

Abstract
Atherosclerosis is the main cause of death in men and women. This so-called “hardening of the arteries” results from advanced atherogenesis, the accumulation and death of subendothelial fat-laden macrophages (vascular plaque). The macrophages are attracted as the result of signals from injured vessels recruiting and activating cells to quell the injury by inflammation. Among the recruited cells are circulating monocytes that may be captured by the formation of neural cell adhesion molecule (nCAM) tethers between the monocytes and vascular endothelium; the tethers are dependent on electrostatic binding between distal segments of apposed nCAM molecules. The capture of monocytes is followed by their entry into the subendothelial area as macrophages, many of which will remain and become the fat-laden foam cells in vascular plaque. Neural cell adhesion molecules are subject to sialylation that blocks their electrostatic binding. We showed that estradiol-induced nCAM sialylases are present in vascular endothelial cells and tested whether sex steroid pretreatment of human vascular endothelium could inhibit the capture of monocytes. Using in vitro techniques, pretreatment of human arterial endothelial cells with estradiol, testosterone, dehydroepiandrosterone and dihydrotestosterone all induced sialylation of endothelial cells and, in a dose–response manner, reduced the capture of monocytes. Steroid hormones are protective against atherogenesis and its sequellae. Sex steroid depletion is associated with atherosclerosis. Based on this knowledge plus our results using sex steroid pretreatment of endothelial cells, we propose that the blockade of the initial step in atherogenesis by sex steroid-induced nCAM sialylation may be crucial to hormonal prevention of atherosclerosis.
Keywords: sex hormones, atherogenesis, prevention, neural cell adhesion molecule, estradiol, cardioprotection, menopause
Cardiovascular disease (CVD) is the leading cause of death in both sexes. Among American women, 30% to 40% die of CVD or its complications. Prior to the onset of menopause, women have a lower incidence of CVD than men; symptomatic CVD (angina and infarction) appear about a decade earlier in men than women.1 After menopause, at which time women become estrogen depleted, the CVD rates become similar to those of men.2 Additionally, women with premature menopause or premature ovarian failure who are not treated with estrogen have a well-documented increased rate of CVD and CVD-related death.3–8
Since coronary events usually follow the cessation of ovarian function in women, it has long been supposed that sex steroids, particularly estrogen, are cardioprotective, and that approximately 10 years were needed to develop clinical disease.9 Observational studies support this conclusion, and we know menopausal women who are treated with hormone replacement therapy within the first several years of menopause onset showed improvement in cardiovascular markers of disease.3,10–16 Age-adjusted subanalysis and long-term follow-up of the estrogen-only arm of the Women’s Health Initiative (WHI) hormone treatment study and other prospective observational studies on women who received conjugated estrogens alone have confirmed that estrogen-treated postmenopausal women have a slower thickening of their internal carotid artery intimal–medial (CIMT), indicating slower vascular plaque formation17; a recent prospective randomized trial of estrogen administration to menopausal women has confirmed that estrogen slows the progression of CIMT.13
Today, we know that prescient studies from Thomas Clarkson’s laboratory explain almost all aspects of the turbulent decades since the first announcement of the WHI’s hormonal cardiovascular prevention trials. The WHI participants were between 50 and 79 years of age, averaging 63 years of age, and ∼12 years postmenopausal. They received 5 or more years of treatment, and the older ones suffered cardiovascular harm, stopping the study.18 Clarkson and colleagues performed elegant and convincing studies in monkeys, showing animals that received human-equivalent doses of estrogen after oophorectomy developed less arterial plaque than those that didn’t, even in the setting of an atherogenic diet.14 However, plaque once developed could not be diminished by estrogen treatment. These studies gave rise to the “timing hypothesis” which postulates that the beneficial effects of hormone replacement therapy, specifically exogenous estrogen, on the progression of atherosclerosis are limited to women who start therapy within several years of menopause, and that if the treatment is delayed until there is underlying disease, the cardioprotective effect is lost.13–15,19,20 The lack of reversibility indicates that prevention of atherogenesis is crucial to prevention of atherosclerosis. In this paper, we propose that such prevention may be provided by sex-steroid blockade of the capture of monocytes by injured blood vessels.
Myriad studies have shown that CVD during the menopausal years is also related to the classic cardiovascular risk factors: elevated lipids, hypertension, lifestyle, diet, metabolic syndrome, and smoking that parallel aging and menopause. Because of space constraints, the specific effects of neither these factors nor their interrelationship with estrogen will be examined in this article.1,4,21–23 However, these risk factors are neither limited to the tight control of sex steroids nor acutely contributing to the accumulation of atherogenic plaque.
While estrogen plays a supporting role in regulating lipids and so on, it is possible that a more direct relationship between estrogen and atherogenesis exists. Specific mechanisms by which estrogen prevents CVD have become increasingly known. Working with the Clarkson group, we demonstrated that human and monkey artery endothelium expresses the enzyme estrogen synthetase (aromatase) and has estrogen receptor immunoreactivity.24 Following the precept that direct vascular action by estrogen could play a role in cardioprotection, we have turned to studying the effect of estrogen and other sex steroids on the initial inflammatory-vascular events that lead to atherogenesis.
The majority of all deaths from CVD are secondary to infarctions/thromboses caused by dislodged atherosclerotic vascular plaque or directly by plaque occlusion of vessels. The accumulation of plaque is a disease of the arterial wall that starts with vascular/perivascular inflammation and is characterized by the formation of lipid-laden plaques beneath the endothelium. This is triggered by subendothelial low-density lipoprotein accumulation which results in endothelial cell activation and chronic inflammation. Hypertensive pulses may also traumatize blood vessels. Cytokine signals (interleukin 1, tumor necrosis factor α) from the injured vascular endothelium recruit circulating monocytes. The activated monocytes adhere to the endothelial cells and penetrate into the intimal layer of the vessel where they transform to macrophages and eventually become foam cells that form plaque.1,25–30
The Role of Sex Steroids in Blocking Monocyte Tethering Capture
Cell adhesion is a general phenomenon that is critical to the activities of all cells. The molecules P-selectin, vascular cell adhesion molecule 1, intercellular adhesion molecule 1, neural cell adhesion molecule (nCAM), and P-selectin glycoprotein ligand 1 have proven to be important in the monocyte recruitment and plaque formation in particular.24–26,30–33 Knowing the importance of monocyte recruitment, we questioned whether atherosclerotic lesions are the result of an overzealous response by “first responders” such as monocytes that could be modulated by estrogen. We already had described ways that estrogen could sabotage the avid responses by estrogen-sensitive immunocytes in the brain.34 However, an even more immediate protective effect could be obtained by avoiding the capture of immunocytes by endothelial cells. Therefore, we specifically investigated the ability of nCAM molecules to achieve the electrostatic bonds necessary to capture monocytes in the face of administered estrogen and other sex steroids.
The basis of cell-to-cell adhesion is the physical attachment of complementary zones of oppositely axial adhesion molecules termed “zippering.” For example, nCAM domains protruding from the membranes of cells that are in proximity become zippered and form a tether for continued intercellular interactions.33 The nCAM-nCAM binding can be prevented by sialylation of nCAM.25,33,35,36 Sialic acid is a hydrophilic extracellular sugar that expands the volume of cellular glycocalyx and diminishes cell-to-cell interactions.37 Forming polysialylated nCAM (PSA-nCAM) prevents the usual electrostatic bonding between nCAMs and the formation of “tethers” between cells (see Figure 1).33,36
Figure 1.

A, Schema illustrating the domains (left) and the posttranslational modifications (right) of nCAM protein. B, Molecular structure of the 3 nCAM isoforms. C, The current model for nCAM interactions includes nCAM cis-dimers interacting in 2 manners, 1 of which results in the binding between cells; the “flat zipper” interaction (green circle). ECD indicates extracellular domain; F3I/II, fibronectin type 3 homology domain I/II; ICD, intracellular domain: IgI-V, Ig-like domain I-V; TMD, transmembrane domain; nCAM, neural cell adhesion molecule. Modified from Gascon, Vutskits, and Kiss33 (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
Monocyte capture from the circulation required nCAM-nCAM tethers that are dependent on the electrostatic forces and proximity. And it was known that nCAM-nCAM binding can be prevented by sialylation of nCAM.25,33,36 We demonstrated that the vascular endothelium expresses PSA-nCAM and demonstrated the presence of the sialylation enzymes in vascular endothelium.18,35 We also showed that these sialylation enzymes are induced by estradiol, as demonstrated in Figures 2 to 4.35 These factors are all consistent with our previous finding that human and monkey vascular endothelium from both males and females could produce estrogen and expressed estrogen receptors.24
Figure 2.
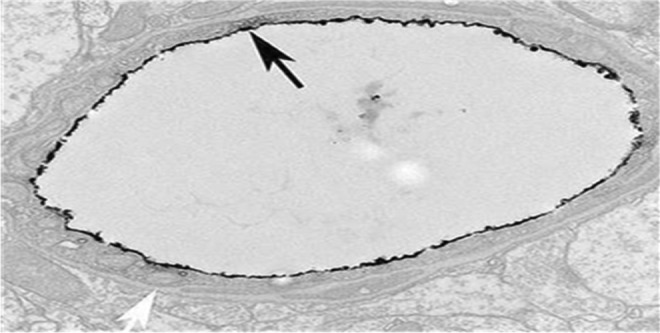
A cross-sectional view of a small arteriole in the rat brain. The black “rind” lining the vessel lumen is (ir-PSA-nCAM. Note its thickness and the full coverage of the lumen (black arrow) and the lack of staining in the abluminal compartment (white arrow). Electron microscope ×5000.18 ir-PSA-nCAM indicates immunoreactive polysialylated nCAM; nCAM, neural cell adhesion molecule.
Figure 3.

Immunostaining for PSA-nCAM of HUVECs grown in (A) no estrogen media, (B) estradiol (E2) 10−8 M, and (C) E2 + fulvestrant. The expression of ir-PSA-nCAM, dark cell staining, induced by estradiol is blocked by the antiestrogen. The E2 effect was dose related.35 HUVECs indicates human umbilical vein endothelial cells; ir-PSA-nCAM, immunoreactive polysialylated nCAM; nCAM, neural cell adhesion molecule.
Figure 4.
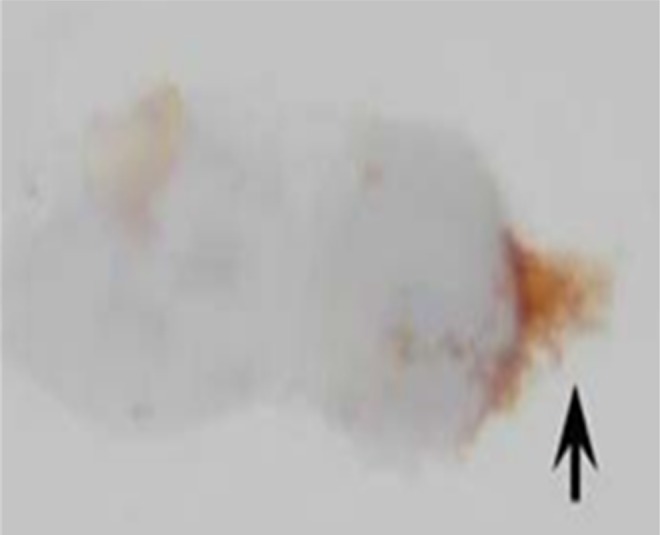
A HUVEC cultured in estrogen-containing medium and stained for ir-PSA-nCAM expression. The black arrow shows the extent of the extracellular domain of the molecule. LM ×100. Modified from Park et al35 HUVEC indicates human umbilical vein endothelial cells; ir-PSA-nCAM, immunoreactive polysialylated nCAM; nCAM, neural cell adhesion molecule.
Since we had shown that the necessary polysialylases exist in vessels and that they are regulated by estrogen, we next sought to abort nCAM-nCAM tethering and monocyte capture by inducing nCAM polysialylation with estradiol. Our hypothesis was that sex steroids can, by inducing PSA-nCAM, abort the electrostatic bonding that is the basis of the nCAM-nCAM tether. This could allow monocytes to successfully pass areas of inflammation without capture.
The experiments were accomplished by culturing a lawn of human arterial endothelial cells, pretreating with estradiol and other steroids, pouring monocytes (phorbol ester-treated THP1 cells) onto the lawn, washing after 90 minutes and then counting adherent monocytes.38 Selected results are shown in Figures 5 and 6. In all cases, the induction of PSA-nCAM was documented by histochemistry.38 Further studies testing monocyte pretreatment and shearing assays are continuing.
Figure 5.
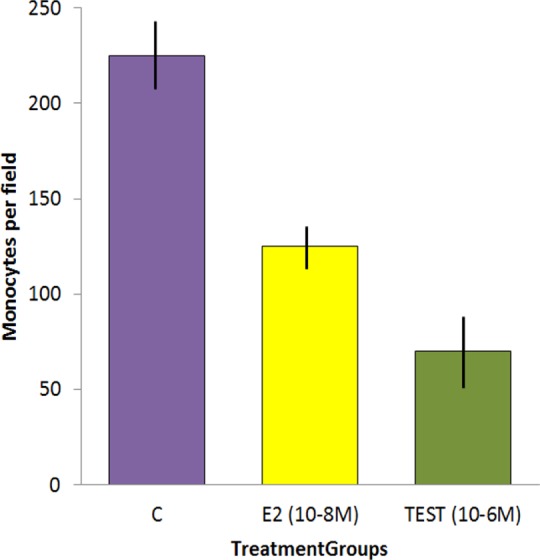
From Curatola et al,38 showing the result of incubating monocytes on a lawn of (female) human arterial endothelial cells (HAECs) and washing away the nonadherent cells after 90 minutes. The antiestrogen SERM fulvestrant blocked the E2 effect but not the testosterone effect. All incubations were performed in triplicate and repeated at least twice. Vertical units: number of monocytes per standard field.
Figure 6.

The application of estradiol, testosterone, and dihydrotestosterone all showed dose–response-related effects. Interestingly, pretreatment with the inflammatory cytokine TNFa did not, in itself, change the number of captured monocytes. TNFa indicates tumor necrosis factor α.
Conclusions
In addition to traditional effects on risk factors for atherogenesis, there is a case for direct effects of estrogen and other sex steroids on the process of atherogenesis. We have shown repeatable, dose-related induction of sialylases and PSA-nCAM in human vascular (arterial and umbilical) endothelial cells. Preincubation of human arterial endothelial cells with estrogen and other sex steroids diminished the number of captured monocytes on the endothelium. Pairing this finding with the observational studies supporting the timing hypothesis and clinical trials of estrogen’s effect on CIMT progression, estrogen’s ability to prevent monocyte adhesion via induction of PSA-nCAM offers a convincing molecular and physiologic explanation for a clinical phenomenon that has been observed for decades. While more study is required, it appears that these effects of sex steroids may break the very nexus of atherogenesis, as portrayed in Figure 7, thereby acting as a cardioprotective agent before the development of atherosclerotic disease.
Figure 7.
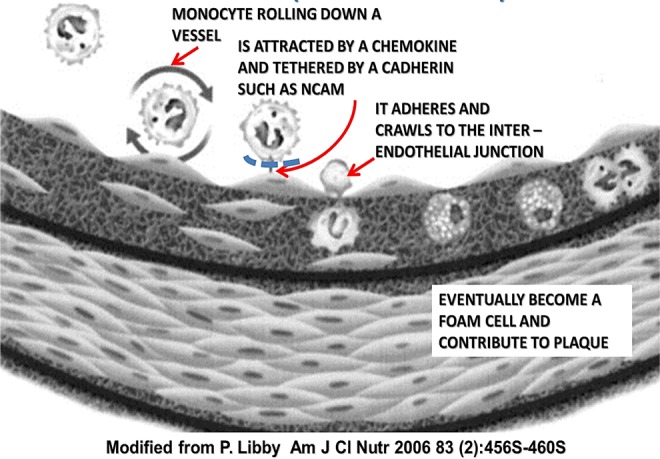
Sex steroid-induced nCAM sialylation blocks the initial step in atherogenesis. nCAM indicates neural cell adhesion molecule.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the NIH HL 796100.
References
- 1. Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308(5728):1583–1587. [DOI] [PubMed] [Google Scholar]
- 2. Tunstall-Pedoe H. Myth and paradox of coronary risk and the menopause. Lancet. 1998;351(9113):1425–427. [DOI] [PubMed] [Google Scholar]
- 3. Colditz GA, Willet WC, Stampfer MJ, Rosner B, Speizer FE, Hennekens CH. Menopause and the risk of coronary heart disease in women. N Engl J Med. 1987;316(18):1105–1110. [DOI] [PubMed] [Google Scholar]
- 4. Salpeter SR, Cheng J, Thabane L, Buckley NS, Salpeter EE. Bayesian meta-analysis of hormone therapy and mortality in younger postmenopausal women. Am J Med. 2009;122(11):1016–1022. [DOI] [PubMed] [Google Scholar]
- 5. Kalantaridou SN, Naka KK, Papanikolaou E, et al. Impaired endothelial function in young women with premature ovarian failure: normalization with hormone therapy. J Clin Endocrinol Metab. 2004;89(8):3907–3913. [DOI] [PubMed] [Google Scholar]
- 6. Perez-Lopez FR, Chedraui P, Gilbert JJ, Perez-Roncero G. Cardiovascular risk in menopausal women and prevalent related co-morbid conditions: facing the post-Women’s health initiative era. Fertil Steril. 2009;92(4):1171–1186. [DOI] [PubMed] [Google Scholar]
- 7. Stevenson JC, Crook D, Godsland IF. Influence of age and menopause on serum lipids and lipoproteins in healthy women. Atherosclerosis. 1993;98(1):83–90. [DOI] [PubMed] [Google Scholar]
- 8. Rosenberg L, Hennekens CH, Rosner B, Belanger C, Rothman KJ, Speizer FE. Early menopause and the risk of myocardial infarction. Am J Obstet Gynecol. 1981;139(1):47–51. [DOI] [PubMed] [Google Scholar]
- 9. Shufelt CL, Merz CN, Prentice RL, et al. Hormone therapy dose, formulation, route of delivery, and risk of cardiovascular events in women: findings from the Women’s Health Initiative Observational Study. Menopause. 2014;21(3):260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stampfer MJ, Willett WC, Colditz GA, Rosner B, Speizer FE, Hennekens CH. A prospective study of postmenopausal estrogen therapy and coronary heart disease. N Engl J Med. 1985;313(17):1044–1049. [DOI] [PubMed] [Google Scholar]
- 11. Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med. 2000;133(12):933–941. [DOI] [PubMed] [Google Scholar]
- 12. Manson JE. Postmenopausal hormone therapy and atherosclerotic disease. Am Heart J. 1994;128(6 pt 2):1337–1343. Review. [DOI] [PubMed] [Google Scholar]
- 13. Ge QS, Tian QJ, Tseng H, Naftolin F. Development of low-dose reproductive hormone therapies in China. Gynecol Endocrinol. 2006;22(11):110. [DOI] [PubMed] [Google Scholar]
- 14. Clarkson TB, Meléndez GC, Appt SE. Timing hypothesis for postmenopausal hormone therapy: its origin, current status, and future. Menopause. 2013;20(3):342–353. [DOI] [PubMed] [Google Scholar]
- 15. Harman SM, Black DM, Naftolin F, et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women in The Kronos Early Estrogen Prevention Study (KEEPS): a randomized controlled trial. Ann Intern Med. 2014;161(4):249–260. [DOI] [PubMed] [Google Scholar]
- 16. Hodis HN, Mack WJ, Shoupe D, et al. Testing the menopausal hormone therapy timing hypothesis: the early Vs late intervention trial with estradiol. Circulation. 2014;130(11):A13283. [Google Scholar]
- 17. Manson JE, Allison MA, Rossouw JE, et al. Estrogen therapy and coronary artery calcification. N Engl J Med. 2007;256(11):2591–2602. [DOI] [PubMed] [Google Scholar]
- 18. Tan O, Harman SM, Naftolin F. What can we learn from design faults in the women’s health initiative randomized clinical trial? Bull NYU Hosp Jt Dis. 2009;67(2):226–229. [PubMed] [Google Scholar]
- 19. Choi S, Steinberg E, Lee H, Naftalin F. The timing hypothesis remains a valid explanation of different cardioprotective effects of menopausal hormone treatment. Menopause. 2011;18(2):230–236. [PubMed] [Google Scholar]
- 20. Marjoribanks J, Farquhar C, Roberts H, Lethaby A. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev. 2012;11(7):CD004143. [DOI] [PubMed] [Google Scholar]
- 21. Garcia M, Miller VM, Gulati M, et al. Focused cardiovascular care for women: the need and role in clinical practice. Mayo Clin Proc. 2016;91(2):226–240. [DOI] [PubMed] [Google Scholar]
- 22. Appelman Y, van Rijn BB, Ten Haaf ME, Boersma E, Peters SA. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis. 2015;241(1):211–218. [DOI] [PubMed] [Google Scholar]
- 23. Matthews KA, Crawford SL, Chae CU, et al. Are changes in cardiovascular disease risk factors in midlife women due to chronological aging or to the menopausal transition? J Am Coll Cardiol. 2009;54(25):2366–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diano S, Horvath TL, Mor G, et al. Aromatase and estrogen receptor immunoreactivity in the coronary arteries of monkeys and human subjects. Menopause. 1999;6(1):21–28. [PubMed] [Google Scholar]
- 25. Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. [DOI] [PubMed] [Google Scholar]
- 26. Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res. 2015;116(2):307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104(3):365–372. [DOI] [PubMed] [Google Scholar]
- 28. Berliner JA, Territo MC, Sevanian A, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85(4):1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berliner JA, Navab M, Fogelman AM, et al. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91(9):2488–2496. [DOI] [PubMed] [Google Scholar]
- 30. Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. The combined role of P- and E-selectins in atherosclerosis. J Clin Invest. 1998;102(1):145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2000;191(1):189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galkina E, Ley E. Vascular adhesion molecules in atherosclerosis. Atherioscler Thromb Vas Biol. 2007;27(11):2292–2301. [DOI] [PubMed] [Google Scholar]
- 33. Gascon E, Vutskits L, Kiss JZ. Polysialic acid-neural cell adhesion molecule in brain plasticity: from synapses to integration of new neurons. Brain Res Rev. 2007;56(1):101–118. [DOI] [PubMed] [Google Scholar]
- 34. Mor G, Nilsen J, Horvath T, et al. Estrogen and microglia: a regulatory system that affects the brain. J Neurobiol. 1999;40(4):484–496. [DOI] [PubMed] [Google Scholar]
- 35. Park H, Pagan L, Tan O, et al. Estradiol regulates expression of polysialated neural cell adhesion molecule by human vascular endothelial cells. Reprod Sci. 2010;17(12):1090–1098. [DOI] [PubMed] [Google Scholar]
- 36. Kiss JZ, Rougon G. Cell biology of polysialic acid. Curr Opin Neurobiol. 1997;7(5):640–646. [DOI] [PubMed] [Google Scholar]
- 37. Rougon G. Structure, metabolism and cell biology of polysialic acids. Eur J Cell Biol. 1993;61(2):197–207. [PubMed] [Google Scholar]
- 38. Curatola AM, Huang K, Naftolin F. Dehydroepiandrosterone (DHEA) inhibition of monocyte binding by vascular endothelium is associated with sialylation of neural cell adhesion molecule. Reprod Sci. 2012;19(1):86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]