancers in Japan. World J. Surg. Oncol., 14, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ogino S., et al. (2006)Epigenetic profiling of synchronous colorectal neoplasias by quantitative DNA methylation analysis. Mod. Pathol., 19, 1083–1090. [DOI] [PubMed] [Google Scholar]
- 7. Weren R.D., et al. (2015)A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet., 47, 668–671. [DOI] [PubMed] [Google Scholar]
- 8. Li H., et al. (2010)Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKenna A., et al. (2010)The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang K., et al. (2010)ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexandrov L.B., et al. (2013)Deciphering signatures of mutational processes operative in human cancer. Cell Rep., 3, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carter S.L., et al. (2012)Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol., 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nariai N., et al. (2015)HLA-VBSeq: accurate HLA typing at full resolution from whole-genome sequencing data. BMC Genomics, 16(Suppl 2), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu G., et al. (2017)PSSMHCpan: a novel PSSM based software for predicting class I peptide-HLA binding affinity. Gigascience, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palles C., et al. ; CORGI Consortium; WGS500 Consortium (2013)Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet., 45, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Network. (2012)Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marchetti A., et al. (2009)Clinical implications of KRAS mutations in lung cancer patients treated with tyrosine kinase inhibitors: an important role for mutations in minor clones. Neoplasia, 11, 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monti P., et al. (2007)Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin. Cancer Res., 13, 3789–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yeh C.H., et al. (2016)Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc. Natl. Acad. Sci. USA, 113, 6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. An O., et al. (2016)NCG 5.0: updates of a manually curated repository of cancer genes and associated properties from cancer mutational screenings. Nucleic Acids Res., 44, D992–D999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sjöblom T., et al. (2006)The consensus coding sequences of human breast and colorectal cancers. Science, 314, 268–274. [DOI] [PubMed] [Google Scholar]
- 22. Walther A., et al. (2009)Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer, 9, 489–499. [DOI] [PubMed] [Google Scholar]
- 23. Alexandrov L.B., et al. ; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain (2013)Signatures of mutational processes in human cancer. Nature, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stoffel E.M., et al. (2015)Genetics and genetic testing in hereditary colorectal cancer. Gastroenterology, 149, 1191–1203.e2. [DOI] [PubMed] [Google Scholar]
- 25. Lek M., et al. ; Exome Aggregation Consortium (2016)Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fabbri G., et al. (2013)Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J. Exp. Med., 210, 2273–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petitjean A., et al. (2007)TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene, 26, 2157–2165. [DOI] [PubMed] [Google Scholar]
- 28. Yu J., et al. (2015)Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut, 64, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDonald C.B., et al. (2011)Biophysical analysis of binding of WW domains of the YAP2 transcriptional regulator to PPXY motifs within WBP1 and WBP2 adaptors. Biochemistry, 50, 9616–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Imajo M., et al. (2015)Dual role of YAP and TAZ in renewal of the intestinal epithelium. Nat. Cell Biol., 17, 7–19. [DOI] [PubMed] [Google Scholar]
- 31. Lu C., et al. (2010)Regulation of tumor angiogenesis by EZH2. Cancer Cell, 18, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imielinski M., et al. (2012)Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell, 150, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grasso C.S., et al. (2012)The mutational landscape of lethal castration-resistant prostate cancer. Nature, 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong I.Y., et al. (2014)Collective and individual migration following the epithelial-mesenchymal transition. Nat. Mater., 13, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng S., et al. ; Cancer Genome Atlas Research Network (2016)Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell, 29, 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagarajan N., et al. (2012)Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol., 13, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., et al. (2009)Signaling to p53: ribosomal proteins find their way. Cancer Cell, 16, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y., et al. (2017)Ribosomal proteins Rpl22 and Rpl22l1 control morphogenesis by regulating pre-mRNA splicing. Cell Rep., 18, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pakneshan S., et al. (2013)Clinicopathological relevance of BRAF mutations in human cancer. Pathology, 45, 346–356. [DOI] [PubMed] [Google Scholar]
- 40. Arteaga C.L., et al. (2014)ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell, 25, 282–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cunningham D., et al. (2004)Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med., 351, 337–345. [DOI] [PubMed] [Google Scholar]
- 42. Le D.T., et al. (2015)PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med., 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.