

Abstract
Background:
Chronic inflammation and elevated basal metabolic rate (BMR) are established features of sickle-cell anemia (SCA). However, there is little information on the possible impacts of these afore-mentioned features on glycemia and insulin sensitivity status of this group of people.
Aim:
This study aims to determine the insulin sensitivity status as well the effect of BMR on glycemia in adults with SCA in steady state.
Materials and Methods:
Fifty participants comprising 30 adults with SCA in steady state and 20 age- and gender-matched apparently healthy adults with hemoglobin genotype AA (HbAA) genotype that served as controls. Anthropometric and clinical indices were obtained using standard methods. After an overnight fast, fasting plasma glucose (FPG), and serum insulin levels were determined using the glucose oxidase method and ELISA, respectively. Indices of insulin sensitivity and β-cell function as well as BMR were appropriately calculated.
Results:
The mean fasting insulin resistance (IR) index, homeostatic model of assessment of IR (HOMA-IR) and of β-cell function (HOMA2-β%), and mean insulin level were significantly lower while the mean HOMA of insulin sensitivity (HOMA2-S%), quantitative insulin sensitivity check index, inverse of insulin sensitivity (1/FI), glucose-insulin ratio, C-reactive protein (CRP), and BMR was significantly higher in patients with SCA compared with the controls. The mean FPG and insulin levels and the mean values of indices of insulin sensitivity and secretion were not significantly different in SCA patients with elevated BMR compared with SCA patients with lower BMR. In addition, BMR had no significant correlation with FPG and HOMA-IR in patients with SCA.
Conclusion:
Despite the established chronic inflammation in SCA patients in steady state, they seem to have better insulin sensitivity status but impaired β-cell activity when compared with adults with HbAA. Furthermore, BMR does not have any pronounced effect on glycemic and insulin sensitivity status in SCA patients in steady state.
Keywords: Basal metabolic rate, inflammation, insulin sensitivity, sickle cell anemia, β-cell activity
Introduction
Sickle-cell anemia (SCA) is an autosomal recessive hemoglobinopathy that is more common in Africa. It is characterized by abnormal hemoglobin production, hemolytic anemia, and intermittent occlusion of small vessels which lead to acute and chronic tissue ischemia, organ dysfunction as well as organ damage.[1] The abnormal hemoglobin production is caused by a single nucleotide mutation that substitutes glutamic acid with valine at the sixth position of the globin protein.[2]
Abnormal erythrocyte membrane and subsequent chronic hemolysis have been reported as the common triggers of inflammation in SCA.[3] Even in the steady state, markers of inflammation such as C-reactive protein (CRP) are usually elevated.[4] Consequently, this persistent chronic inflammation in patients with SCA causes slow damage of the β-cell of pancreas which plays a prominent role in glycemic control.[5] Furthermore, iron overload, as a result of multiple transfusion, has been associated with insulin resistance (IR), β-cell damage and decreased insulin production which can all cause defective glycemic control.[6] Interestingly, despite these SCA features that have a strong potential in causing dysglycemia and diabetes mellitus (DM), concurrence of SCA with DM continues to be a rare finding even in tropical countries where SCA is prevalent.
Reports have shown that there is an association between SCA, inflammation, altered basal metabolic rate (BMRs), and impaired glucose metabolism.[7,8] Despite this association, DM continues to be a rare finding in individuals with SCA. A number of studies detected no single case of DM in patients with SCA while others detected a very low prevalence.[6,9] This unexpected rare association has been linked with low body mass index (BMI), hypermetabolism and possibly, other yet to be identified genetic factors.[10,11]
Inflammatory mediators play significant roles in the initiation and progression of complications of SCA. Similarly, it is well established that inflammation plays an important role in the pathogenesis of chronic diseases such as obesity and DM. Alsultan et al.[12] reported that fasting blood glucose and insulin levels are elevated in SCA patients. In contrast, Akinlade et al.[13] observed similar level of insulin and a relatively lower level of fasting plasma glucose (FPG) in SCA patients compared with controls. They also showed that patients with SCA have similar insulin sensitivity status as individuals with hemoglobin genotype AA (HbAA). Therefore, understanding the mechanism responsible for this rare concurrence of SCA and DM is of endocrinology importance as it could help further elucidate the pathogenesis of DM especially, type 2 DM (T2DM).
Hypermetabolism is one of the factors suggested to be responsible for the protection of patients with SCA against diseases such as DM.[10,11] This is usually assessed by determining the BMR, the rate of energy expenditure at rest in a thermally neutral environment and postabsorptive state. BMR reflects the energy requirements to maintain and conduct normal metabolic activity of muscle, brain, liver, kidney, and other organs.[14] It has been used as a measure of physiological function with increased level documented in adults, adolescents, and children with SCA.[8,15] This hypermetabolism was attributed to the chronic illness, anemia, increased cardiac workload, hyperactive erythropoiesis, increased protein turnover, inflammation, and oxidative stress associated with SCA.[16]
At present, there is little information on the possible effect of BMR on the steady state glycemic control in patients with SCA, this study, therefore, determined the BMR and factors involved in glycemic control in adults with SCA in steady state.
Materials and Methods
The study was approved by the University of Ibadan/University College Hospital Joint Ethics Review Committee (UI/EC/15/0123). It was explained to all the study participants and written informed consent was obtained before the commencement of the study. Fifty participants were enrolled into this case–control study using convenient sampling. They comprised of 30 adults with SCA in steady state and 20 age- and gender-matched apparently healthy adults with HbAA genotype that served as controls. Steady state was defined as earlier reported as absence of acute complicating factors or acute clinical symptoms or crisis for at least 3 months.[4] Having confirmed the genotype of the participants with hemoglobin electrophoresis, individuals whose genotypes were not HbAA and HbSS were excluded from the study. In addition, pregnant and lactating mothers as well as participants with DM, hypertension, human immunodeficiency virus, hepatitis, cancer, established endocrine dysfunctions, and those in vaso-occlusive crisis were excluded from the study.
Body weight, BMI, waist and hip circumferences (HCs), waist–hip ratio, blood pressure, and liver span were determined using standard methods. After an overnight fast of about 10 h, 10 ml of venous blood was obtained, and thereafter, plasma and serum were appropriately obtained and kept at −20°C until time of analysis.
Serum levels of insulin, CRP and ferritin were determined using ELISA (Genway Biotechnology, USA and Wkea Med Supplies Corp, China) following the manufacturers' instructions. The plasma glucose level was determined using the glucose oxidase method.
Insulin sensitivity indices and beta-cell function were calculated as – (a) computer-based homeostatic model assessment (HOMA) index of insulin sensitivity (HOMA2-S%) and computer-based HOMA index of beta-cell function (HOMA2-B%), were calculated using homeostasis model assessment-2 (HOMA-2) calculator (www.dtu.ox.ac.uk/homa); (b) homeostasis model assessment-estimated IR (HOMA-IR) was calculated as the product of fasting insulin (μU/ml) and fasting glucose (mmol/l) divided by 22.5; (c) quantitative insulin sensitivity check index (QUICKI) was calculated as 1/[log fasting insulin(μU/ml) + log fasting glucose (mg/dl)]; (d) 1/FI was calculated as the reciprocal of fasting insulin; (e) fasting IR index (FIRI) was calculated as (fasting glucose × fasting insulin)/25; and (f) glucose-insulin (G/I) ratio was calculated as the ratio of FPG concentration (mg/dl) to the concentration of insulin (U/ml).
BMR was calculated in participants with HbAA using the Harris-Benedict formula [17,18] and the Schofield equations.[19] Since standard equations have been shown not to be reliable in individuals with SCA,[20] the formula developed by Buchowski et al.[21] was used to determine BMR in the study participants with SCA.
Statistical analysis
Statistical analysis was done using SPSS version 20.0 (IBM Corporation, Armonk, New York, United States). Results are presented as mean ± standard error of mean. Differences between variables were determined using the independent Student's t-test while correlations between the variables were determined using the Spearman's correlation. P < 0.05 was considered as statistically significant.
Results
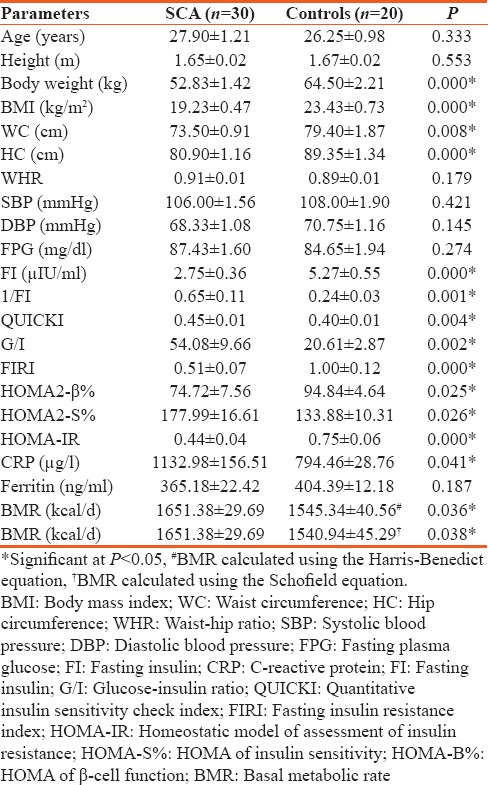
Table 1 shows the anthropometry, clinical and metabolic characteristics of the study participants. The mean body weight, BMI, waist circumference (WC), HC, FIRI, HOMA-IR, HOMA2-β%, and mean insulin level were significantly lower while the mean QUICKI, HOMA2-S%, 1/FI, G/I, CRP, and BMR were significantly higher in patients with SCA compared with the controls.
Table 1.
Anthropometry, clinical and metabolic characteristics of the study participants
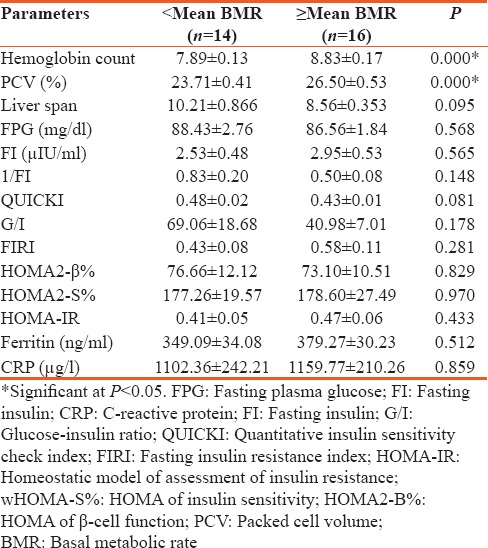
To understand the possible effect of BMR on the clinical and metabolic factors in patients with SCA, the mean value of the BMR was used to classify them into 2 groups: patients whose BMR values were less than the mean (<mean BMR) and patients whose BMR values were equal or greater than the mean BMR (≥mean BMR). Only hemoglobin count and packed cell volume (PCV) were significantly higher in SCA patients with ≥mean BMR compared with SCA patients with <mean BMR [Table 2]. The mean FPG and insulin levels and the mean values of indices of insulin sensitivity and secretion were not significantly different when the two groups were compared with each other.
Table 2.
Clinical and metabolic parameters in patients with sickle cell anemia based on the basal metabolic rate
As shown in Figures 1 and 2, BMR had no significant correlation with FPG (r = −0.077, P = 0.685) and HOMA-IR (r = −0.068, P = 0.722) in patients with SCA.
Figure 1.
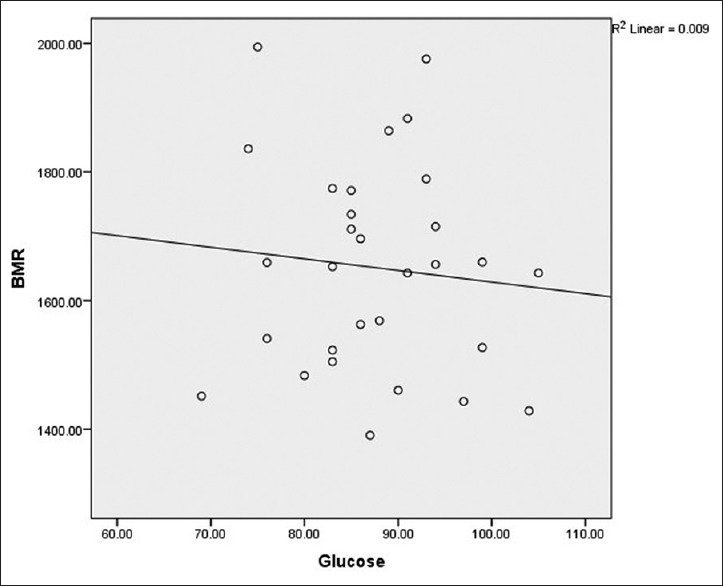
Correlation between basal metabolic rate and fasting plasma glucose in patients with sickle cell anemia
Figure 2.
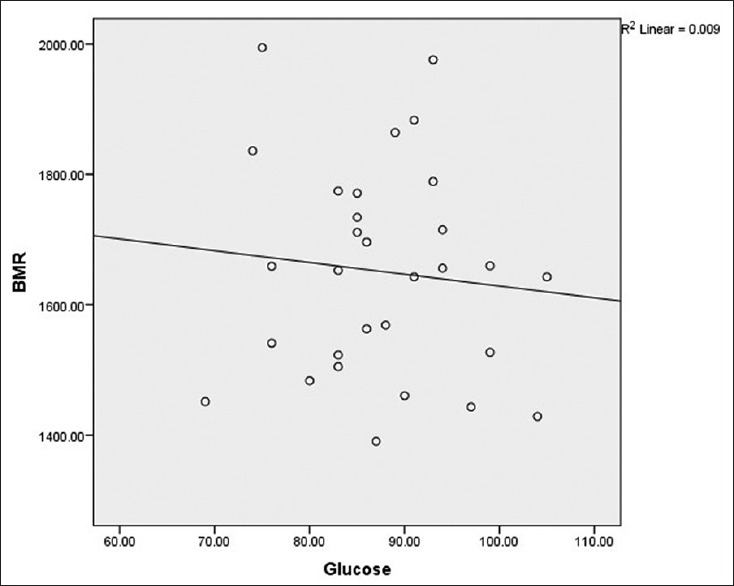
Correlation between basal metabolic rate and homeostatic model of assessment of insulin resistance in patients with sickle cell anemia
Discussion
SCA is characterized by low-grade chronic inflammation which is an important factor in induction of IR and β-cell dysfunction in patients with obesity and T2DM.[22,23] Despite this established SCA-associated inflammation even, in steady state, the prevalence of DM in SCA remains very low [9,24] and factors responsible for this are presently not yet identified.
The observed significantly lower insulin level in SCA compared with the controls contradicts the report of Alsultan et al.[12] but is in line with our earlier report.[13] This observation might suggest that the main role of β-cell, which is to synthesize and secrete insulin, is impaired in patients with SCA. This suggestion is further alluded to by the observed significantly lower HOMA2-β% in patients with SCA. HOMA2-β% is a measure of β-cell activity which reflects insulin secretion but not of β-cell health or pathology.[25] The reduction in β-cell activity and insulin secretion could be explained by the observed elevated CRP in patients with SCA. Inflammation has been shown to be involved in early induction and amplification of immune assault against β-cells causing suppression of their functions and apoptosis of the cells.[26,27]
A number of indices derived from the steady-state and dynamic testing have been validated for the assessment of insulin sensitivity and resistance during DM. The observed better insulin sensitivity, exhibited by the observed significantly higher 1/FI, G/I, QUICKI, and HOMA2-S% values as well as lower FIRI and HOMA-IR value, in patients with SCA compared with controls might indicate that patients with SCA in steady state enjoy some form of protection against IR. This better insulin sensitivity could be a compensatory liver and peripheral sensitivity due to the observed diminished activity of β-cell with its attendant low insulin secretion.
Elevated BMR has been documented in individuals with SCA. Our observed significantly higher BMR in SCA compared with the controls supports the reports of Borel et al.[8] and Barden et al.[15] The inclusion of BMR in this study was sequel to our earlier report [13] where it was observed that FPG although within the reference range, in patients with SCA was lower compared with the controls. We, therefore, sought to know the possible effect of BMR on glucose homeostasis in adults with SCA. As observed in this study, BMR had no significant effect on the levels of glucose and insulin and IR status in patients with SCA. Our earlier observed lower FPG in SCA [13] was also not observed in this present study as the mean FPG levels were similar in SCA and controls.
The observed lower body weight and BMI in SCA compared with controls support the report of Alsultan et al.[12] This observation could be due to reduced nutrients intake possibly, as a result of anorexia resulting from SCA comorbidities and increased nutrients requirements/utilization due to elevated BMR.[15,28,29] These factors could also explain the significantly lower WC, HC, and waist–hip ratio in SCA compared with controls. The observed significantly higher hemoglobin count and PCV in patients with elevated BMR might be consequent to poor tissue oxygenation which in turn stimulates the production of erythropoietin resulting in increased production of red blood cells by the bone marrow.[30,31] Our observation indicates that tissue oxygenation status in patients with SCA could be predicted using estimated BMR.
Small sample size was a major limitation of this study. Therefore, a large population study is suggested to confirm the findings of this study.
Conclusion
It could be concluded from this study that adults with SCA have better insulin sensitivity status but impaired β-cell activity when compared with adults with HbAA. In addition, BMR does not have any pronounced effect on glycemic and insulin sensitivity status in adults with SCA in steady state. Therefore, adults with SCA do not seem to be more prone to developing T2DM despite heightened inflammation associated with the disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors would like to appreciate the understanding and cooperation of the patients with SCA who participated in the study. The contribution of the entire resident Doctors of the Department of Haematology, University College Hospital, Ibadan is also appreciated.
References
- 1.Hand L. Sickle Cell Treatment Guideline Released. Medscape. 2014. [Last accessed on 2017 Mar 17]. Available from: http://www.medscape.com/viewarticle/831603.
- 2.Frenette PS, Atweh GF. Sickle cell disease: Old discoveries, new concepts, and future promise. J Clin Invest. 2007;117:850–8. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–20. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akinlade KS, Atere AD, Olaniyi JA, Rahamon SK, Adewale CO. Serum copeptin and cortisol do not accurately predict sickle cell anaemia vaso-occlusive crisis as C-reactive protein. PLoS One. 2013;8:e77913. doi: 10.1371/journal.pone.0077913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39:1005–29. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 6.Fung EB, Harmatz PR, Lee PD, Milet M, Bellevue R, Jeng MR, et al. Increased prevalence of iron-overload associated endocrinopathy in thalassaemia versus sickle-cell disease. Br J Haematol. 2006;135:574–82. doi: 10.1111/j.1365-2141.2006.06332.x. [DOI] [PubMed] [Google Scholar]
- 7.Okafor L, Osamo N. Pancreatic function in sickle cell anaemia. West Afr J Med. 1982;1:9–12. [Google Scholar]
- 8.Borel MJ, Buchowski MS, Turner EA, Goldstein RE, Flakoll PJ. Protein turnover and energy expenditure increase during exogenous nutrient availability in sickle cell disease. Am J Clin Nutr. 1998;68:607–14. doi: 10.1093/ajcn/68.3.607. [DOI] [PubMed] [Google Scholar]
- 9.Morrison JC, Schneider JM, Kraus AP, Kitabchi AE. The prevalence of diabetes mellitus in sickle cell hemoglobinopathies. J Clin Endocrinol Metab. 1979;48:192–5. doi: 10.1210/jcem-48-2-192. [DOI] [PubMed] [Google Scholar]
- 10.Reid HL, Ene MD, Photiades DP, Famodu AA. Insulin-dependent diabetes mellitus in homozygous sickle-cell anaemia. Trop Geogr Med. 1990;42:172–3. [PubMed] [Google Scholar]
- 11.Mohapatra MK. Type 1 diabetes mellitus in homozygous sickle cell anaemia. J Assoc Physicians India. 2005;53:895–6. [PubMed] [Google Scholar]
- 12.Alsultan AI, Seif MA, Amin TT, Naboli M, Alsuliman AM. Relationship between oxidative stress, ferritin and insulin resistance in sickle cell disease. Eur Rev Med Pharmacol Sci. 2010;14:527–38. [PubMed] [Google Scholar]
- 13.Akinlade K, Adewale C, Fasola F, Rahamon S, Dada V. Indices of insulin sensitivity and oral disposition index in adult Nigerians with sickle cell anaemia: A pilot study. BJMMR. 2014;4:4972–81. [Google Scholar]
- 14.Sardesai VM, editor. Introduction to Clinical Nutrition. New York: Marcel Dekker; 1998. Fundamentals of nutrition; pp. 1–13. [Google Scholar]
- 15.Barden EM, Zemel BS, Kawchak DA, Goran MI, Ohene-Frempong K, Stallings VA, et al. Total and resting energy expenditure in children with sickle cell disease. J Pediatr. 2000;136:73–9. doi: 10.1016/s0022-3476(00)90053-2. [DOI] [PubMed] [Google Scholar]
- 16.Akohoue SA, Shankar S, Milne GL, Morrow J, Chen KY, Ajayi WU, et al. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res. 2007;61:233–8. doi: 10.1203/pdr.0b013e31802d7754. [DOI] [PubMed] [Google Scholar]
- 17.Harris JA, Benedict FG. A biometric study of human basal metabolism. Proc Natl Acad Sci U S A. 1918;4:370–3. doi: 10.1073/pnas.4.12.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris JA, Benedict FG. A Biometric Study of Basal Metabolism in Man. Publication 279. Washington, DC: Carnegie Institute of Washington; 1919. [Google Scholar]
- 19.Schofield WN. Predicting Basal metabolic rate, new standards and review of previous work. Hum Nutr Clin Nutr. 1985;39(Suppl 1):5–41. [PubMed] [Google Scholar]
- 20.Kopp-Hoolihan LE, van Loan MD, Mentzer WC, Heyman MB. Elevated resting energy expenditure in adolescents with sickle cell anemia. J Am Diet Assoc. 1999;99:195–9. doi: 10.1016/S0002-8223(99)00047-4. [DOI] [PubMed] [Google Scholar]
- 21.Buchowski MS, Chen KY, Byrne D, Wang WC. Equation to estimate resting energy expenditure in adolescents with sickle cell anemia. Am J Clin Nutr. 2002;76:1335–44. doi: 10.1093/ajcn/76.6.1335. [DOI] [PubMed] [Google Scholar]
- 22.Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: A comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation. 2005;111:1448–54. doi: 10.1161/01.CIR.0000158483.13093.9D. [DOI] [PubMed] [Google Scholar]
- 23.van Greevenbroek MM, Schalkwijk CG, Stehouwer CD. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: Causes and consequences. Neth J Med. 2013;71:174–87. [PubMed] [Google Scholar]
- 24.Reid HL, Photiades DP, Oli JM, Kaine W. Concurrent sickle cell disease and diabetes mellitus. Trop Geogr Med. 1988;40:201–4. [PubMed] [Google Scholar]
- 25.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–95. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 26.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 27.Khodabandehloo H, Gorgani-Firuzjaee S, Panahi G, Meshkani R. Molecular and cellular mechanisms linking inflammation to insulin resistance and beta-cell dysfunction. Transl Res. 2016;167:228–56. doi: 10.1016/j.trsl.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Fung EB, Malinauskas BM, Kawchak DA, Koh BY, Zemel BS, Gropper SS, et al. Energy expenditure and intake in children with sickle cell disease during acute illness. Clin Nutr. 2001;20:131–8. doi: 10.1054/clnu.2000.0367. [DOI] [PubMed] [Google Scholar]
- 29.Kawchak DA, Schall JI, Zemel BS, Ohene-Frempong K, Stallings VA. Adequacy of dietary intake declines with age in children with sickle cell disease. J Am Diet Assoc. 2007;107:843–8. doi: 10.1016/j.jada.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 30.Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol. 2010;299:F1–13. doi: 10.1152/ajprenal.00174.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27:41–53. doi: 10.1016/j.blre.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]