

Abstract
Eukaryotic genomes are replete with repeated sequences in the form of transposable elements (TEs) dispersed across the genome or as satellite arrays, large stretches of tandemly repeated sequences. Many satellites clearly originated as TEs, but it is unclear how mobile genetic parasites can transform into megabase-sized tandem arrays. Comprehensive population genomic sampling is needed to determine the frequency and generative mechanisms of tandem TEs, at all stages from their initial formation to their subsequent expansion and maintenance as satellites. The best available population resources, short-read DNA sequences, are often considered to be of limited utility for analyzing repetitive DNA due to the challenge of mapping individual repeats to unique genomic locations. Here we develop a new pipeline called ConTExt that demonstrates that paired-end Illumina data can be successfully leveraged to identify a wide range of structural variation within repetitive sequence, including tandem elements. By analyzing 85 genomes from five populations of Drosophila melanogaster, we discover that TEs commonly form tandem dimers. Our results further suggest that insertion site preference is the major mechanism by which dimers arise and that, consequently, dimers form rapidly during periods of active transposition. This abundance of TE dimers has the potential to provide source material for future expansion into satellite arrays, and we discover one such copy number expansion of the DNA transposon hobo to approximately 16 tandem copies in a single line. The very process that defines TEs—transposition—thus regularly generates sequences from which new satellites can arise.
Eukaryotic genomes are inundated with two types of repetitive sequences: transposable elements (TEs), which are dispersed by a variety of transposition mechanisms, and satellite sequences, which are tandemly repeated sequences that expand, contract, and are homogenized by recombination events. Both types of repeats are enriched in the heterochromatin surrounding the telomeres and centromeres, likely because the low frequency of recombination in heterochromatin permits their persistence (Charlesworth et al. 1986).
The essential roles played by telomeres and centromeres in genome integrity and chromosome segregation suggest that some repetitive sequences are of functional significance (Blackburn et al. 2006; Mason et al. 2008; Malik and Henikoff 2009). Examples supporting functional roles for repetitive sequences mostly follow from observations of phenotypes associated with repeat variation. Contractions of the human subtelomeric satellite D4Z4 cause facioscapulohumeral muscular dystrophy by altering the chromatin state of nearby genes (Zeng et al. 2009). Sequence variation in a human centromeric satellite is associated with aneuploidy (Aldrup-MacDonald et al. 2016). Variants of the mostly repetitive Drosophila melanogaster Y Chromosome have global impacts on gene expression, possibly by titrating chromatin binding factors (Francisco and Lemos 2014). Satellites can also engage in meiotic drive and gamete competition (Hardy et al. 1984; Fishman and Saunders 2008; Larracuente 2014), selfish processes whereby alleles bias meiotic segregation or gamete survival to gain a transmission advantage. Finally, the structural importance of constitutive heterochromatin means that changes in repeat composition between species can cause reproductive barriers (Ferree and Barbash 2009).
Despite the potential consequences of satellite variation, many satellite sequences turnover rapidly between closely related species (Lohe and Roberts 2000). Partially explaining this is the potential of satellite sequence to recombine out of register via unequal exchange. In the absence of selection acting on copy number, evolution by unequal exchange leads to (1) dramatic changes in copy number from relatively few exchange events and (2) the eventual contraction of the array to a single repeat unit (Charlesworth et al. 1986). The long-term persistence of some conserved satellites (Strachan et al. 1982) may therefore reflect functional importance. Given their ubiquity, however, unless all satellites are functional, mechanisms to generate new satellites must exist to counter the inevitable loss of neutrally evolving ones (Charlesworth et al. 1986).
Models of satellite evolution suggest two stages in the emergence of new satellites: (1) Amplification processes generate small tandem sequences, and (2) some of these sequences expand to large arrays by unequal exchange (Stephan and Cho 1994). Thus, any process that generates sequence upon which unequal exchange can act is a potential source of new satellites. Simple satellites (those with monomer units of approximately <10 bp), for example, can readily arise by polymerase slippage and subsequent copy number expansion. These simple satellites can transition to more complex satellite types by the interplay of unequal exchange and mutations (Prosser et al. 1986; Stephan and Cho 1994).
More enigmatic mechanisms to generate new satellites also exist. TEs are found as tandem arrays in many species, including as centromeric satellites (Meštrović et al. 2015). The easiest to understand are satellites derived from TEs with intrinsic repeats, such as long terminal repeats (LTRs) and tandemly repeated regulatory elements, which provide substrates for expansion by unequal exchange (Fig. 1A; Ke and Voytas 1997; Macas et al. 2009; Gong et al. 2012; Dias et al. 2014; Zhang et al. 2014).
Figure 1.
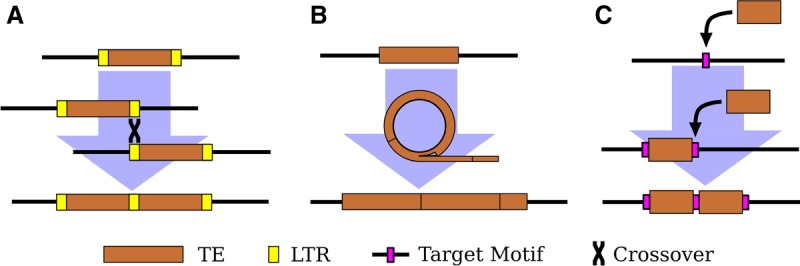
Three mechanisms of tandem TE formation. (A) Ectopic recombination between long-terminal repeats (LTRs; shown in yellow) generates tandem LTR retrotransposons with shared LTRs. (B) Circularization and rolling circle replication of a TE, followed by insertion of the resulting concatemer. The possible mechanism(s) of circularization remains unclear. (C) Two insertions of a TE at the same target site (shown in magenta). Note the preservation of the target site within the tandem junction.
Yet, TEs without intrinsic repeats also form tandem arrays of complete elements (Miller et al. 1992; Caizzi et al. 1993). One proposed mechanism is rolling circle replication (RCR) wherein an element is circularized and then replicated to form a concatemer that is subsequently reinserted into the genome (Fig. 1B; Marsano et al. 2003; Meštrović et al. 2015). Alternatively, double insertion of the same element into a single site is possible for TEs that create target site duplications (TSDs) upon insertion. One example is a tandem array of the non-LTR retrotransposon R1 on the X Chromosome in D. melanogaster (Kidd and Glover 1980; Peacock et al. 1981). R1 has the unusual property of only inserting at a specific site in the multicopy ribosomal RNA genes (rDNA). The tandem elements are separated by identical 33-nt duplications of rDNA sequence, consistent with the tandem originating when two R1 elements inserted in the same rDNA unit and then subsequently expanded by unequal exchange (Fig. 1C; Roiha et al. 1981). Tandem dimers of DNA transposons have been found in bacterial genomes, and these also contain target-site duplications between the tandem elements (Dalrymple 1987; Prudhomme et al. 2002), hinting that double insertions may not be limited to elements with insertion site preferences as extreme as in R1.
Whatever the generative process is, that two TEs transitioned to satellite sequence in D. melanogaster (Kidd and Glover 1980; Caizzi et al. 1993) suggests that a survey of population variation might reveal its early stages. A few tandem TEs, mostly LTR retrotransposons or complex nested insertions, were identified in analyses of the D. melanogaster genome assembly (Kaminker et al. 2002; Bergman et al. 2006). However, a full assessment of the mechanisms and frequency with which TEs generate tandem arrays remains unexplored.
Largely this is due to the wider challenge of applying the most comprehensive population genomic resource available—short-read Illumina data—to investigating the evolutionary dynamics of repetitive DNA. But the existence of TE-derived satellites provides a potential opportunity: Rather than searching for the emergence of satellites from all possible single-copy sequences, tandems arising from repeats that are normally dispersed rather than tandemly arranged might yield a tractable model for studying the early stages of satellite evolution.
Results
ConTExt identifies repeat structures from paired-end Illumina data
Paired-end reads are powerful for detecting the junctions arising from structural rearrangements in unique sequences such as deletions, inversions, translocations, and tandem duplications (Bashir et al. 2008; Rogers et al. 2014). We define a junction, following Bashir et al. (2008), as a pair of adjacent positions in a sequenced genome that are nonadjacent in a reference sequence. Conceptually the problem of identifying junctions in repetitive DNA is identical but is complicated by the fact that repeat-derived reads can rarely be mapped to a unique locus in the reference genome. However, such reads can generally be uniquely mapped to a specific repeat family, and this property has been leveraged to identify TEs inserted into unique sequence (Hormozdiari et al. 2010; Kofler et al. 2012). We extend this idea to identify all types of junctions involving repetitive sequence, including insertions into unique and repetitive sequence, deletions and inversions internal to a repeat, and tandem duplications.
Aligning to the set of all individual repeats present in a reference genome provides the power to detect reads originating from highly divergent variants but results in reads being distributed across many different sequences. On the other hand, aligning reads to repeat consensus sequences is less powerful in detecting divergent copies but organizes all reads from a repeat family in the same place, greatly simplifying visualization and downstream analyses. We therefore combine these two approaches into a single pipeline (Fig. 2A). In trial runs, we recovered ∼20% more repeat-derived reads using this two-step procedure than when we aligned only to consensus sequences. We also aligned reads to the repeat-masked reference genome, allowing the detection of junctions between repeats and unique sequence.
Figure 2.

An outline of the ConTExt pipeline and examples of identified structures. Thin and thick bars of repeats represent noncoding and coding sequences, respectively. (A) Reads are derived from genomic DNA, with many copies of a particular repeat family (black) dispersed among single-copy sequence (orange); some repeat copies have polymorphisms relative to the consensus (yellow bars), especially those in heterochromatin (purple bar). The reads are aligned to individual repeats identified in the reference genome, including divergent elements; three examples are shown. Alignments to these individual elements are then collapsed onto a consensus sequence for that repeat family. Inverted arrowheads indicate short terminal inverted repeats (TIRs) that are common to many DNA transposons. (B) Schematics of paired-end reads spanning sequence concordant with the consensus (i), the junction of an internal deletion (ii), and the junction of a head-to-tail tandem (iii). (C) A two-dimensional scatterplot of paired-end alignments from strain I03 to the hobo element. Each dot represents a single read pair. Its position on the x- and y-axes corresponds to the 3′ ends of the reads aligning to the minus and plus strands of the hobo consensus, respectively. For example, the red arrow indicates a read pair where the 5′ end of the forward read aligns to the beginning of the consensus (as in panel B,i). Both reads are 70 bp and the gap is 330 bp, so the corresponding dot is located at position 70 on the y-axis (the location of the 3′ end of the forward read) and at position 400 on the x-axis (70 + 330). The Roman numerals indicate how the three types of structures shown in B correspond to patterns in the scatter plot and where the reads map on each of the axes. (i) Concordant reads (black dots) that form the main diagonal. (ii) Reads spanning internal deletions. (iii) Reads spanning head-to-tail tandem junctions. The nonblack colors correspond to nonconcordant clusters identified by the EM algorithm, and gray squares are potential artifacts. The plus symbols are the estimated junction for the cluster with the corresponding color. Note that some colors are used twice to indicate distinct widely separated clusters. Read pairs where both ends map to the same strand (e.g., head-to-head tandems) require a different scatterplot to detect. (D) A scatter plot of all junctions involving hobo across all GDL strains. Each dot represents a junction estimated from a cluster in a specific strain (the plus symbols in C). The red arrowhead indicates the location of the deletion identified previously in the Th hobo variant (Periquet et al. 1994). At some rate, concordant read pairs are misclassified as discordant and may generate spurious junctions along the main diagonal; we excluded these from the analysis (see Methods, “Categorizing Tandem Junctions”) and colored these junctions in gray. (E) A scatter plot depicting all junctions across all GDL strains between the minus-strand of the R2 retrotransposon and the plus-strand of rDNA. The thick black bar on the rDNA schematic represents the transcribed rRNAs. The first ∼1500 bp of the rDNA cistron is not shown because only a few low-frequency R2 junctions are present there. The plot successfully identifies that most R2 insertions occur at the same position in the 28S rDNA subunit, as previously demonstrated (Kojima and Fujiwara 2005; Stage and Eickbush 2009).
Once reads are organized relative to consensus sequences, we consider the alignment patterns to identify junctions (Fig. 2B) and use mixture modeling to cluster the reads and resolve the many junctions that map to each consensus (Fig. 2C; Supplemental Fig. S1D). This clustering strategy had >97% recall for junctions supported by at least three reads (Supplemental Fig. S3C) and the ability to resolve nearby junctions was consistent across the Global Diversity Lines (GDL), speaking to the uniformity of the sequencing library preparations (Supplemental Fig. S3A,B; Supplemental Table S1). Once we identify these clusters, we estimate the underlying junctions (Fig. 2C) and visualize their distribution across all samples in the data set (Fig. 2D). We cannot accurately infer tandem structures that contain intervening sequence larger than the insert size of the sequencing libraries (on average 338 bp), as we will detect only the junctions between the elements and the intervening sequence, not the elements themselves; this limitation applies mainly to tandem LTR retrotransposons that have large LTRs.
Our pipeline detects various structures involving repeats, including tandem junctions (Fig. 2B,C), insertions into unique and repetitive sequence (Fig. 2E), and internal deletions (Fig. 2B,C). While we focus here on tandems, we note that we successfully identified known internally deleted elements, such as the Th hobo variants (Fig. 2D; Periquet et al. 1994) and the KP nonautonomous P-element (Black et al. 1987). We also identified known nested repeats such as R-element insertions into the ribosomal RNA genes, including both full-length and distinct 5′-truncated insertions (Fig. 2E).
TEs of all three major types frequently form tandems
Most TEs can be detected in tandem in at least one strain, but the three major types of TEs show distinct patterns of tandem junctions (Fig. 3; Supplemental Fig. S5). We divide tandems into three types, with head-to-tail tandems being likely to involve full-length elements and/or have intact termini, while tail-to-internal and internal-to-internal tandems are likely to involve 5′-truncated or internally deleted elements. Some tail-to-internal tandems may also reflect nested insertions.
Figure 3.

The proportion of GDL strains in which a tandem junction was identified for LTR retrotransposon families (A) and non-LTR retrotransposon families and DNA transposon families (B). Head-to-tail tandems have junctions involving the first and last 200 nt of the consensus sequence. Tail-to-internal junctions have junctions between the last 200 nt of the consensus sequence and internal sequence; these are consistent with tandems involving 5′-truncated elements, though they can also be formed by nested insertions. We do not depict the frequency of internal-to-internal tandems because they are present in most strains, but generally at low copy number; Supplemental Figure S5 provides a more informative visualization of internal-to-internal tandem variation. A does not include LTR–LTR junctions shown in Supplemental Table S2. The scatter plot inset in A depicts the relationship between LTR length and the frequency of detecting head-to-tail tandems for each LTR retrotransposon family.
LTR retrotransposons
LTR retrotransposons have a high propensity to form tandems because they are flanked by direct repeats that are prone to recombination events, yielding structures where adjacent TEs share an LTR (Fig. 1A; Ke and Voytas 1997). We detect the majority of LTR retrotransposons in tandem (Fig. 3A), though many involve internal sequence and are present at low-copy number (Supplemental Fig. S5A). These internal-to-internal tandems are consistent with deletions that span the junctions of head-to-tail tandems. We less frequently observe head-to-tail tandems, likely because we have limited power to detect tandems when the LTR is longer than the average gap size (∼330 nt) (Fig. 3A). Notably, however, all LTR retrotransposons with LTRs shorter than this detection limit are detected as head-to-tail tandems in at least one strain (Fig. 3A, inset). This suggests that the absence of head-to-tail tandems of elements with longer LTRs is due to the detection limit rather than their true absence and that it is reasonable to extrapolate the frequency of tandems observed for elements with short LTRs to all LTR elements. Given this and the abundance of internal-to-internal tandems, we conclude that most LTR retrotransposons frequently form tandems, generally by recombination between LTRs. At a lower frequency, we do detect some tandem junctions between LTRs themselves (Supplemental Table S2), suggesting that a fraction of LTR element tandems arise by a mechanism other than unequal exchange.
Non-LTR retrotransposons
Unlike LTR retrotransposons, non-LTR retrotransposons and DNA transposons do not provide their own substrates for unequal exchange, yet most can be detected as tandems, demonstrating that additional mechanisms allow tandem formation (Fig. 3B; Supplemental Fig. S5B,C). These include the Drosophila telomeric TEs, which form head-to-tail tandems whenever two elements of the same family insert consecutively at the same telomere (George et al. 2006). Non-LTR retrotransposons are prone to 5′ truncation due to incomplete reverse transcription during transposition, and consistent with these tandems arising through consecutive insertion events at the same telomere, many telomeric TE tandem junctions are tail-to-internal (Fig. 3B). Most other non-LTR retrotransposons also can be detected as tail-to-internal tandems in at least one strain, suggesting that transposition is a widespread process generating tandems among non-LTR retrotransposons (Fig. 4A,B,E).
Figure 4.

Junction distributions from all strains in the GDL for two non-LTR retrotransposons (A,B) and two DNA transposons (C,D). Note that C and D only show head-to-tail tandem distributions, and thus, the axes only include the terminal regions. Each dot represents a junction identified from a single strain. A junction present in multiple strains will generate a diagonal distribution around the true coordinate due to estimation errors. In A and B, head-to-tail and tail-to-internal tandem junctions are highlighted in red, internal-to-internal tandems and deletions are colored in blue, and probable artifacts are colored in gray (see Methods, “Categorizing Tandem Junctions”); all junctions in C and D are head-to-tail. The distribution of tandem junctions of jockey (A) are dispersed, with few distinct diagonal clusters, indicating that most individual tandem junctions are low-frequency. In contrast, the four distinct diagonal clusters of DMRT1B (B) indicate junctions at moderate to high population frequency, suggesting that they represent older tandems. While not the focus of our analysis, internal deletions ranging from low to high frequency are also evident in both A and B as junctions below the main diagonal, with several distinct deletion variants of jockey sharing similar sequence coordinates and with many distinct deletions identifiable in DMRT1B. (C) For the P-element, most junctions fall within a single tight diagonal cluster, consistent with their representing tandem P-elements separated by an 8-bp target site duplication. Several junctions are dispersed above this cluster, consistent with additional sequence of variable length within the junction. (D) In contrast, only a few hobo junctions form a tight diagonal cluster, while most are dispersed below the cluster, consistent with small internal deletions spanning most of the tandem junctions. (E) Schematics of the head-to-tail and tail-to-internal DMRT1B tandems denoted with i–iii in B.
The population frequency of tandem junctions across all strains provides insight into the dynamics of tandem TE formation (Fig. 3). For jockey, we find junctions between the 3′ end (i.e., tail) and many different internal locations (Figs. 3B, 4A), and these junctions are at low frequency (between 1/85 and 7/85 strains) (Fig. 4A). These results strongly indicate many recent and independent tandem forming events. In contrast, DMRT1B shows tail-to-internal junctions involving four distinct internal truncations (Fig. 4B), found at intermediate frequencies (between 7/85 to 40/85 strains), indicating that four independent events that occurred further in the past than the jockey tandems and that DMRT1B tandem formation subsequently ceased. Taken together, these results suggest that non-LTR tandems are regularly generated within D. melanogaster, but by different elements at different periods of time.
DNA transposons
DNA transposons are primarily detected as head-to-tail tandems (Fig. 3B; Supplemental Fig. S5C), but the pattern of junction estimates suggests that small deletions frequently span the tandem junctions. The junction estimates for P-element dimers form a tight diagonal distribution, suggesting recently formed dimers with intact termini (Fig. 4C). For hobo, we observe a more diffuse distribution, consistent with small deletions near or spanning the tandem junction that are specific to each genome (Fig. 4D).
Inverted tandems
We did not extensively consider tandems in an inverted orientation, as they cannot expand by unequal exchange and so are unlikely to give rise to satellite sequence. We did search, though, for TEs with clear junctions indicative of head-to-head or tail-to-tail inverted orientations. Several elements were detected as inverted tandems in many strains, but, in general, inverted tandems were present in only a few strains (Supplemental Table S2). Three of the high-frequency inverted tandems were LTR retrotransposons, and an inverted tandem of one can be identified in the reference, two Rover insertions (Chr 2R 580,417–595,299). Notably, a 6-nt palindromic motif (ATATAT) resides in the tandem junction, consistent with the possibility that it formed by double insertion (Fig. 1C). Additionally, many elements had inversion junctions involving internal rather than terminal sequence. In some cases, these clearly reflected nested insertions in opposite orientations, as both insertion junctions were identifiable. Other inversion junctions likely reflect complex sequence rearrangements and fragmented elements that are readily apparent when examining heterochromatic regions of the reference genome.
Multiple insertion drives rapid formation of TE tandems during periods of active transposition
P-elements swept through D. melanogaster populations during the mid-20th century (Bingham et al. 1982; Engels 1992; Kelleher 2016). Given this recent invasion, it is striking that we find tandem P-elements in over half of the GDL strains, indicating that tandem TEs form rapidly during periods of high transpositional activity (Fig. 5A). Head-to-head P-element tandems were frequently generated during a genetic screen (Tower et al. 1993), but the majority of strains we analyzed harbor head-to-tail (55/85) rather than head-to-head tandems (8/85). Selection removing head-to-head tandems from natural populations could underlie this, as long inverted repeats are prone to forming cruciform DNA secondary structures (Leach 1994). Alternatively, this may reflect technical bias, if amplicons containing head-to-head tandem junctions form hairpin secondary structures that decrease their PCR amplification efficiency, as suggested by Yang et al. (2014).
Figure 5.

Copy number, location, and sequence of TE tandem junctions. (A) Copy number (CN) distributions for the P-element. The dots are maximum a posteriori estimates in a particular strain, while the gray lines indicate 98%-credible intervals. (B,C) Sequence logos constructed from the 8-nt motifs found within the junctions of P-element tandem dimers (B) and the P-element TSDs described by Liao et al. (2000) (C). (D) A boxplot depicting the distances to the nearest TSS for P-element dimers and single insertions. (N) Counts of insertions in each category; (p) Kolmogorov–Smirnov test. (E) A similar plot for jockey elements; there is no significant difference between singles and dimers. (F) A UCSC Genome Browser view of the region on Chromosome 2L inferred to contain the hobo tandem array in strain I03, with the site of the hobo tandem added in as a black triangle. (G–I) CN distributions for hobo (G), Bari1 (H) and R1 (I) tandems. The dots are maximum a posteriori estimates in a particular strain, while the gray lines indicate 98%-credible intervals.
To identify the mechanism generating P-element tandems, we reasoned that if TE tandems are driven by multiple insertions at the same site, the tandem junction should contain a TSD of the insertion site (Fig. 1C), as has been observed in bacterial DNA transposon dimers (Dalrymple 1987). We examined reads containing fully intact head-to-tail junctions and found that the majority contain 8 nt of intervening sequence, the same length of the known P-element TSD. We generated a consensus motif (GTCTAGAG) and found that it is nearly identical to the TSD consensus motif previously identified from 1469 single P-element insertions (Fig. 5B,C; Liao 2000). We conclude that P-element tandems are formed by double insertion at the same site. Importantly, the motifs found at tandem junctions have a higher sequence specificity at each position than single-insertion sites, particularly at the first 2 nt, (sign test, P = 0.008) (Fig. 5B,C), suggesting that P-element tandems are more likely to form at sites that more closely match its preferred target sequence.
Genomic distribution of tandem dimers
The TSDs found in many tandem dimers originate from the locus into which the TEs inserted, and thus contain information about the location of the dimer. We reasoned that we should be able to infer the location of some dimers by identifying every instance of the TSD in the reference genome and asking which ones also contain evidence of a TE insertion at that site in that GDL strain (Supplemental Fig. S6A–F). Because TSDs are short and contain limited information, we imposed a number of filtering steps to restrict ourselves to dimers that could be confidently mapped to a single locus (see Methods, “Mapping Tandem Dimers to Specific TE Insertions”). This strategy may miss alternative mappings in sequence missing from the assembly, but the patterns of mapped dimers we observe for P-elements are consistent with what is known about the element's insertion preferences, so we do not believe this issue biases our inference.
We successfully mapped 72 dimers, 47 of which are euchromatic using the heterochromatin boundaries defined by Riddle et al. (2011) (Supplemental Table S3). P-elements comprise the majority of mapped dimers (46/72 mapped dimers), followed by jockey elements (11/72). P-element dimers are significantly closer to the transcription start sites of genes (median distance = 169 bp) than are single insertions (median distance = 430 bp) (Fig. 5D). Because P-elements preferentially insert near the promoters of genes (Spradling et al. 2011), the enrichment of dimers near transcription start sites supports the idea that dimers form at strong insertion sites. This is consistent with the higher information content we observed at TSDs within P-elements dimer junctions (Fig. 5B). Indeed, several of the tandems we mapped are adjacent to genes previously identified as among the strongest P-element insertion hotspots: apt, RapGAP1, Hers, Hsromega, Men, and mir-282 (Spradling et al. 2011). Furthermore, we identified dimers near the transcription start site of Hers in three strains (I26, N17, and T29A) and adjacent to Hsromega in two strains (B11 and B23), all containing different TSDs, strongly suggesting that they formed independently. The distance between the 11 jockey dimers we mapped and the nearest gene was comparable to that of single insertions, indicating again that the contrasting result with P-elements reflects its insertion site preference near promoters (Fig. 5D,E). Moreover, among the 10 mapped jockey dimers where the 5′ ends of both elements could be identified, six dimers involved clearly distinct 5′ trunctations (>500 nt difference), further supporting our conclusion that these dimers arise by double insertions (Supplemental Fig. S6A–F).
TE dimers can expand into larger arrays
The abundance of TE tandems that we discovered potentially provides the substrate for expansion by unequal exchange. We therefore searched for higher copy (greater than 10) TE tandems that may be polymorphic among the GDL populations and discovered one such expansion for the DNA transposon hobo. Most strains contain no or only one hobo tandem, but Ithacan line I03 has an estimated 13–19 tandem copies (Figs. 2C, 5G). To determine if this represents multiple independently formed tandems or a single expanded tandem array, we again searched all lines for reads containing fully intact head-to-tail tandem junctions. Only four strains contained fully intact head-to-tail hobo tandems, consistent with the distribution of junction estimates that suggested that many hobo tandems involve elements with deleted terminal sequence (Fig. 4F). We found, uniquely in I03, many reads containing an identical 8-nt motif (GTGGGGAC) between the TIRs of the tandem hobos. Using the mapping strategy outlined above, we determined that there is only one locus in the I03 genome that contains both the 8-bp motif and a hobo insertion, suggesting that the hobo tandem array is found on 2L at position 17,943,032 of the reference, well outside the pericentric heterochromatin and approximately 19.5 kb away from the protein coding gene beethoven (Fig. 5F). I03 is the only strain containing a hobo insertion at this position, indicating that the tandem likely formed from a recent hobo double insertion. Together, these observations strongly suggest that all elements of the array descend from a single tandem dimer. Multiple independent insertions would instead likely involve distinct motifs unless hobo has an extremely specific insertion motif. But, if that were the case, we would observe multiple sites in I03 with this motif harboring hobo insertions, which we do not.
Copy number variation in TE-derived satellites
Expansion events like we observed with hobo can eventually give rise to very large arrays and become fixed. ConTExt successfully identified the two known TE-derived satellites in D. melanogaster, which we further investigated to understand the dynamics of established satellites. One satellite is comprised of tandemly arrayed copies of the 1.7-kb DNA transposon Bari1 and is located in two blocks, with the majority of copies in the pericentromeric heterochromatin of the right arm of Chromosome 2 (Caizzi et al. 1993; Marsano et al. 2003; Palazzo et al. 2016). The second block is nested in a Stalker4 element on an unmapped scaffold (JSAE01000184) suggested to reside in the X or Y heterochromatin based on the presence of rDNA and R-elements (Palazzo et al. 2016). We identify both expected junctions between Bari1 and Stalker4 in the all-female GDL sequences (Supplemental Table S4), indicating that it cannot reside on the Y Chromosome. Previous analyses identified the Bari1 tandems in all strains examined (n = 10) and estimated its copy number at approximately 80 repeat units (Caggese et al. 1995). We find the Bari1 array in all 85 GDL strains, ranging from 32 to 130 copies (∼54,000–220,000 bp) (Fig. 5H).
The second known TE-derived satellite is comprised of R1 elements, which generally insert only at a specific site in the 28S ribosomal RNA gene. A fragment of this satellite was mapped to the X heterochromatin but inferred to not be directly within the rDNA array (Kidd and Glover 1980; Peacock et al. 1981). Subsequent analyses suggested it has a high copy number, but its overall size and organization was not known (Eickbush and Eickbush 1995; Stage and Eickbush 2009). We found that the array is enormous and fixed in the GDL, ranging from 91 to 273 head-to-tail tandem junctions (∼500,000 to ∼1,300,000 bp) (Fig. 5I). Intriguingly, the scaffold containing the smaller Bari1 tandem described above ends with five tandem R1 elements, and the junction between the first R1 and an rDNA unit is evident. We suggest that this scaffold also contains the boundary of the megabase-sized R1 array.
We also found that R1 tandem dimers are still being generated. First, we discovered several low-copy R1 tandems that have rDNA TSDs longer or shorter than 33 nt. These are independent of the R1 array, as we confirmed the previous observation that the array contains junctions with a 33-nt TSD of rDNA sequence. Second, we find many strains also contain 5′-truncated tandems (Supplemental Fig. S7B), suggesting the continuous production of R1 dimers distinct from those in the large array. We conclude that our population survey captures R1 elements in both nascent and fixed arrays.
The R1 array is more heterogenous than the Bari1 array
In addition to copy number variation, we discovered many R1 junctions corresponding to internal deletions, internal-to-internal tandems, and TEs inserted into R1 elements, many of which might reside in the tandem array. Theory predicts that evolution by unequal exchange can organize such structural variation into higher-order repeats (HORs) (Stephan 1989), as is found for the centromeric human alpha satellites. We could not determine the exact organization of the array due to the impossibility of assembling from Illumina data, but we reasoned that the copy number of junctions interspersed across the array should correlate with the overall size of the R1 array across lines. As this requires comparing the copy number distributions of specific junctions rather than general categories of structures as we have done above, we needed a principled strategy for matching junctions across strains. To this end, we employed a fuzzy C-means-like algorithm to match junctions across strains, using the uncertainty around each junction estimate to inform cluster assignments (see Supplemental Methods; Supplemental Fig. S4A–E). We then assessed each junction for a copy number correlation with the head-to-tail tandem junction, determining significance with the Benjamini–Hochberg procedure at a FDR of 1%.
Using this approach, we found 92 junctions involving R1 to have significant positive correlations with array size (Supplemental Table S4). Neither negative correlations nor any of the many junctions corresponding to R1 insertions in the rDNA were identified as significant, either of which would likely reflect false positives, suggesting that technical bias is rare. Among the 92 junctions, we found 20 tandem junctions and 24 internal deletions (Supplemental Fig. S7B). One of these tandem junctions is a high copy junction consistent with a small deletion near or spanning the head-to-tail tandem junction, which is present in about one quarter of tandem R1 units; an examination of ISO-1 Pacific Biosciences (PacBio) long reads confirms its presence in the array (Supplemental Fig. S7A; Kim et al. 2014). We also found a number of TE insertions into R1 elements that are correlated with the copy number of the array. The highest copy examples are specific fw, circe, and accord2 insertions averaging 18, six, and six copies, respectively (Supplemental Table S4). circe has previously been described within the array (Losada et al. 1999). The copy numbers and degrees of positive correlation we observe also indicate that these structures either are dispersed throughout the entire array or constitute subarrays that may be arranged as HORs, as expansion and contraction events that change the array's copy number also alter the copy number of these junctions. For comparison, we looked for junctions involving Bari1 that were correlated in copy number with the Bari1 head-to-tail tandem junction, and found only eight junctions, none of which had amplified to multiple copies in any strain (Supplemental Table S4). The reference genome indicates a Max LTR retrotransposon is inserted into the Bari1 array, but we find no evidence of this in the GDL strains, suggesting it is specific to the reference strain (Marsano et al. 2004; Hoskins et al. 2015). Taken together, these observations suggest that the R1 array, but not the Bari1 array, is heterogenous with respect to deletions and TE insertions, some of which may be arranged into HORs.
Discussion
ConTExt successfully identifies repetitive structures in NGS data
Leveraging NGS population genomic data sets to learn about highly repeated sequence is a challenging problem. We employed an alignment strategy that maps repeat-derived reads to repeat consensus sequences and used mixture modeling to interpret the alignment patterns. By applying this method to a panel of five populations, we observed multiple stages of TE-derived satellite evolution ongoing within a single species. We successfully detected previously known tandem structures, including tandem junctions among all telomeric TE families and large tandem arrays of the Bari1 and R1 elements. We also identified internally deleted TEs as well as nested insertions (Fig. 2D,E), highlighting the large amount of information about these understudied structures present in the thousands of publicly available short-read data sets. Furthermore, some of the strategies used in ConTExt may be applied to long-read technologies. ConTExt can retain the long-range information provided by the GemCode barcoding strategy and thus could be supplemented with long-range information for greater power in mapping structures. The visualization strategies employed may also be adapted to PacBio traces for summarizing regions that cannot be assembled even with PacBio data, though the statistical model for clustering the data would require modification.
Our strategies have some limitations imposed by our reliance upon sequence alignments. Repeat-derived reads rarely align uniquely to the reference genome, meaning we cannot locate most structures we identify. Further, reliance on consensus sequences limits our survey to known repeat families. However, as the TE families in D. melanogaster are well characterized, this is unlikely to have strongly biased our analysis. For less well characterized species, tools exist to extract repeat consensus sequences out of NGS reads (Novak et al. 2013).
Structure inference from paired-end alignments also has limitations. First, we cannot detect junctions containing intervening sequences longer than the mate pair distance of the library, such as LTRs exceeding 338 bp. Second, chimeric inserts can produce spurious structure discovery calls. We mitigate this by only considering structures supported by multiple read pairs with distinct coordinates. Further, false positives would be dispersed across the consensus sequences rather than concentrated in biologically plausible patterns that we observed, such as the tendency of non-LTR retrotransposon dimers to involve 5′-truncated elements. Moreover, the tandems we find for LTR elements are largely restricted to elements with LTRs shorter than the length cutoff expected based on the library insert size. False positives resulting from mapping or library preparation artifacts would instead appear as head-to-tail dimers regardless of LTR length.
Transposition drives continuous production of tandem formation
We discovered that the processes by which TEs transition to satellites are actively ongoing in D. melanogaster. Multiple tandem TE dimers from which large satellite arrays can expand are common in most D. melanogaster genomes. The observed patterns of LTR retrotransposon tandem junctions are consistent with most arising from ectopic recombination between LTRs, as has been previously observed (Ke and Voytas 1997). In contrast, several observations strongly suggest non-LTR retrotransposon, DNA transposon, and some LTR tandems are formed by multiple insertions at the same locus. First, it is well documented that repair mechanisms generate direct repeats flanking most TE insertions (Craig 1997). Thus, a tandem formed by double insertion should contain a duplicate of the target site within its tandem junction. We found this pattern for many of the tandems, in particular the majority of P-element dimers. Second, the patterns of 5′-truncated tandems we observe in non-LTR retrotransposons support multiple insertion events. Non-LTR retrotransposons are prone to losing sequence from their 5′ ends during integration, and most non-LTR retrotransposon tandem junctions we found are between the intact 3′ end of one element and the 5′-truncated end of the adjacent element. We also discovered several non-LTR retrotransposon dimers involving elements with distinct 5′ truncations, clear evidence of two independent insertion events.
Third, if TE dimers form through transposition events then periods of high TE activity should also have high rates of dimer formation. Indeed, despite only invading the species in the last century, we find that most strains in the GDL contain tandem P-elements. We further note that the population frequency of particular dimers varies among elements, suggesting discrete periods of dimer formation. Thus, bursts of TE activity likely correspond to bursts of tandem formation.
We emphasize that the mechanism of transposition can almost guarantee dimer formation. Dimer formation requires only that an element inserts preferentially at certain motifs and generates TSDs. If so, then a new TE insertion preserves its target site while generating a new one, enabling subsequent insertions at that locus (Fig. 1C). This is conceptually similar to the mechanism by which plasmids occasionally integrate in tandem during transformation, where the homologous sequence at the integration site is preserved upon integration, thus permitting subsequent integrating events at the same site (Orr-Weaver and Szostak 1983). The propensity of most TE families to form dimers highlights the degree of insertion site preference: A TE family that inserted at random sequence would almost never be detected in tandem.
Tandem dimers expand into large arrays
Having discovered that TE dimers are common in natural populations, we suggest that their subsequent amplification is the major mechanism generating TE-derived satellites. We observed one such event, a copy number expansion of a hobo dimer to approximately 16 copies in a single line (Fig. 5G). We also confirmed earlier suggestions that the previously discovered array of R1 elements is large, finding that it varies between ∼530,000–1,300,000 bp in length. Our analysis suggest that it likely originated when an R1 dimer formed within an rDNA unit, and then expanded. We further found that many independent deletions and TE insertions occurred within the array subsequent to its expansion, some of which also expanded in copy number. The obvious candidate for causing such expansions is unequal exchange.
While the R1 and hobo arrays contain TSDs clearly indicating that they originated as a dimer, the junctions within the two Bari1 arrays instead display several unusual features: They are missing sequence from their terminal inverted repeats, each ends with a partial element at its 5′ edge, and the smaller array is flanked by a ∼500-nt TSD, inconsistent with Bari1's usual transposition mechanism (Marsano et al. 2003). RCR is a plausible explanation, with TIR sequence being lost during circle formation and the partial terminal elements resulting from utilization of a random cut site due to the incomplete TIRs (Marsano et al. 2003). However, the absence of TSD at the tandem junctions does not preclude the alternative possibility that the Bari1 arrays arose by tandem insertion of two Bari1 elements, followed by partial deletion of the terminal inverted repeats at the junction and then expansion by unequal crossing over. We suggest that deletions of TIRs may be common for DNA transposon dimers, as most hobo dimers (24/36) harbored similar deletions. The expansion of such a dimer would result in an array where each junction contains an identical deletion, as found in the Bari1 arrays.
Tandem persistence and TIR status
P-element dimers, which are younger than 100 yr old, generally have intact TIRs. In contrast, a large P-element related array in Drosophila guanche is comprised of elements missing ∼100 nt of terminal sequence (Miller et al. 1992), and all elements in the fixed Bari1 tandem array have incomplete TIRs (Marsano et al. 2003). The terminal inverted repeats at the ends of DNA transposons are endonuclease cut sites, which should expose the tandem to elevated rates of double-strand breaks (DSBs) and unequal exchange. In contrast, tandems lacking intact TIRs will experience a reduced rate of DSBs and fewer recombination events over time. Charlesworth et al. (1986) proposed that tandem arrays in regions with high recombination rates should be lost more rapidly than those in low recombination regions, shaping the genome-wide distribution of satellite sequence. More generally, they proposed that this applies to any satellite with features that reduce the rate of unequal exchange. We suggest that presence or absence of intact TIRs may shape the rate at which tandem DNA transposons persist, with tandems harboring intact TIRs being lost more rapidly. A second possibility is that the palindrome formed by inverted repeats at the tandem junction leads to hairpin secondary structures, which may be prone to deletions.
Heterogeneity differences between arrays
The R1 array is substantially more heterogeneous than the Bari1 array, harboring a number of deletions and TE insertions residing within the array. Such organization is typical of many satellite arrays where TEs tend to accrete to the edges of the array (McAllister and Werren 1999; Khost et al. 2017). One explanation is that the R1 array is older than the Bari1 array. Consistent with the R1 array being relatively old, we find that most of its variant structures are at relatively high population frequency, indicating that they were present when the GDL populations diverged. Alternatively, differences in the recombination rates at the two arrays might account for their structural differences, as heterogeneity and higher-order structure arise naturally when the rate of unequal exchange is low relative to the mutation rate (Stephan and Cho 1994). A third possibility is that homogeneity of the Bari1 array is maintained by purifying selection, perhaps as a source of piRNAs or as a structural element.
Implications of tandem TEs
While the structures we describe are present in most genomes, they cannot be detected by the standard tools for structural variant discovery. They have thus been largely ignored in previous analyses of TE structural variation despite having known biological effects, such as on gene expression. For example, tandem P-element transgenes induce position-effect variegation, the strength of which increases with copy number (Dorer and Henikoff 1994). This is likely because TE insertions are silenced by the piRNA pathway (Brennecke et al. 2007), and this can impact nearby genes (Shpiz et al. 2014; Lee 2015). TEs also frequently carry internal regulatory elements that can be recruited into gene regulatory networks and even alter the three-dimensional organization of the genome (Byrd and Corces 2003; Feschotte 2008). Loehlin and Carroll (2016) recently described synergistic increases in the expression of recently duplicated genes which may result from concentrating regulatory elements. We suggest therefore that future studies on the functional impacts of TE variation should consider whether the insertions in question are single elements or tandemly arrayed. This is particularly important for elements with strong site preferences such as P-elements, with insertional hotspots being most likely to harbor tandem structures.
Methods
Overview
First, we align reads to the consensus sequences of known repeats. Second, we employ a clustering strategy to infer structures from the distributions of discordant read pairs in each library. Specifically, we seek to identify junctions: sequence coordinates that are nonneighboring in the reference genome but that neighbor each other in the sequenced genome (Bashir et al. 2008). We use mixture modeling to identify a generative model that explains the observed distribution of aligned read pairs. This general approach allows us to identify not only the presence and copy number of tandem structures but also deletions internal to repeats and insertions into both unique and repeated sequence. Importantly, it can be applied to any genome for which the repeat families are known.
Constructing the repeat index
We used Repbase repeat annotations (release 19.06) (Bao et al. 2015) for D. melanogaster and supplemented these with additional repeats. To remove redundant entries, we manually curated the index by performing all pairwise alignments to identify entries that share considerable homology (for details, see Supplemental Methods; Supplemental File 1). While we subsequently refer to the entries as consensus sequences, not all are true consensus sequences; rather, some are representative examples. We use the Repbase nomenclature for repeats, with the exception of the Bari transposon, which we refer to as Bari1 (Kaminker et al. 2002).
Read preprocessing
We used Trimmomatic for read quality control (Bolger et al. 2014). We removed all sequence from the 3′ ends such that the average Phred score was ≥20 in all the remaining 4-nt windows, and discarded any trimmed reads <40-nt long. Because we can only detect a junction if it falls in the gap of a read pair, we trimmed all remaining reads to 70-bp from their 3′ ends to increase the size of this gap.
Aligning reads to repeats
We employed a two-step alignment procedure, first aligning the reads to the set of all individual repeats extracted from the reference genome (including the unmapped contigs) and then collapsing these alignments onto the corresponding consensus sequence.
We first used RepeatMasker (Smit et al. 2015) to both hard-mask release 6.01 of the D. melanogaster reference genome (Hoskins et al. 2015) for repeats (ensuring repeat-derived reads are assigned to the repeat index) and to identify the location of all instances of each repeat family, using the most sensitive seed setting. We extracted these repeats from the reference to construct an index of individual insertions. We then used Bowtie 2 (version 2.1.0) to align the reads in each read pair as single-end reads to both the repeat-masked reference genome and the index of individual repeats (Langmead and Salzberg 2012).
Alignments were then collapsed onto the corresponding repeat consensus sequences guided by BLASTN alignments between the individual insertions and the consensus sequences (see Supplement). We filtered out any non-repeat-derived reads aligning to the reference genome with mapping quality scores <20. Because the index of TE insertions is highly repetitive, mapping quality scores are not informative of alignment quality, and so, we do not apply the same filter to repeat aligned reads.
Estimating the gap size distribution
The distribution of reads spanning a junction depends upon the size distribution of read pairs in the library. We refer to the distance between the 5′ ends of a concordant read pair as the insert size, and the interval of sequence between the 3′ ends of a read pair as the gap. For a junction to be detected, it must be spanned by the gap (junctions interrupting a read will likely prevent its alignment), so for each library we estimated the gap size distribution with a kernel density estimate, choosing the bandwidth by twofold cross-validation (for more details, see Supplemental Methods; Supplemental Fig. S1A). For simulations of read distributions, we use the kernel density estimate conditioned on the reads spanning a junction, which accounts for small inserts being less likely to span a junction and is approximately given by
where G is the gap size distribution.
In all subsequent sections, we consider a read pair concordant when its two reads map to opposite strands and are oriented toward each other and when its gap size falls between the 0.5% percentile and the 99.5% percentile of the gap size distribution.
Representing paired-end alignments as two-dimensional scatterplots
A read pair can be represented as a point in a two-dimensional space, where the x-axis represents the sequence and strand to which one read maps, and the y-axis represents the sequence and strand to which the other read maps (Fig. 2C; Supplemental Fig. S1B,C). This is an effective visualization strategy for manually examining the patterns of TE insertions into unique sequence and into repetitive sequences. Organizing reads where both ends map to the same sequence requires additional constraints. For read pairs that map to opposite strands of the same sequence, we assign the reverse strand to the x-axis and the forward strand to the y-axis. For read pairs that map to the same strands of the same sequence, there is ambiguity as to which axes the reads should be assigned. In the case of forward–forward read pairs, we assign the read with the higher sequence coordinate to the x-axis, and for reverse–reverse, we assign the read with the lower sequence coordinate to the x-axis.
Discovering structures with mixture modeling
The problem of structural variant discovery can be framed as trying to identify clusters of read pairs that span junctions. While agglomerative clustering strategies are successful at identifying structural variation in unique sequence (Medvedev et al. 2009), the alignment patterns of repeat-derived reads are more challenging to resolve. This is because one is collapsing reads derived from up to megabases of sequence onto consensus sequences <10 kb in length, and so read pairs representing distinct junctions are often crowded and sometimes interspersed. Mixture modeling, however, provides tools for clustering data, especially when clusters are partially overlapping. Therefore, we model the distribution of discordant read pairs within a scatterplot with a Gaussian mixture model (GMM) (Supplemental Fig. S1D). A junction involving a repeat will generate a distribution of discordant read pairs (Supplemental Fig. S1B,C), and so, the set of discordant read pairs, X, in a scatterplot can be thought of as arising from a mixture of many distributions, each corresponding to a junction (Supplemental Fig. S1D):
Thus, each component, k, in the GMM corresponds to a junction, with the mean, μk, relating to the junction's sequence coordinates; the mixing proportion, ϕk, relating to the number of read pairs spanning that junction; and the covariance, Σ, reflecting the library's gap size distribution. The actual distribution of read pairs spanning a junction is not Gaussian (Supplemental Fig. S1C); however, the approximation makes the problem tractable, and we use Gaussians with sufficiently large covariances to cluster the read pair distributions. We fit the GMM using an accelerated expectation–maximization algorithm (Dempster et al. 1977; Varadhan and Roland 2008). We then use the fitted GMM to group read pairs into clusters that correspond to junctions, assigning each read pair to the most likely component (Fig. 2C; Supplemental Figs. S1D, S2A,B). Once clusters are identified, we remove clusters that are possibly technical artifacts and estimate the sequence coordinates and copy number of the underlying junctions in a manner that accounts for GC bias in read depth. For further details of the EM implementation, covariance selection (Supplemental Fig. S1E), and post-processing of the identified clusters, see the Supplemental Methods. Summaries of clustering parameters and performance can be found in Supplemental Table S1.
Mapping tandem dimers to specific TE insertions
To infer the location of a tandem dimer, we first identified sequencing reads in that strain containing the tandem junction (Supplemental Methods; Supplemental Fig. S6B). From these reads, we identified any intervening sequence at the junction and identified every locus in the reference genome matching this motif. For motifs ≥9 bp, we used BLASTN with an e-value cutoff of 10 and accepted the top hit and all other hits whose e-values were within two orders of magnitude of the best e-value. For motifs 7 or 8 bp in length, we required an exact match to either the sequence or its reverse complement and employed string matching. We did not attempt to map any dimer whose intervening sequence was <7 bp.
An 8-nt motif will occur hundreds of times in the genome, but we reasoned that if only one matching locus contained an insertion of that TE family, it was the likely location of the dimer. We therefore identified the location of each insertion of that TE family in the strain with the motif-containing tandem junction (Supplemental Methods; Supplemental Fig. S6C–F). We considered the locus of an intervening sequence to match a TE insertion junction if its location estimates were within 150 nt. We only considered dimers that could be uniquely mapped to a single location. We note that while we putatively mapped R1 tandems to two locations in autosomal heterochromatin, these were driven by partial alignments with distinct TSDs, and we believe these are likely artifacts; thus, we excluded R1 from our efforts to map dimers.
Gene annotation
We downloaded the RefSeq gene annotations for D. melanogaster from UCSC's Table Browser and excluded all computed genes and RNAs (entries beginning with CG or CR).
Aligning PacBio reads
We aligned PacBio reads to the repeat index using BLASTN, using a linear gap penalty of 2 to account for the high rate of indels and imposed an e-value cutoff of 0.01.
Categorizing tandem junctions
We divide the tandem junctions we observe into three broad categories based upon their sequence coordinates: head-to-tail, tail-to-internal, and internal-to-internal. We define head-to-tail junctions as those within 200 nt of both the 5′ and 3′ ends. We define tail-to-internal as junctions within 200 nt of the 3′ end, but not the 5′ end. All other tandem junctions are classified as internal-to-internal. We exclude junctions where the coordinates are within 400 nt of each other, as these potentially reflect groups of concordant reads misidentified as discordant. We restrict this analysis to TE families estimated to contribute at least 20 kb of sequence to at least one genome, based on coverage of the consensus normalized by GC-corrected read depth.
Matching junctions across samples
Fitting the GMM to the data allows us to identify junctions within each sample, but for some questions, we needed to match these junctions across samples. To do this automatically, we employ a second fuzzy clustering step that uses the estimated uncertainty around each junction estimate to define cluster membership weights based on the probability that multiple inferred junctions reflect the same structure. For more details, see Supplemental Methods.
Software availability
Scripts were written in Python 2.7 and made use of SciPy (Jones et al. 2001), NumPy (van der Walt et al. 2011), Scikit-Learn (Pedregosa et al. 2011), and Biopython (Cock et al. 2009). Figures were generated with Matplotlib (Hunter 2007) and Seaborn.
The ConTExt pipeline is located at https://github.com/LaptopBiologist/ConTExt. We include the Python scripts in Supplemental File 2 but refer readers to GitHub for the most up-to-date versions of the package.
Supplementary Material
Acknowledgments
We thank Dean Castillo, Andy Clark, Anne-Marie Dion-Côté, Jullien Flynn, Sarah Lower, Kevin Wei, and the anonymous reviewers for helpful comments and discussions. Support was provided by National Institute of General Medical Sciences grants R01-119125 and R01-074737 to D.A.B.
Author contributions: D.A.B and M.P.M. conceived the experiments and wrote the paper; M.P.M developed and implemented the ConTExt pipeline, performed the experiments, and analyzed the data.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.231472.117.
References
- Aldrup-MacDonald ME, Kuo ME, Sullivan LL, Chew K, Sullivan BA. 2016. Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res. 26: 1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir A, Volik S, Collins C, Bafna V, Raphael BJ. 2008. Evaluation of paired-end sequencing strategies for detection of genome rearrangements in cancer. PLoS Comput Biol 4: e1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, Quesneville H, Anxolabéhère D, Ashburner M. 2006. Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 7: R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham PM, Kidwell MG, Rubin GM. 1982. The molecular basis of P–M hybrid dysgenesis: the role of the P element, a P-strain-specific transposon family. Cell 29: 995–1004. [DOI] [PubMed] [Google Scholar]
- Black DM, Jackson MS, Kidwell MG, Dover GA. 1987. KP elements repress P-induced hybrid dysgenesis in Drosophila melanogaster. EMBO J 6: 4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH, Greider CW, Szostak JW. 2006. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 12: 1133–1138. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. [DOI] [PubMed] [Google Scholar]
- Byrd K, Corces VG. 2003. Visualization of chromatin domains created by the gypsy insulator of Drosophila. J Cell Biol 162: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggese C, Pimpinelli S, Barsanti P, Caizzi R. 1995. The distribution of the transposable element Bari-1 in the Drosophila melanogaster and Drosophila simulans genomes. Genetica 96: 269–283. [DOI] [PubMed] [Google Scholar]
- Caizzi R, Caggese C, Pimpinelli S. 1993. Bari-1, a new transposon-like family in Drosophila melanogaster with a unique heterochromatic organization. Genetics 133: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Langley CH, Stephan W. 1986. The evolution of restricted recombination and the accumulation of repeated DNA sequences. Genetics 112: 947–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25: 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig NL. 1997. Target site selection in transposition. Annu Rev Biochem 66: 437–474. [DOI] [PubMed] [Google Scholar]
- Dalrymple B. 1987. Novel rearrangements of IS30 carrying plasmids leading to the reactivation of gene expression. Mol Gen Genet 207: 413–420. [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. 1977. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B Methodol 39: 1–38. [Google Scholar]
- Dias GB, Svartman M, Delprat A, Ruiz A, Kuhn GC. 2014. Tetris is a foldback transposon that provided the building blocks for an emerging satellite DNA of Drosophila virilis. Genome Biol Evol 6: 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer DR, Henikoff S. 1994. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 77: 993–1002. [DOI] [PubMed] [Google Scholar]
- Eickbush DG, Eickbush TH. 1995. Vertical transmission of the retrotransposable elements R1 and R2 during the evolution of the Drosophila melanogaster species subgroup. Genetics 139: 671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels WR. 1992. The origin of P elements in Drosophila melanogaster. BioEssays 14: 681–686. [DOI] [PubMed] [Google Scholar]
- Ferree PM, Barbash DA. 2009. Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol 7: e1000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C. 2008. Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman L, Saunders A. 2008. Centromere-associated female meiotic drive entails male fitness costs in monkeyflowers. Science 322: 1559–1562. [DOI] [PubMed] [Google Scholar]
- Francisco FO, Lemos B. 2014. How do Y-chromosomes modulate genome-wide epigenetic states: genome folding, chromatin sinks, and gene expression. J Genomics 2: 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JA, DeBaryshe PG, Traverse KL, Celniker SE, Pardue M-L. 2006. Genomic organization of the Drosophila telomere retrotransposable elements. Genome Res 16: 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Wu Y, Koblížková A, Torres GA, Wang K, Iovene M, Neumann P, Zhang W, Novák P, Buell CR, et al. 2012. Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell 24: 3559–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RW, Lindsley DL, Livak KJ, Lewis B, Siversten AL, Joslyn GL, Edwards J, Bonaccorsi S. 1984. Cytogenetic analysis of a segment of the Y chromosome of Drosophila melanogaster. Genetics 107: 591–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, Eichler EE, Sahinalp SC. 2010. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics 26: i350–i357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, Booth BW, Pfeiffer BD, George RA, Svirskas R, et al. 2015. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD. 2007. Matplotlib: a 2D graphics environment. Comput Sci Eng 9: 90–95. [Google Scholar]
- Jones E, Oliphant T, Peterson P. 2001. SciPy: open source scientific tools for Python. http://www.scipy.org/.
- Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, et al. 2002. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 3: research0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke N, Voytas DF. 1997. High frequency cDNA recombination of the Saccharomyces retrotransposon Ty5: the LTR mediates formation of tandem elements. Genetics 147: 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher ES. 2016. Reexamining the P-element invasion of Drosophila melanogaster through the lens of piRNA silencing. Genetics 203: 1513–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khost DE, Eickbush DG, Larracuente AM. 2017. Single-molecule sequencing resolves the detailed structure of complex satellite DNA loci in Drosophila melanogaster. Genome Res 27: 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd SJ, Glover DM. 1980. A DNA segment from D. melanogaster which contains five tandemly repeating units homologous to the major rDNA insertion. Cell 19: 103–119. [DOI] [PubMed] [Google Scholar]
- Kim KE, Peluso P, Baybayan P, Yeadon PJ, Yu C, Fisher W, Chin C-S, Rapicavoli NA, Rank DR, Li J, et al. 2014. Long-read, whole genome shotgun sequence data for five model organisms. Sci Data 1: 140045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Betancourt AJ, Schlötterer C. 2012. Sequencing of pooled DNA samples (Pool-Seq) uncovers complex dynamics of transposable element insertions in Drosophila melanogaster. PLoS Genet 8: e1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima KK, Fujiwara H. 2005. Long-term inheritance of the 28S rDNA-specific retrotransposon R2. Mol Biol Evol 22: 2157–2165. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM. 2014. The organization and evolution of the Responder satellite in species of the Drosophila melanogaster group: dynamic evolution of a target of meiotic drive. BMC Evol Biol 14: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DR. 1994. Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. Bioessays 16: 893–900. [DOI] [PubMed] [Google Scholar]
- Lee YCG. 2015. The role of piRNA-mediated epigenetic silencing in the population dynamics of transposable elements in Drosophila melanogaster. PLoS Genet 11: e1005269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G-c. 2000. Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc Natl Acad Sci 97: 3347–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehlin DW, Carroll SB. 2016. Expression of tandem gene duplicates is often greater than twofold. Proc Natl Acad Sci 113: 5988–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Roberts PA. 2000. Evolution of DNA in heterochromatin: the Drosophila melanogaster sibling species subgroup as a resource. Genetica 109: 125–130. [DOI] [PubMed] [Google Scholar]
- Losada A, Abad JP, Agudo M, Villasante A. 1999. The analysis of Circe, an LTR retrotransposon of Drosophila melanogaster, suggests that an insertion of non-LTR retrotransposons into LTR elements can create chimeric retroelements. Mol Biol Evol 16: 1341–1346. [DOI] [PubMed] [Google Scholar]
- Macas J, Koblížková A, Navrátilová A, Neumann P. 2009. Hypervariable 3′ UTR region of plant LTR-retrotransposons as a source of novel satellite repeats. Gene 448: 198–206. [DOI] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. 2009. Major evolutionary transitions in centromere complexity. Cell 138: 1067–1082. [DOI] [PubMed] [Google Scholar]
- Marsano RM, Milano R, Minervini C, Moschetti R, Caggese C, Barsanti P, Caizzi R. 2003. Organization and possible origin of the Bari-1 cluster in the heterochromatic h39 region of Drosophila melanogaster. Genetica 117: 281–289. [DOI] [PubMed] [Google Scholar]
- Marsano RM, Marconi S, Moschetti R, Barsanti P, Caggese C, Caizzi R. 2004. MAX, a novel retrotransposon of the BEL-Pao family, is nested within the Bari 1 cluster at the heterochromatic h39 region of chromosome 2 in Drosophila melanogaster. Mol Genet Genomics 270: 477–484. [DOI] [PubMed] [Google Scholar]
- Mason JM, Frydrychova RC, Biessmann H. 2008. Drosophila telomeres: an exception providing new insights. Bioessays 30: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF, Werren JH. 1999. Evolution of tandemly repeated sequences: what happens at the end of an array? J Mol Evol 48: 469–481. [DOI] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M. 2009. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6: S13–S20. [DOI] [PubMed] [Google Scholar]
- Meštrović N, Mravinac B, Pavlek M, Vojvoda-Zeljko T, Šatović E, Plohl M. 2015. Structural and functional liaisons between transposable elements and satellite DNAs. Chromosome Res 23: 583–596. [DOI] [PubMed] [Google Scholar]
- Miller WJ, Hagemann S, Reiter E, Pinsker W. 1992. P-element homologous sequences are tandemly repeated in the genome of Drosophila guanche. Proc Natl Acad Sci 89: 4018–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak P, Neumann P, Pech J, Steinhaisl J, Macas J. 2013. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29: 792–793. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW. 1983. Multiple, tandem plasmid integration in Saccharomyces cerevisiae. Mol Cell Biol 3: 747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo A, Lovero D, D'Addabbo P, Caizzi R, Marsano RM. 2016. Identification of Bari transposons in 23 sequenced Drosophila genomes reveals novel structural variants, MITEs and horizontal transfer. PLoS One 11: e0156014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock WJ, Appels R, Endow S, Glover D. 1981. Chromosomal distribution of the major insert in Drosophila melanogaster 28S rRNA genes. Genet Res 37: 209–214. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. 2011. Scikit-learn: Machine learning in Python. J Mach Learn Res 12: 2825–2830. [Google Scholar]
- Periquet G, Lemeunier F, Bigot Y, Hamelin MH, Bazin C, Ladevèze V, Eeken J, Galindo MI, Pascual L, Boussy I. 1994. The evolutionary genetics of the hobo transposable element in the Drosophila melanogaster complex. Genetica 93: 79–90. [DOI] [PubMed] [Google Scholar]
- Prosser J, Frommer M, Paul C, Vincent PC. 1986. Sequence relationships of three human satellite DNAs. J Mol Biol 187: 145–155. [DOI] [PubMed] [Google Scholar]
- Prudhomme M, Turlan C, Claverys J-P, Chandler M. 2002. Diversity of Tn4001 transposition products: the flanking IS256 elements can form tandem dimers and IS circles. J Bacteriol 184: 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, et al. 2011. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21: 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Cridland JM, Shao L, Hu TT, Andolfatto P, Thornton KR. 2014. Landscape of standing variation for tandem duplications in Drosophila yakuba and Drosophila simulans. Mol Biol Evol 31: 1750–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiha H, Miller JR, Woods LC, Glover DM. 1981. Arrangements and rearrangements of sequences flanking the two types of rDNA insertion in D. melanogaster. Nature 290: 749–754. [DOI] [PubMed] [Google Scholar]
- Shpiz S, Ryazansky S, Olovnikov I, Abramov Y, Kalmykova A. 2014. Euchromatic transposon insertions trigger production of novel Pi- and endo-siRNAs at the target sites in the Drosophila germline. PLoS Genet 10: e1004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2015. RepeatMasker Open-4.0. 2013–2015. www.repeatmasker.org.
- Spradling AC, Bellen HJ, Hoskins RA. 2011. Drosophila P elements preferentially transpose to replication origins. Proc Natl Acad Sci 108: 15948–15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stage DE, Eickbush TH. 2009. Origin of nascent lineages and the mechanisms used to prime second-strand DNA synthesis in the R1 and R2 retrotransposons of Drosophila. Genome Biol 10: R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W. 1989. Tandem-repetitive noncoding DNA: forms and forces. Mol Biol Evol 6: 198–212. [DOI] [PubMed] [Google Scholar]
- Stephan W, Cho S. 1994. Possible role of natural selection in the formation of tandem-repetitive noncoding DNA. Genetics 136: 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan T, Coen E, Webb D, Dover G. 1982. Modes and rates of change of complex DNA families of Drosophila. J Mol Biol 158: 37–54. [DOI] [PubMed] [Google Scholar]
- Tower J, Karpen GH, Craig N, Spradling AC. 1993. Preferential transposition of Drosophila P elements to nearby chromosomal sites. Genetics 133: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S, Colbert SC, Varoquaux G. 2011. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng 13: 22–30. [Google Scholar]
- Varadhan R, Roland C. 2008. Simple and globally convergent methods for accelerating the convergence of any EM algorithm. Scand J Stat 35: 335–353. [Google Scholar]
- Yang H, Volfovsky N, Rattray A, Chen X, Tanaka H, Strathern J. 2014. GAP-Seq: a method for identification of DNA palindromes. BMC Genomics 15: 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, de Greef JC, Chen Y-Y, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, et al. 2009. Specific loss of histone H3 lysine 9 trimethylation and HP1γ/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 5: e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Koblížková A, Wang K, Gong Z, Oliveira L, Torres GA, Wu Y, Zhang W, Novák P, Buell CR, et al. 2014. Boom-bust turnovers of megabase-sized centromeric DNA in Solanum species: rapid evolution of DNA sequences associated with centromeres. Plant Cell 26: 1436–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.