

Abstract
Suboptimal cellular conditions result in the accumulation of unfolded proteins in the endoplasmic reticulum (ER) and trigger ER stress. In this study, we investigated the effects of follicle stimulating hormone (FSH) on ER stress in granulosa cells (GCs) obtained from 3-week-old female C57BL6 mice 24 or 48 hours after intraperitoneal injection of 5 IU pregnant mare’s serum gonadotropin (PMSG), and in primary mouse GCs in culture treated with FSH (10-100 mIU/mL) for 24 or 48 hours. Moreover, mouse GCs in culture were treated with tunicamycin (Tm) or thapsigargin (Tp), which induce ER stress by inhibiting N-glycosylation of ER proteins and ER calcium adenosine triphosphatase, respectively, and their response to FSH was evaluated. We found that FSH attenuated ER stress in mouse GCs in vivo and in vitro; messenger RNA levels of ER stress-associated genes Xbp1s, Atf6, Chop, and Casp12 were decreased upon exposure to FSH/PMSG. Activating transcription factor 4 protein levels also demonstrated consistent decrease following FSH stimulation. Both Tm and Tp treatments inhibited FSH response, ER stress-induced cells did not show any change in estradiol levels in response to FSH, whereas in untreated GCs, estradiol production increased 3-fold after incubation with FSH for 60 hours. Furthermore, ER stress-induced cells failed to demonstrate aromatase (Cyp19a1) expression upon exposure to FSH. Importantly, under high-ER stress conditions FSH stimulation was unable to downregulate the expression of ER stress-associated genes. Our findings suggest that FSH decreases ER stress in GCs under physiologic conditions. However, under conditions that cause a significant increase in ER stress, FSH response is attenuated.
Keywords: FSH, granulosa cell, endoplasmic reticulum stress, unfolded protein response
Introduction
Protein synthesis is an essential process required for cell growth and survival. Proteins are synthesized in ribosomes that are either freely dispersed in the cytoplasm or attached to the endoplasmic reticulum (ER), forming the rough ER (RER). Secretory and transmembrane proteins are synthesized on the RER and enter the RER lumen to undergo folding and posttranslational modifications prior to being transported to the Golgi complex.1–3 Perturbations in these processes triggered by impairment of various cellular functions, such as redox regulation, ion homeostasis, protein degradation, autophagy, or by insults such as viral infections and nutrient deprivation may lead to the accumulation of unfolded proteins in the RER and trigger a cellular stress known as ER stress.4,5
Cells cope with ER stress via activation of the unfolded protein response (UPR), which aims to reestablish homeostasis.6–9 Several proteins that reside on the ER membrane participate in UPR, including protein kinase RNA-like ER kinase (PERK), inositol requiring enzyme-1 (IRE1α), and activating transcription factor 6 (ATF6). These proteins serve as sensors and trigger UPR in response to ER stress.2,10 During ER stress, PERK undergoes autophosphorylation and phosphorylates several target proteins including elongation initiation factor 2α (eIF2α). Phosphorylation of eIF2α leads to the attenuation of global protein synthesis,11,12 decreasing the workload of the ER to allow recovery. Interestingly, an important protein in UPR, activating transcription factor 4 (ATF4), is not affected by this global inhibition of translation.13 Another mediator of UPR, IRE1α, oligomerizes and autophosohorylates upon induction of ER stress.14,15 Following autophosphorylation, IRE1α unconventionally splices X-box binding protein-1 (XBP1) messenger RNA (mRNA) by removing a 26 base pair long intron; spliced XBP1 (XBP1s) encodes an active transcription factor.16,17 Another transcription factor, ATF6, translocates to the Golgi complex during ER stress and is cleaved into ATF6F, its active form, via serine site 1 and 2 proteases.18–20
Unfolded protein response-associated transcription factors (XBP1s, ATF4, and ATF6) translocate to the nucleus and mediate UPR via increasing their own expression and the expression of the downstream genes that promote protein folding, protein transport from ER to proteasomes for ER-associated degradation, and RNA degradation in a process referred to as IRE1-dependent decay of mRNAs (RIDD). In summary, UPR consists of a plethora of intracellular mechanisms including transcription and translation modulation, alternative splicing, (auto)phosphorylation, and protein and RNA decay that all together aim at reducing ER workload and improve protein folding.
Importantly, if the stress is chronic or severe, the transcription factors that mediate UPR trigger the expression of proapoptotic genes.21–24 CCAAT/enhancer-binding protein homologous protein (CHOP) is a proapoptotic molecule and harbors binding sites for all 3 major transcription factors of UPR (XBP1s, ATF4, and ATF6) in its promoter.8 The expression of CHOP is significantly upregulated during ER stress and increases the expression of proapoptotic genes, while downregulating antiapoptotic gene expression.25,26 Caspase-12, an ER membrane cysteine protease activated during prolonged ER stress, is thought to play a key role in mediating ER stress-induced apoptosis in rodents.27 Following prolonged ER stress, it is cleaved into active caspase-12 and activates downstream caspases, such as caspase-3 and caspase-9.28–30
Endoplasmic reticulum stress and its associated genes are implicated in glucose and lipid metabolism,31–34 pancreatic β-cell function,35–38 insulin and leptin resistance,39–41 liver disease,42–44 atherosclerosis, and Alzheimer disease.10,45,46 Emerging evidence also demonstrates that ER stress affects many aspects of follicle development and oocyte and embryo viability. Endoplasmic reticulum stress induction in mouse cumulus oocyte complexes (COCs) with thapsigargin (Tp), a well-studied agent that causes ER stress, increases Xbp1s, Atf4, and Atf6 expression and results in reduced vitro fertilization and poor embryo development.47 Mice fed high-fat diet demonstrate increased anovulation and decreased fertilization rates that can be explained by lipotoxicity-induced ER stress, which subsequently impairs mitochondrial function and increases apoptosis in cumulus and granulosa cells (GCs).48 Similarly, exposure to lipid-rich follicular fluid leads to increased levels of Atf4 and Atf6 mRNAs and impairs oocyte maturation in mouse COCs.49 Apoptotic cell death is the central mechanism underlying follicular atresia,50–54 and recent evidence demonstrates that ER stress is one of the mechanisms that triggers GC apoptosis. Pharmacological induction of ER stress in mouse GC culture increases the expression of Chop and Casp12 and leads to apoptosis.30 The levels of Atf4, Atf6, and Chop mRNA are higher in goat GCs from atretic follicles.55 These observations strongly suggest that conditions associated with high ER stress may promote cumulus/GC apoptosis and impair oocyte and embryo viability.
Since follicle stimulating hormone (FSH) promotes follicle viability, we hypothesized that FSH may reduce ER stress in GCs, an effect that could at least in part explain FSH’s antiapoptotic role. We investigated the effect of FSH on the expression of ER stress-associated genes and demonstrated that FSH downregulates the expression of ER stress-related genes in murine GCs. Importantly, we also found that excess ER stress inhibits FSH response of mouse GCs. Our findings indicate an intricate relationship between FSH responsiveness and cellular stress, and have implications for human fertility under normal and pathologic conditions.
Materials and Methods
Mouse Ovary and GC Collection
C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, Maine). Mice were bred and maintained according to the Yale University animal research requirements. Food and water were provided ad libitum and animals were housed under a 12-hour light–dark cycle. The Institutional Animal Care and Use Committee approval was obtained prior to the initiation of the study (protocol number 2011–11207).
To assess the effect of FSH in vivo on the whole ovary, 3-week-old female mice were intraperitoneally (ip) injected with 5 IU of pregnant mare’s serum gonadotropin (PMSG, Sigma-Aldrich, St Louis, Missouri). Mice were euthanized via CO2 inhalation 48 hours after injection, and ovaries were dissected in phosphate-buffered saline (PBS), cleaned from surrounding fat tissue, and stored at −80°C until RNA extraction. Respective controls of the same age did not receive PMSG injections. Eight mice were studied per group (stimulated and unstimulated) and both ovaries from each mouse were used for RNA extraction.
To collect mouse GCs, freshly dissected ovaries from unstimulated or PMSG-injected 3-week-old mice were incubated for 15 to 20 minutes in M-199 media containing 0.5 mol/L sucrose. Antral follicles were then punctured with 26.5-gauge needle under a dissecting microscope (Olympus SZH-ILLK, Japan) in M-199 medium, and GCs were separated from ovarian tissue using a 0.40-μm strainer and isolated by centrifugation at 1500g for 5 minutes. This method allows efficient collection of GCs from ovaries of young mice without the need for ovarian hyperstimulation.56,57 These cells were either used for RNA extraction or in vitro GC culture/FSH stimulation experiments.
For GC experiments, GCs from both ovaries of 2 different mice were pooled per sample. Four samples for each group were used for each experiment, and each experiment was repeated 4 times.
GC Culture, FSH Stimulations, and ER Stress Induction
For in vitro experiments, GCs from unstimulated 3-week-old mice were resuspended in Dulbecco’s modified Eagle medium (DMEM)/F12 media (Gibco, Carlsbad, CA, USA) supplemented with 5% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1% antimycotic-antibiotic (Gibco, Carlsbad, CA, USA). Cells were grown in 24-well plates at 37°C with 5% CO2 for 2 to 3 days, until they reached 50% to 70% confluency. Following 6 to 8 hours of serum starvation, cells were incubated with ovine FSH (National Hormone and Peptide Program, Harbor-UCLA, California) for 24 or 48 hours at 10, 30, and 100 mIU/mL concentration to reflect the serum levels achieved under physiological conditions and following controlled ovarian hyperstimulation in women undergoing infertility treatment.58
To induce ER stress, GCs in culture were treated with tunicamycin (Tm; 5 μg/mL; Sigma-Aldrich) or Tp (1.5 μg/mL; Sigma-Aldrich) for 24, 48, or 60 hours, and compared with controls treated with media only.
RNA Extraction, Reverse Transcription, Polymerase Chain Reaction, and Quantitative Real-Time Polymerase Chain Reaction
Total RNA was isolated from ovaries and GCs using Trizol (Invitrogen) according to the manufacturer’s instructions, dissolved in 50 µL of diethylpyrocarbonate–water, and kept at −80°C until use (500 µL of Trizol per ovary or well were used). Ovaries were homogenized in Trizol using a Kontes Pellet Pestle Motor (Fisher Scientific, Pittsburgh, Pennsylvania). The quality and concentration of the RNA were determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Rockford, Illinois). Two micrograms of total RNA were reverse transcribed to complementary DNA using oligo-dT priming at 37°C for 1 hour (Omniscript; Qiagen, the Netherlands).
Quantitative real-time polymerase chain reaction (qPCR) was used to detect the expression of Atf4, Atf6, Chop, Caspase-12, and Xbp1s (for the list of primers see Table 1). Primers used for Xbp1s detect only the spliced-active variant of Xbp1. Quantitative reverse transcription-PCRs were carried out on a CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, California). Complementary DNA was assayed in triplicate. Each 15 μL reaction contained 7.5 μL of SYBR Green supermix (Bio-Rad Laboratories), 0.4 μmol/L of each primer, and 1 μmol/L of cDNA (1:5 diluted). Amplifications were carried out using 40 cycles of PCR, in which the initial 5 minutes for denaturation at 94°C was followed by a touch-down program for 7 to 10 cycles of 92°C/20 s, 65 to 63°C/20 s (−1°C per cycle), and 72°C/30 s, and then 30 to 33 cycles of 92°C/20 s, 53 to 58°C/20 s, and 72°C/30 s. Expression of the target gene was normalized to β-actin levels. A standard curve for each set of primers was first used to determine the linear dynamic range of each reaction and PCR efficiency. A melting curve analysis was used to exclude nonspecific amplifications. The 2-ΔΔCT method was used to calculate relative gene expression.
Table 1.
The List of Primers Used for Endpoint and Quantitative RT-PCR.
| Gene | Primer Sequences (5′ to 3′; F; R) |
|---|---|
| Atf4 | F: TATGGATGATGGCTTGGCCAG R: TTCCAGGTCATCCATTCGAAAC |
| Atf6 | F: TCGAGGCTGGGTTCATAGACATG R: TACTGGACAGCCATCAGCTGAG |
| β-actin | F: TGCGTGACATCAAAGAGAAG R: CGGATGTCAACGTCACACTT |
| Casp12 | F: CTGGCTCTCATCATCTGCAACAA R: CGGCCAGCAAACTGCATTAAC |
| Chop | F: AGCTGGAAGCCTGGTATGAGGA R: AGCTAGGGACGCAGGGTCAA |
| Cyp19a1 | F: TGTGTTGACCCTCATGAGACA R: CTTGACGGATCGTTCATACTTTC |
| Xbp1s | F: GGTCTGCTGAGTCCGCAGCAGG R: GAAAGGGAGGCTGGTAAGGAAC |
Abbreviation: F, forward; R, reverse; RT-PCR, reverse transcriptase polymerase chain reaction.
End-point PCR was used to detect aromatase (Cyp19a1) expression in a 25 μL reaction mix containing 3 μL cDNA, 1X PCR buffer (Ambion, Carlsbad, CA, USA), 0.25 mmol/L of each deoxynucleotide triphosphate, 0.5 μmol/L of each primer, and 1 U of SuperTaq Polymerase (Ambion, Carlsbad, CA, USA). Polymerase chain reaction products were run on a 3% agarose gel and visualized by ethidium bromide staining. β-Actin was used as an endogenous control.
Western Blot Analysis
Granulosa cells in culture were washed in PBS several times and lysed in radioimmunoprecipitation assay buffer (sc-364162; Santa Cruz, Dallas, Texas) containing 1 mmol/L sodium orthovanadate and 1:100 protease inhibitor cocktail in dimethyl sulfoxide. Lysates were cleared by centrifugation at 12 000 × g for 30 minutes at 4°C and supernatant was stored at −80°C. Protein concentrations were determined using bicinchoninic acid assay (Thermo Scientific) with a Nanodrop 2000 spectrophotometer (Thermo Scientific). Lysates were mixed with Laemmli Sample Buffer (Bio-Rad Laboratories), boiled at 95°C for 5 minutes, and fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% Tris-hydrochloric acid Ready gels [Bio-Rad Laboratories]). Six micrograms of total protein per lane was used. Proteins were then transferred on to polyvinylidene fluoride membranes (Bio-Rad Laboratories), and membranes were blocked with 2% (w/v) nonfat dry milk powder in Tris-buffered saline and tween 20 mixture (TBS-T; American Bioanalytical, Natick, Massachusetts) for 1 hour at room temperature. Membranes were then incubated with rabbit polyclonal anti-ATF4 primary antibodies (ARP37017 [1:2500 dilution in 2% (w/v) nonfat dry milk powder in TBS-T]; Aviva, San Diego, California) overnight at 4°C. Following washes with TBS-T, membranes were incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (sc-2030; Santa Cruz) for 1 hour. Signals were detected using Supersignal West Pico Chemiluminescent substrate (Thermo Scientific). Glyceraldehyde-3-phosphate dehydrogenase was used for normalization (Cell Signaling Technology, Beverly, Massachusetts). The experiments were repeated at least 3 times. Films with different exposures were scanned, and band intensities were measured using ImageJ (National Institutes of Health [NIH], New York City, New York).
Estradiol Measurements
Mouse GCs were harvested and cultured as above in 24-well plates. Cells were serum starved for approximately 8 hours and then incubated with FSH (100 mIU/mL) in the presence of androstenedione (10−6 mol/L) and in the presence or absence of Tm (5 μg/mL; Sigma-Aldrich) or Tp (1.5 μg/mL; Sigma-Aldrich) for 60 hours. 17β-Estradiol levels in the media were determined using enzyme-linked immunosorbent assay (Cayman Chemical Co, Ann Arbor, Michigan) according to the manufacturer’s instructions. Measurements were performed in undiluted and 1:3 dilutions (with DMEM/F12 medium) of the cell culture supernatant.
Statistical Analysis
Unpaired t test or one-way analysis of variance with Tukey’s or Dunnett’s post tests were performed using GraphPad Prism software version 6.0 to compare 2 or more groups, respectively. Statistical significance was defined as P < .05.
Results
Follicle Stimulating Hormone Modulates the Expression of ER Stress-Associated Genes in Mouse Ovary and GCs
Expression of UPR genes Xbp1s, Atf4, and Atf6 in the whole ovary of mice treated with PMSG was determined by qPCR. We detected an approximately 2-fold decrease in the expression of Xbp1s in mouse ovaries after 48 hours of PMSG stimulation, whereas the expression of Atf4 and Atf6 did not change (Figure 1).
Figure 1.

Expression of endoplasmic reticulum (ER) stress-associated genes in mouse ovaries following pregnant mare’s serum gonadotropin (PMSG) stimulation. Three weeks old female mice were stimulated with PMSG. Ovaries were collected 48 hours later, total RNA was extracted, and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed for indicated genes. Data are the mean ± standard error of the mean (SEM). Asterisk indicates statistical significance (P < .05). Unpaired t test was used to determine statistical significance. Xbp1s indicates X-box binding protein (spliced); Atf4, activating transcription factor 4; Atf6, activating transcription factor 6.
Granulosa cells express FSH receptor (FSHR) and are one of the main targets of FSH action in the ovary. Therefore, we examined the expression of UPR genes in FSH stimulated mouse GCs. Ovaries were removed 24 or 48 hours after ip injection of 5 IU PMSG and GCs were isolated. At 24 hours, qPCR analysis showed significant decreases in the expression of Xbp1s, Atf6, Chop, and Caspase-12, approximately 2.5-, 1.5-, 2.5-, and 7-8-fold, respectively. Conversely, Atf4 expression was increased by 2-fold. Similarly, 48 hours after PMSG stimulation in vivo, Xbp1s, Atf6, Chop, and Caspase-12 demonstrated significant decreases in expression of about 2.5-, 1.5-, 2-, and 6 to 7-fold, respectively, whereas Atf4 levels did not change (Figure 2).
Figure 2.

Expression of endoplasmic reticulum (ER) stress-associated genes in mouse granulosa cells following pregnant mare’s serum gonadotropin (PMSG) stimulation. Three weeks old female mice were stimulated with PMSG. Ovaries were collected 24 and 48 hours later and granulosa cells were isolated. Total RNA was extracted from granulosa cells and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed for indicated genes. Data are the mean ± standard error of the mean (SEM). Column bars with asterisk are significantly different from control (P < .05). One-way analysis of variance (ANOVA) was used to determine statistical significance. Xbp1s indicates X-box binding protein (spliced); Atf4, activating transcription factor 4; Atf6, activating transcription factor 6; Chop, C/EBP homologous protein; Casp12, Caspase-12.
It is noteworthy that ovarian surface epithelium harbors stem cells, which express FSHR and respond to FSH.59–63 However, these cells were not independently assessed for ER stress response.
Follicle Stimulating Hormone Modulates the Expression of ER Stress-Associated Genes in Cultured Mouse GCs
To further explore the effects of FSH on ER stress-associated gene expression in mouse GCs, we isolated and cultured mouse GCs in vitro in the presence or absence of FSH. The Xbp1s, Atf6, and Caspase-12 transcripts demonstrated significant decrease, of about 2-fold each, after stimulation with 100 mIU/mL FSH for 24 or 48 hours. At lower concentrations, there was a trend toward reduction, but it did not reach statistical significance. The most robust results were obtained for Chop, which showed significant decrease in expression of approximately 2-fold with 30 mIU/mL FSH stimulation for 24 hours with 10, 30, and 100 mIU/mL FSH stimulation for 48 hours (Figure 3A-D). Although Atf4 mRNA levels did not demonstrate any change at any dose or time point, 100 mIU/mL FSH stimulation significantly downregulated ATF4 protein levels after 48 hours (Figure 3E and F).
Figure 3.

Expression of endoplasmic reticulum (ER) stress-associated genes in mouse granulosa cells treated with follicle stimulating hormone (FSH). A-E, Mouse granulosa cells in culture were treated with indicated doses of FSH for 24 or 48 hours. Total RNA was extracted and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed for ER stress associated genes. F, Mouse granulosa cells in culture were treated with 100 mIU/mL FSH for 48 hours. Activating transcription factor 4 (ATF4) Western blot band intensity is normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are the mean ± standard error of the mean (SEM). Gene names are as in Figure 2. Column bars with asterisk are significantly different from control (P < .05). One-way analysis of variance (ANOVA) was used to determine statistical significance of quantitative polymerase chain reaction (qPCR) experiments. Unpaired t test was used to determine statistical significance of the difference in ATF4 Western blot band intensity.
Induction of ER Stress Inhibits the Effect of FSH on the Expression of ER Stress Markers in Mouse GCs
After finding out that FSH can modulate the expression of ER stress-associated genes, we aimed to explore whether FSH is able to affect the expression of these genes under high ER stress conditions. In cultured GCs, ER stress was induced with Tm and the expression of ER stress-associated genes was assessed in the presence or absence of 100 mIU/mL FSH. As expected, Tm treatment successfully induced ER stress as demonstrated by the significant increase in expression of associated genes. However, addition of FSH did not have any effect on their expression following 24 or 48 hours of incubation (Figure 4).
Figure 4.

Expression of endoplasmic reticulum (ER) stress-associated genes in mouse granulosa cells treated with tunicamycin and follicle stimulating hormone (FSH). Mouse granulosa cells in culture were treated with tunicamycin alone or with tunicamycin and FSH for 24 or 48 hours. Total RNA was extracted and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed for ER stress associated genes. Data are the mean ± standard error of mean (SEM). Gene names are as in Figure 2 and 3. Tm: tunicamycin. Column bars with different letters on top are significantly different from each other (P < .05). One-way analysis of variance (ANOVA) was used to determine statistical significance.
Endoplasmic Reticulum Stress Inhibits FSH Response in Mouse GCs
Since FSH treatment failed to downregulate the expression of ER stress-associated genes under high-ER stress conditions, we hypothesized that high ER stress may interfere with the response to FSH in mouse GCs. We therefore tested how ER stress inducers Tm and Tp affect the ability of FSH to stimulate aromatase (Cyp19a1) expression and estradiol production by GCs in culture.
Follicle stimulating hormone treatment led to a 3-fold increase in estradiol production in mouse GCs following 60 hours of incubation (Figure 5B), while ER stress induction with Tm inhibited estradiol production in GCs treated with FSH (Figure 5A and B). Similarly, while aromatase expression was upregulated in GCs in response to FSH in the absence of Tm, this increase was completely obliterated by Tm treatment (Figure 5C). To demonstrate that the decrease in estradiol production and aromatase expression was caused by ER stress induction and was not due to nonspecific effects of Tm, we treated GCs with another ER stress inducer—Tp and evaluated the response to FSH. Similar to that observed in Tm-treated cells, Tp-treated GCs failed to upregulate aromatase expression and estradiol production in response to FSH (Figure 6A-C). Based on our observations, we propose a model highlighting the interaction between ER stress and FSH response in mouse GCs (Figure 7).
Figure 5.

The effect of endoplasmic reticulum (ER) stress induction with tunicamycin on granulosa cell follicle stimulating hormone (FSH) response. Mouse granulosa cells in culture were treated with tunicamycin and/or FSH for 60 hours. A, Total RNA was extracted and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed for ER stress associated genes. Data are the mean ± standard error of mean (SEM). B, Estradiol production was assessed by enzyme-linked immunosorbent assay (ELISA). Data are the mean ± standard error of the mean (SEM). C, Aromatase expression was assessed by PCR. Actin was used as a control. Gene names are as in Figures 2 and 3. Asterisks indicate statistical significance (P < .05). Unpaired t test was used to determine statistical significance of quantitative polymerase chain reaction (qPCR) and estradiol production experiments. Cyp19a1 indicates aromatase; E2, estradiol; N, negative control.
Figure 6.

The effect of endoplasmic reticulum (ER) stress induction with thapsigargin on granulosa cell follicle stimulating hormone (FSH) response. Mouse granulosa cells in culture were treated with thapsigargin and/or FSH for 60 hours. A, Total RNA was extracted and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed for ER stress associated genes. Data are the mean ± standard error of the mean (SEM). B, Estradiol production was assessed by enzyme-linked immunosorbent assay (ELISA). Data are the mean ± standard error of the mean (SEM). C, Aromatase expression was assessed by PCR. Actin was used as a control. Gene names are as in Figures 2, 3, and 5. Asterisks indicate statistical significance (P < .05). Unpaired t test was used to determine statistical significance of quantitative polymerase chain reaction (qPCR) and estradiol production experiments. Tp indicates thapsigargin.
Figure 7.
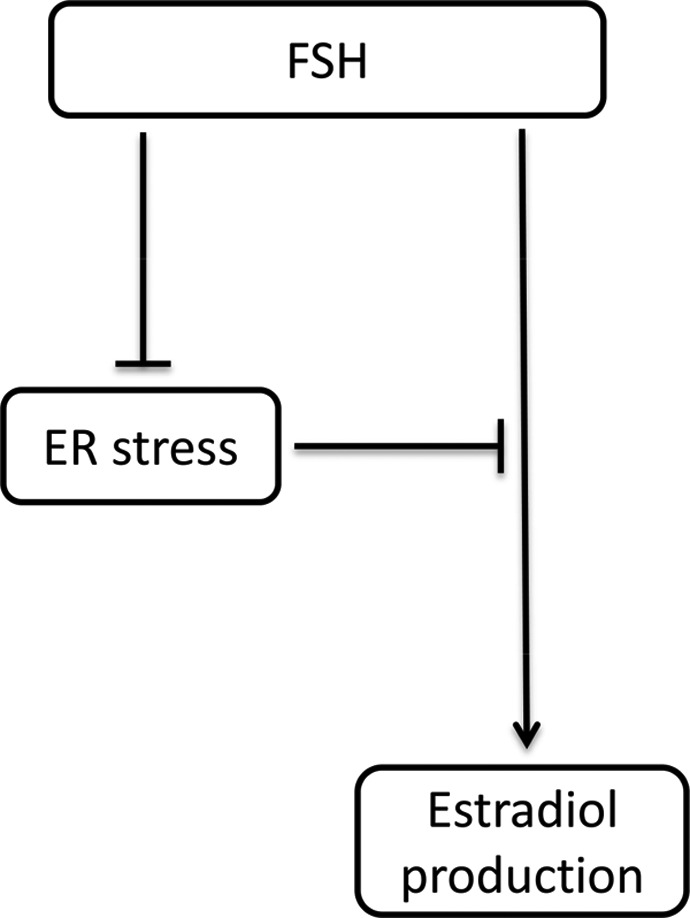
Schematic representation of observed interactions between follicle stimulating hormone (FSH) and endoplasmic reticulum (ER) stress in mouse granulosa cells. FSH attenuates ER stress and high ER stress inhibits FSH response in mouse granulosa cells.
Discussion
Protein synthesis, folding, modification, accurate cellular localization, and/or excretion are important metabolic processes, and their impairment may result in cell death. Endoplasmic reticulum stress is a perturbation in protein metabolism where the amount of unfolded protein exceeds the folding capacity of the ER. Cells utilize UPR to decrease ER stress and revert back to a homeostatic state. However, in cases of chronic or high levels of stress, UPR induces apoptosis.2,8,10,21 In recent years, emerging evidence demonstrated that ER stress is involved in the pathogenesis of many disorders, while our understating of the role of ER stress in reproductive disorders remains limited. In this study, we demonstrate that FSH attenuates ER stress in mouse GCs in vivo and in vitro. Conversely, ER stress induction inhibits FSH response and obliterates FSH’s effects on ER stress-associated gene expression in GCs.
In order to determine whether FSH affects ER stress in GCs, we chose targets and experimental procedures, which would reliably represent the ER stress status of the cell. The strength of our study is that we utilized multiple approaches to evaluate the role of FSH in modulating ER stress, both in vivo and in vitro, and used 2 different ER stress inducers. We studied 5 genes, 3 of these encode key transcription factors that mediate response to UPR, which is a consequence of ER stress, while 2 are important genes implicated in ER stress-associated apoptosis. These genes were all previously shown to be altered at the transcript level in reproductive tissues and/or GCs upon ER stress induction,30,47–49,55 so we tested them with qPCR. Atf4 is primarily regulated at the translational level during ER stress,13 and therefore, we did Western blotting for this target to complement our qPCR experiments. Four of these genes had reduced expression following PMSG treatment in vivo, with only Atf4 demonstrating increased transcript levels after 24 hours of stimulation. Moreover, the expression levels of all these 5 genes were reduced after FSH treatment in vitro: Xbp1s, Atf6, Casp12, and Chop at the transcript level and Atf4 at the protein level. Since all genes chosen as surrogates of ER stress status of the cell had altered expression, we concluded that FSH modulates ER stress in GCs. However, it is noteworthy that even if only some of these genes showed altered expression in response to FSH treatment, it could still be possible to conclude that FSH modulates ER stress. This is because many cell signaling pathways are complex networks with various key molecular players, and a certain treatment may affect some elements of a signaling pathway, while others remain unaltered. In other words, not all elements of a signaling pathway need to be dysregulated for that pathway to be referred as altered. This concept is an integral part of microarray pathway analysis algorithms, and there are a myriad of studies in the literature where a certain treatment (hormonal or other experimental intervention) affects some, but not all parts of the signaling cascade. As an example, in the liver, ER stress inhibitor tauroursodeoxycholic acid (TUDCA) alters the expression of 2 of the 3 ER stress pathways without affecting ATF6 expression.64 However, in the current study, all genes we have tested for had altered expression.
Our findings demonstrate that FSH downregulates the expression of ER stress-associated genes under basal conditions in vivo and in vitro. In accordance with our results, higher blastocyst formation rates in porcine embryos cultured in the presence of ER stress inhibitor TUDCA were reported.65 Moreover, TUDCA treatment resulted in a higher number of cells in the inner cell mass and trophectoderm, and decreased expression of proapoptotic genes, including caspase-3. The same study also reported that ER stress inhibition increases maturation rate of pig oocytes.65 Similarly, mouse embryos cultured in the presence of an ER stress inhibitor had higher cleavage and blastocyst formation rates, and increased cell number.66 These studies demonstrate that the attenuation of “baseline” ER stress is beneficial for embryonic development. Therefore, FSH’s antiapoptotic and growth-promoting role in follicles under normal physiological conditions and during controlled ovarian hyperstimulation67,68 might at least be partially explained by its ability to downregulate ER stress-associated genes.
We found that high ER stress inhibits FSH response in mouse GCs in culture. In a similar manner, it was previously demonstrated that ER stress affects receptor-mediated signaling cascades such as insulin and leptin signaling.39–41 The ER stress induction leads to increased serine phosphorylation of insulin receptor substrate 1, which results in downregulation of insulin receptor signaling and contributes to insulin resistance in hepatocytes and adipocytes, an effect mediated via IRE-1α activation of c-Jun N-terminal kinase.40 In addition, inhibition of ER stress with chemical chaperons increases insulin sensitivity.41 Endoplasmic reticulum stress induction also inhibits leptin signaling by inhibiting tyrosine phosphorylation of leptin receptor, Janus kinase (JAK2), and signal transducer and activator of transcription 3. Moreover, ER stress inhibitors act as leptin sensitizers.39
In the ovary, the main outcome of FSH signaling is upregulation of aromatase expression and estradiol production. We observed that estradiol production and aromatase expression in response to FSH were inhibited in GCs treated with Tm or Tp. Tunicamycin inhibits N-glycosylation of ER proteins and induces ER stress by inhibiting posttranslational modifications and folding.69 Thapsigargin induces ER stress by primarily inhibiting ER calcium (Ca2+)-adenosine triphosphatase (ATPase) and ER uptake of calcium, which affects the function of Ca2+-binding chaperones.70,71 In addition, Tp was also demonstrated to inhibit the fusion of autophagosomes with lysosomes, induce secretion of ER chaperones, and decrease protein levels of ER chaperone GRP78/BIP.72–74 These 2 ER stress inducers, which act through different mechanisms, resulted in suppression of FSH response, suggesting that the observed effect is caused by ER stress induction and is not due to nonspecific effects of these molecules. The inability of FSH to “reverse” ER stress or induce aromatase expression and estradiol production in Tm and/or Tp-treated cells can be explained by the inhibitory effects of high levels of stress on FSH signaling. Our findings suggest that there might be a threshold level of ER stress, above which FSH does not exert stress-reducing action.
Ovaries are the main source of estrogen in mammals. Androgens synthesized in ovarian theca cells are transported into GCs, where they are converted into estrogens by aromatase.75 Follicle stimulating hormone binding to FSH receptor (FSHR) upregulates the transcription of the CYP19A1 (aromatase) gene,76,77 which is regulated by tissue-specific promoters.78 In our experiments, reduced expression of aromatase and subsequent decrease in estradiol production could be due to the effects of ER stress on Cyp19a1 transcription.
Another possible mechanism responsible for suppression of FSH action in ER stress-induced GCs could be the inhibition of posttranscriptional modulation of protein expression. For example, overexpression of glucose-regulated protein 78 (GRP78), a heat shock protein upregulated during ER stress, in HEK-293 cells stably expressing FSHR, leads to decreased FSHR protein.79 We observed an at least 20-fold increase in Grp78 levels in ER stress-induced GCs (data not shown), which could lead to decreased FSHR levels and decreased FSH response.
Moreover, it is known that ER stress leads to the degradation of mRNAs via IRE1α activation (RIDD).22,23 During this process, the total amount of mRNA that is eventually translated into protein in the RER is reduced in order to decrease ER protein load. So et al have recently demonstrated that RIDD regulates lipid metabolism in liver cells by decreasing the expression of genes that regulate sterol biosynthesis.80 In a parallel study, Hur et al demonstrated that RIDD leads to the degradation of mRNAs of several members of the cytochrome P450 enzyme family in the liver.81 It is noteworthy that aromatase is also a member of the cytochrome P450 superfamily,78 and therefore, it is possible that aromatase mRNA is degraded via RIDD upon ER stress induction in GCs, leading to decreased aromatase expression and estradiol synthesis. However, the inhibitory effects of ER stress on FSH signaling upstream of aromatase expression and downstream of FSHR cannot be excluded. Moreover, it is possible that ER stress can lead to decreased FSH response by acting at multiple levels of the FSH signaling cascade, as in the case of insulin and leptin signaling.
In the ovary, the optimal response to FSH is regulated via a well-orchestrated temporal interplay of negative and positive feedback loops. During the follicular phase, FSH treatment of rat ovaries upregulates FSHR expression and presumably makes them more sensitive to FSH.82–84 In our study, FSH decreases baseline ER stress. Perhaps, this decrease in baseline ER stress in mouse GCs upon FSH treatment contributes to increased sensitivity of these cells to FSH. Our model highlights the interactions between ER stress and FSH in mouse GCs. Whether or not the same is true in human GCs and whether these interactions could be exploited for therapeutic benefits in reproductive disorders and/or in women undergoing IVF remain to be elucidated.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: E.S. was supported by award no. R01HD059909 from the NIH. M.D.L. was supported by the National Center for Research Resources (NCRR) and the National Center for Advancing Translational Science (NCATS; KL2 RR024138]). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
References
- 1. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86(4):1133–1149. [DOI] [PubMed] [Google Scholar]
- 2. Park SW, Ozcan U. Potential for therapeutic manipulation of the UPR in disease. Semin Immunopathol. 2013;35(3):351–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. [DOI] [PubMed] [Google Scholar]
- 4. Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica. 2012;2012:857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hampton RY. ER stress response: getting the UPR hand on misfolded proteins. Curr Biol. 2000;CB10(14):R518–R521. [DOI] [PubMed] [Google Scholar]
- 7. Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. [DOI] [PubMed] [Google Scholar]
- 8. Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013;18(5):537–546. [DOI] [PubMed] [Google Scholar]
- 9. Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101(5):451–454. [DOI] [PubMed] [Google Scholar]
- 10. Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. J Biol Chem. 2014;289(3):1203–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular Cell. 2000;5(5):897–904. [DOI] [PubMed] [Google Scholar]
- 12. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. [DOI] [PubMed] [Google Scholar]
- 13. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(31):11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (New York, NY). 2000;287(5453):664–666. [DOI] [PubMed] [Google Scholar]
- 15. Welihinda AA, Kaufman RJ. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J Biol Chem. 1996;271(30):18181–18187. [DOI] [PubMed] [Google Scholar]
- 16. Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. [DOI] [PubMed] [Google Scholar]
- 17. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kokame K, Kato H, Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J Biol Chem. 2001;276(12):9199–9205. [DOI] [PubMed] [Google Scholar]
- 20. Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275(35):27013–27020. [DOI] [PubMed] [Google Scholar]
- 21. Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108(12):2777–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science (New York, NY). 2006;313(5783):104–107. [DOI] [PubMed] [Google Scholar]
- 24. Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science (New York, NY). 2011;334(6059):1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. [DOI] [PubMed] [Google Scholar]
- 27. Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. [DOI] [PubMed] [Google Scholar]
- 28. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277(37):34287–34294. [DOI] [PubMed] [Google Scholar]
- 29. Vannuvel K, Renard P, Raes M, Arnould T. Functional and morphological impact of ER stress on mitochondria. J Cell Physiol. 2013;228(9):1802–1818. [DOI] [PubMed] [Google Scholar]
- 30. Yang Y, Lin P, Chen F, et al. Luman recruiting factor regulates endoplasmic reticulum stress in mouse ovarian granulosa cell apoptosis. Theriogenology. 2013;79(4):633–639 e631–633. [DOI] [PubMed] [Google Scholar]
- 31. Bailly-Maitre B, Belgardt BF, Jordan SD, et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem. 2010;285(9):6198–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mao T, Shao M, Qiu Y, et al. PKA phosphorylation couples hepatic inositol-requiring enzyme 1alpha to glucagon signaling in glucose metabolism. Proc Natl Acad Sci U S A. 2011;108(38):15852–15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang S, Chen Z, Lam V, et al. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012;16(4):473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460(7254):534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25(4):406–409. [DOI] [PubMed] [Google Scholar]
- 36. Gao Y, Sartori DJ, Li C, et al. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol. 2012;32(24):5129–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Molecular Cell. 2001;7(6):1153–1163. [DOI] [PubMed] [Google Scholar]
- 38. Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4(6):491–497. [DOI] [PubMed] [Google Scholar]
- 39. Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35–51. [DOI] [PubMed] [Google Scholar]
- 40. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Sci (New York, NY). 2004;306(5695):457–461. [DOI] [PubMed] [Google Scholar]
- 41. Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Sci (New York, NY). 2006;313(5790):1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kammoun HL, Chabanon H, Hainault I, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119(5):1201–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rutkowski DT, Wu J, Back SH, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15(6):829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang K, Wang S, Malhotra J, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30(7):1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5(9):781–792. [DOI] [PubMed] [Google Scholar]
- 46. Thorp E, Iwawaki T, Miura M, Tabas I. A reporter for tracking the UPR in vivo reveals patterns of temporal and cellular stress during atherosclerotic progression. J Lipid Res. 2011;52(5):1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu LL, Russell DL, Norman RJ, Robker RL. Endoplasmic reticulum (ER) stress in cumulus-oocyte complexes impairs pentraxin-3 secretion, mitochondrial membrane potential (DeltaPsi m), and embryo development. Mol Endocrinol (Baltimore, Md). 2012;26(4):562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu LL, Dunning KR, Yang X, et al. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology. 2010;151(11):5438–5445. [DOI] [PubMed] [Google Scholar]
- 49. Yang X, Wu LL, Chura LR, et al. Exposure to lipid-rich follicular fluid is associated with endoplasmic reticulum stress and impaired oocyte maturation in cumulus-oocyte complexes. Fertil Steril. 2012;97(6):1438–1443. [DOI] [PubMed] [Google Scholar]
- 50. Hsueh AJ, Billig H, Tsafriri A. Ovarian follicle atresia: a hormonally controlled apoptotic process. Endocr Rev. 1994;15(6):707–724. [DOI] [PubMed] [Google Scholar]
- 51. Jolly PD, Tisdall DJ, Heath DA, Lun S, McNatty KP. Apoptosis in bovine granulosa cells in relation to steroid synthesis, cyclic adenosine 3′,5′-monophosphate response to follicle-stimulating hormone and luteinizing hormone, and follicular atresia. Biol Reprod. 1994;51(5):934–944. [DOI] [PubMed] [Google Scholar]
- 52. Quirk SM, Cowan RG, Joshi SG, Henrikson KP. Fas antigen-mediated apoptosis in human granulosa/luteal cells. Biol Reprod. 1995;52(2):279–287. [DOI] [PubMed] [Google Scholar]
- 53. Tilly JL, Kowalski KI, Johnson AL, Hsueh AJ. Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology. 1991;129(5):2799–2801. [DOI] [PubMed] [Google Scholar]
- 54. Tilly JL, Kowalski KI, Schomberg DW, Hsueh AJ. Apoptosis in atretic ovarian follicles is associated with selective decreases in messenger ribonucleic acid transcripts for gonadotropin receptors and cytochrome P450 aromatase. Endocrinology. 1992;131(4):1670–1676. [DOI] [PubMed] [Google Scholar]
- 55. Lin P, Yang Y, Li X, et al. Endoplasmic reticulum stress is involved in granulosa cell apoptosis during follicular atresia in goat ovaries. Mol Reprod Dev. 2012;79(6):423–432. [DOI] [PubMed] [Google Scholar]
- 56. Campbell KL. Ovarian granulosa cells isolated with EGTA and hypertonic sucrose: cellular integrity and function. Biol Reprod. 1979;21(4):773–786. [DOI] [PubMed] [Google Scholar]
- 57. Carletti MZ, Fiedler SD, Christenson LK. MicroRNA 21 blocks apoptosis in mouse periovulatory granulosa cells. Biol Reprod. 2010;83(2):286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cedrin-Durnerin I, Massin N, Galey-Fontaine J, et al. Timing of FSH administration for ovarian stimulation in normo-ovulatory women: comparison of an early or a mid follicular phase initiation of a short-term treatment. Human Reprod (Oxford, England). 2006;21(11):2941–2947. [DOI] [PubMed] [Google Scholar]
- 59. Bhartiya D, Singh J. FSH-FSHR3-stem cells in ovary surface epithelium: basis for adult ovarian biology, failure, aging, and cancer. Reproduction (Cambridge, England). 2015;149(1):R35–R48. [DOI] [PubMed] [Google Scholar]
- 60. Bhartiya D, Sriraman K, Gunjal P, Modak H. Gonadotropin treatment augments postnatal oogenesis and primordial follicle assembly in adult mouse ovaries? J Ovarian Res. 2012;5(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parte S, Bhartiya D, Manjramkar DD, Chauhan A, Joshi A. Stimulation of ovarian stem cells by follicle stimulating hormone and basic fibroblast growth factor during cortical tissue culture. J Ovarian Res. 2013;6(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Patel H, Bhartiya D, Parte S, Gunjal P, Yedurkar S, Bhatt M. Follicle stimulating hormone modulates ovarian stem cells through alternately spliced receptor variant FSH-R3. J Ovarian Res. 2013;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Sriraman K, Bhartiya D, Anand S, Bhutda S. Mouse ovarian very small embryonic-like stem cells resist chemotherapy and retain ability to initiate oocyte-specific differentiation. Reprod Sci (Thousand Oaks, Calif.). 2015;22(7):884–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ben Mosbah I, Alfany-Fernandez I, Martel C, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang JY, Diao YF, Oqani RK, Han RX, Jin DI. Effect of endoplasmic reticulum stress on porcine oocyte maturation and parthenogenetic embryonic development in vitro. Biol Reprod. 2012;86(4):128. [DOI] [PubMed] [Google Scholar]
- 66. Zhang JY, Diao YF, Kim HR, Jin DI. Inhibition of endoplasmic reticulum stress improves mouse embryo development. PloS One. 2012;7(7):e40433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. Annu Rev Physiol. 1997;59:349–363. [DOI] [PubMed] [Google Scholar]
- 68. Chun SY, Billig H, Tilly JL, Furuta I, Tsafriri A, Hsueh AJ. Gonadotropin suppression of apoptosis in cultured preovulatory follicles: mediatory role of endogenous insulin-like growth factor I. Endocrinology. 1994;135(5):1845–1853. [DOI] [PubMed] [Google Scholar]
- 69. Mahoney WC, Duksin D. Biological activities of the two major components of tunicamycin. J Biol Chem. 1979;254(14):6572–6576. [PubMed] [Google Scholar]
- 70. Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266(26):17067–17071. [PubMed] [Google Scholar]
- 72. Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42(6):731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peters LR, Raghavan M. Endoplasmic reticulum calcium depletion impacts chaperone secretion, innate immunity, and phagocytic uptake of cells. J Immunol (Baltimore, Md.: 1950). 2011;187(2):919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rosengren V, Johansson H, Lehtio J, Fransson L, Sjoholm A, Ortsater H. Thapsigargin down-regulates protein levels of GRP78/BiP in INS-1E cells. J Cell Biochem. 2012;113(5):1635–1644. [DOI] [PubMed] [Google Scholar]
- 75. Findlay JK, Britt K, Kerr JB, et al. The road to ovulation: the role of oestrogens. Reprod Fertil Dev. 2001;13(7-8):543–547. [DOI] [PubMed] [Google Scholar]
- 76. Parakh TN, Hernandez JA, Grammer JC, et al. Follicle-stimulating hormone/cAMP regulation of aromatase gene expression requires beta-catenin. Proc Natl Acad Sci U S A. 2006;103(33):12435–12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Thompson EA, Jr, Siiteri PK. The involvement of human placental microsomal cytochrome P-450 in aromatization. J Biol Chem. 1974;249(17):5373–5378. [PubMed] [Google Scholar]
- 78. Bulun SE, Lin Z, Imir G, et al. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev. 2005;57(3):359–383. [DOI] [PubMed] [Google Scholar]
- 79. Kogure K, Nakamura K, Ikeda S, et al. Glucose-regulated protein, 78-kilodalton is a modulator of luteinizing hormone receptor expression in luteinizing granulosa cells in rats. Biol Reprod. 2013;88(1):8. [DOI] [PubMed] [Google Scholar]
- 80. So JS, Hur KY, Tarrio M, et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16(4):487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hur KY, So JS, Ruda V, et al. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209(2):307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. LaPolt PS, Tilly JL, Aihara T, Nishimori K, Hsueh AJ. Gonadotropin-induced up- and down-regulation of ovarian follicle-stimulating hormone (FSH) receptor gene expression in immature rats: effects of pregnant mare’s serum gonadotropin, human chorionic gonadotropin, and recombinant FSH. Endocrinology. 1992;130(2):1289–1295. [DOI] [PubMed] [Google Scholar]
- 83. Nakamura K, Minegishi T, Takakura Y, et al. Hormonal regulation of gonadotropin receptor mRNA in rat ovary during follicular growth and luteinization. Mol Cell Endocrinol. 1991;82(2-3):259–263. [DOI] [PubMed] [Google Scholar]
- 84. Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocrine reviews. 1997;18(6):739–773. [DOI] [PubMed] [Google Scholar]