

Abstract
Identification of modifiable risk factors is gravely needed to prevent adverse prostate health outcomes. We previously developed a murine precancer model in which exposure to maternal obesity stimulated prostate hyperplasia in offspring. Here, we used generalized linear modeling to evaluate the influence of additional environmental covariates on prostate hyperplasia. As expected from our previous work, the model revealed that aging and maternal diet-induced obesity (DIO) each correlated with prostate hyperplasia. However, prostate hyperplasia was not correlated with the length of maternal DIO. Cage density positively associated with both prostate hyperplasia and offspring body weight. Expression of the glucocorticoid receptor in prostates also positively correlated with cage density and negatively correlated with age of the animal. Together, these findings suggest that prostate tissue was adversely patterned during early life by maternal overnutrition and was susceptible to alteration by environmental factors such as cage density. Additionally, prostate hyperplasia may be acutely influenced by exposure to DIO, rather than occurring as a response to worsening obesity and comorbidities experienced by the mother. Finally, cage density correlated with both corticosteroid receptor abundance and prostate hyperplasia, suggesting that overcrowding influenced offspring prostate hyperplasia. These results emphasize the need for multivariate regression models to evaluate the influence of coordinated variables in complicated animal systems.
Keywords: prostate hyperplasia, cage overcrowding, maternal obesity, developmental programming, generalized linear modeling
Introduction
In 2014, prostate cancer was predicted to be the most frequently diagnosed male cancer in the United States.1 Unfortunately, preventative care is limited by a lack of known modifiable risk factors for this disease. Given that prostate development occurs many decades before disease presentation,2 we and others have suggested that environmental exposures during fetal and early childhood developmental stages may predispose precancerous prostate phenotypes that appear in late adult life.3–5
To test the idea that maternal nutritional state is one such exposure, we previously gave female C57Bl/6J mice ad libitum access to a high-sugar, high-fat diet beginning at 4 weeks of age. After at least 4 weeks, when the dams had diet-induced obesity (DIO), we mated them to control chow-fed males and maintained the mothers on the high-sugar, high-fat diet through gestation and weaning. Control mothers were given ad libitum access to control chow diet the entire time. After weaning we maintained the maternal control- and DIO-exposed offspring on chow until sacrifice at approximately 16, 26, or 63 weeks of age. After sacrifice, we examined the dorsolateral prostate (DLP) epithelial ductal tubules (denoted here as ducts) of the offspring for signs of hyperplasia. We found that, at 26 and 63 weeks of age, the prostates of DIO-exposed offspring were more likely to have hyperplastic ducts than were those of control-exposed offspring.3
Given that the offspring in our previous study was exposed to maternal DIO during early life (gestation until weaning), we hypothesized that other environmental exposures during early or adult life also influenced hyperplasia in prostates. Additionally, the mothers in the study were exposed to DIO for varying lengths of time before pregnancy. Thus, we did not know whether the length of time the mothers were obese before pregnancy influenced prostate hyperplasia in the adult offspring. However, it was impractical to perform a second exploratory 14-month long screen to identify additional potentially relevant environmental covariates and determine how long the mothers needed to be obese to predispose the offspring to prostate hyperplasia. Instead, we describe here statistical modeling of previously collected data to identify environmental exposures that influence prostate hyperplasia.
Bayesian statistical multivariate regression methodologies allow factors in the environmental milieu to be simultaneously considered for their combined effects on a given outcome. This approach has not traditionally been utilized in biological studies with genetically near-identical mice because the number of subjects is small and would limit such analysis. However, Nelder and Lee previously demonstrated that a Poisson (log-linear) generalized linear model (GLM) can be applied to “small n” studies.6 Here, a GLM provides evidence that the number of male littermates in a cage (cage density) contributes to prostate hyperplasia in mice. We additionally show that cage density correlates with glucocorticoid receptor (GR) protein abundance in DLP. Together the data suggest that overcrowding may contribute to murine prostate ductal hyperplasia.
Methods
Mouse Husbandry and Collection
The data used here were derived from a study that was performed with approval from the Washington University Animal Studies Committee, Animal Welfare Assurance protocol # A-3381-01. Methods for mouse husbandry and evaluation were as published previously.3 The 4-week-old C57Bl/6J female mice were purchased from The Jackson Laboratories (Bar Harbor, Maine) and housed 5 per cage in standard 12-hour light–dark conditions with ad libitum access to food and water. For each breeding cycle, 5 age-matched females were fed either control chow diet (PicoLab Rodent Diet 20; 13.2% of calories from fat) or standard high-sugar/high-fat diet (TestDiet, 58R3; 59.4% of calories from fat7; referred to here as DIO). Starting at 8 weeks of age, females were mated to age-matched males. Resulting offspring remained with their mothers until weaning (21 days) and then the male littermates were removed to individual cages of ≤5 animals and fed chow diet until killing. We analyzed data from each of the 54 mice of the following maternal diet exposures and age cohorts: (1) 10 control- and 8 DIO-exposed offspring, 116 to 144 days old (the approximately 16-week-old cohort); (2) 7 control- and 7 DIO-exposed offspring, 177 to 192 days old (the approximately 26-week-old cohort); and (3) 12 control- and 10 DIO-exposed offspring, 432 to 446 days old (the approximately 63-week-old cohort). At sacrifice, Urogenital Sinus (UGS) regions were removed en bloc to cold phosphate-buffered saline (PBS; pH 7.4) and DLP regions were disassociated (as previously described8,9). After fixing overnight in 4% paraformaldehyde, prostates were dehydrated to 70% ethanol and processed for paraffin-embedding overnight. The DLP were oriented with the ventral face down in paraffin. The 6-μm thick longitudinal sections were cut through the DLP (adult murine DLP exhibit morphological and proliferative heterogeneity in the distal and proximal regions of epithelial ducts10,11; this orientation allowed evaluation of both regions). Two planes were selected from each animal, 1 ventral and 1 dorsal, and planes were hematoxylin and eosin stained with standard methods (described in Benesh et al3). An expert prostate pathologist (Humphrey12,13) performed blinded evaluation of prostate ductal morphology.
Statistical Model
In our previous publication, mouse prostates were scored blindly for percentage (denoted here as “counts”) of hyperplastic ducts by an expert prostate pathologist.3 Here, these data were modeled as a function of a set of explanatory variables in a regression setting. These were stored in the vector y with individual counts denoted y i = 1, 54 for 54 cases. The explanatory variables were given in the 54 × 7 − matrix C, with a leading column of 1s and individual-level vectors denoted Ci for the ith mouse. The standard linear regression model was not appropriate because the outcome variable (hyperplasia counts) took on only nonnegative integer values. Therefore, a GLM14 with a Poisson (log-linear) link function was used to associate the expected counts on the left-hand side of the model with the standard linear-additive right-hand side treatment of the covariates:
where b was the estimated set of regression parameters and ei was the ith residual. Thus, the b values represented the expected change in the number of counts, given changes in the explanatory variables, although this relationship was nonlinear because of the log link function; the model mentioned earlier was equivalent to . There was mild evidence of overdispersion with this specification, but a negative binomial model gave almost identical results. A small number (3.1%) of the values were missing at random. Because deleting records (cases) with missing data can lead to biased model results,15,16 the missing values were imputed using multiple imputation with chained equations in R and the tools for discrete missing values.17 This gave a set of 10 fully filled-in data sets (standard for this method) that require replicated modeling and averaging of inferential results. See the review essay by Rubin18 or the text by Schafer19 for details.
Immunofluorescence
Prostate sections were processed in 3 batches by age range as described previously,20 and data collected were normalized to a negative (no primary antibody) control. Briefly, sections were deparaffinized, rehydrated, and boiled for 30 minutes in 10 mmol/L sodium citrate solution for antigen retrieval. Sections were permeabilized in 0.2% TritonX-100 in PBS for 15 minutes, blocked in 10% goat serum/5% bovine serum albumin in PBS, and incubated with antiglucocorticoid receptor-α (anti-GRα; Thermo Fisher Scientific, Waltham, MA; 1:250) overnight at 4°C. Slides were washed in PBS and incubated in Goat anti-Mouse IgG (H+L) Alexa Fluor 568 conjugated secondary antibody for 1 hour (Life Technologies, Grand Island, NY; 1:1000). Sections were counterstained in TOPRO-3 iodide-642 (Life Technologies, Grand Island, NY; 1:500) for 5 minutes and mounted with Prolong Gold antifade mounting medium (Life Technologies, Grand Island, NY).
Images were collected by confocal microscopy (Nikon D-Eclipse-C1 E800 [Melville, NY]; Nikon EZ-C1si image Capture software Bronze Version 3.80 [Melville, NY]). Prostate epithelium regions were selected, brightness/contrast adjustments were uniformly made, and pixel gray values were measured in Photoshop CS6 (Adobe Systems, San Jose, CA).
Results
Identifying Explanatory Variables of Prostate Hyperplasia With Statistical Modeling
We used a statistical method, GLM, to evaluate the relevant contributions of covariates to prostate hyperplasia. The outcome modeled by the statistical analysis was the count of the number of hyperplastic ducts per prostate. The 6 covariates evaluated were as follows:
Maternal diet: This dichotomous covariate indicated whether or not each mouse was exposed to maternal DIO (diet = 1) or control (diet = 0). Because we showed that this variable influences prostate hyperplasia in offspring,3 it was treated as the critical control in the regression model.
Age cohort: Male offspring ages were dichotomized into 2 cohorts: those 16 weeks old or younger and those 26 weeks old or older.
Days old when analyzed: This was the exact age in days when the mouse was killed for analysis (distribution in Table 1).
Body weight: Body weights of the fully grown adult offspring at killing ranged from 24.3 to 45.3 g; weight may be relevant to prostate hyperplasia because obesity correlates with poor outcomes for patients with aggressive prostate cancer.21
Cage density: Offspring were maintained in cages with 1 to 5 male littermates from weaning until sacrifice. We hypothesized that the number of cage mates could affect the prostate health of the subject. The number of representative animals exposed to each cage density is given in Table 2.
Maternal exposure length: Mothers were exposed to the DIO diet for different lengths of time before mating, and the distribution is shown in Table 3. Maternal DIO exposure length has been shown to affect other aspects of offspring health.22
Table 1.
The Age of Male Offspring at Sacrifice (Days) and the Number of Offspring at Each Age.
| Age of Offspring | |
|---|---|
| Days | # Males |
| 116 | 4 |
| 119 | 4 |
| 122 | 1 |
| 123 | 1 |
| 124 | 2 |
| 126 | 4 |
| 144 | 2 |
| 177 | 2 |
| 180 | 2 |
| 187 | 2 |
| 189 | 3 |
| 194 | 2 |
| 218 | 1 |
| 292 | 2 |
| 432 | 2 |
| 438 | 4 |
| 441 | 8 |
| 444 | 2 |
| 445 | 4 |
| 446 | 2 |
Table 2.
The Number of Adult Males per Cage and the Number of Males in Each Cage Density Exposure Group.
| Cage Density | |
|---|---|
| # Per Cage | n |
| 1 | 2 |
| 2 | 14 |
| 3 | 5 |
| 4 | 26 |
| 5 | 7 |
Table 3.
The Length of DIO (Days) of the Dams and the Number of Offspring with Dams of that Exposure Length.
| Maternal DIO Length | |
|---|---|
| Days | # Males |
| 57 | 2 |
| 63 | 3 |
| 69 | 4 |
| 72 | 4 |
| 93 | 2 |
| 103 | 2 |
| 105 | 2 |
| 107 | 1 |
| 111 | 4 |
| 113 | 2 |
| 115 | 1 |
| 128 | 1 |
| 146 | 4 |
| 153 | 1 |
| 176 | 1 |
| 179 | 2 |
| 181 | 2 |
| 194 | 8 |
| 196 | 4 |
| 203 | 2 |
| 209 | 2 |
Abbreviation: DIO, diet-induced obesity.
The resulting model is shown in Table 4, which gives the estimated effect sizes of each explanatory variable on prostate hyperplasia in this cohort. Of the 6 potential explanatory covariates, 4 were statistically significant (P < .05), and the GLM was statistically distinct from the null model, as evidenced by the fact that the difference between the null and residual deviances was 30.293, which is in the tail of a chi-square distribution with 6 (52-46) degrees of freedom. Thus, the Poisson GLM regression model was an exceptionally good fit to the data, despite the modest sample size. Most importantly, the coefficient for Maternal diet was positive and statistically significant, which was evidence that the GLM appropriately reflected the biological context of the experiment. We conclude that this mix of explanatory variables reliably predicted the percentage of hyperplastic ducts (denoted here as “counts”).
Table 4.
A Multivariate Analysis of Exposures That Could Contribute to Prostate Hyperplasia Outcomes in Offspring.
| Multivariate GLMa of Offspring Prostate Hyperplasia | ||||
|---|---|---|---|---|
| Estimated Effect Size | Std Error | t Value | Pr(>|t|) | |
| (Intercept) | −0.3012 | 1.2642 | −0.2383 | 0.8117 |
| Maternal diet | 0.8261 | 0.2261 | 3.6542 | 0.0003 |
| Age cohort | 1.2585 | 0.3292 | 3.8233 | 0.0001 |
| Days old when analyzed | −0.0039 | 0.0015 | −2.6206 | 0.0094 |
| Body weight | −0.0202 | 0.0336 | −0.6007 | 0.5513 |
| Cage density | 0.2844 | 0.1380 | 2.0609 | 0.0394 |
| Log (maternal exposure length) | 0.0957 | 0.2631 | 0.3636 | 0.7162 |
Abbreviation: AIC, Akaike information criterion; GLM, generalized linear model; SE, standard error.
aNull deviance: 106.662 on 52 degrees of freedom; residual deviance: 76.369 on 46 degrees of freedom; AIC: 200.4. Estimated effect size = b. t value = b/SE(b).
Age of Subjects Predicted Prostate Hyperplasia Counts
In the GLM for prostate hyperplasia, the coefficient estimate for Age cohort was positive and statistically significant (Table 4). Thus, the older mouse cohort (26 and 63 weeks) had higher expected counts of hyperplastic ducts than the younger mouse cohort (16 weeks). This result was anticipated, as age is the most highly correlated risk factor for changes in prostate health, and we observed that aged prostates exhibited increased hyperplasia in the previous analysis.3 In contrast, the coefficient estimate for Days old when analyzed demonstrated a small, negative, and statistically significant effect. However, the absolute value of the Days old when analyzed coefficient was 3 orders of magnitude smaller than the positive effect of belonging to the older Age cohort (effect size −0.0039, P = .0094 vs 1.2585, P = .0001). This suggested that the Days old when analyzed effect was an anomaly of the model and biologically irrelevant.
The GLM was next used to obtain predictions for hyperplasia counts across the range of age cohorts. To do this, all other explanatory variables were set at means except for Maternal diet, multiplied by the coefficient estimates, and the inverse link function was applied. The effect of Age cohort on both DIO- and control-exposed groups indicated that the DIO-exposed mice had higher expected hyperplasia counts than the control-exposed mice (Figure 1A). This was important as it confirmed the expected effect of the key treatment variable, Maternal diet. Belonging to the older cohort appeared to exacerbate the DIO-induced hyperplasia (older cohort DIO: 4.6% hyperprolific and older cohort control: 1.9% hyperprolific). Thus, these data indicated that both aging and DIO exposure were both strong predictors of prostate hyperplasia.
Figure 1.

Graphical predictions of prostate hyperplasia outcomes for 3 covariates from the generalized linear model (GLM). A, Age cohorts are graphed bimodally, representing members of the young cohort (16 weeks, 0) or old cohort (26 and 63 weeks, 1). B, Hyperplasia counts predicted relative to maternal (high sugar/high fat) exposure length (graphed in months). C, Predicted hyperplasia counts relative to the number of littermates in the cage with the subject. In all panels, means and 95% confidence intervals (CI) are plotted.
Maternal Exposure Length Did Not Predict Prostate Hyperplasia Counts
Longer exposures to obesogenic diets are associated with worsening overall body composition, cardiometabolic health, and obesity in exposed dams.22,23 Consistent with this, we previously reported that females maintained on the high-sugar/high-fat diet for 16 weeks weighed 46.92 g, whereas chow-exposed animals weighed 22.04 g, with fasting serum glucose of 145.7 to 97 mg/dL, respectively.24 Weights of our chow-exposed animals were consistent with published values for C57Bl/6J females (20.7 and 23.6 g at 3 and 5 months, respectively).3 Thus, we hypothesized that longer maternal exposure would positively correlate with increased prostate hyperplasia in offspring. Although mothers were exposed to the high-sugar/high-fat diet for 57 to 209 days before the birth of offspring, increased Maternal exposure length demonstrated no effect on offspring prostate hyperplasia counts (Table 4; 0.0957, P = .7162). The data were logged in this model specification for a better overall fit, but no permutations of the data points demonstrated a significant effect on offspring hyperplasia. When a range of predictions based on the model was generated for Maternal exposure length, the hyperplasia outcome did not vary over time (Figure 1B). It is important to note that while increased Maternal exposure length failed to exacerbate the DIO exposure effect, any exposure to maternal DIO predicted higher prostate hyperplasia outcomes than chow exposure. This suggests that prostate hyperplasia is not worsened in response to increasing obesity and comorbidities in the mother but is set by an acute exposure occurring during gestation or lactation.
Cage Density Positively Predicts Prostate Hyperplasia Outcomes and Body Weight
In the GLM, Cage density exhibited a strong positive correlation with hyperplastic duct counts (Table 4). Transformation of the data indicated that the differences in hyperplasia between control- and DIO-exposed offspring became increasingly distinct as the number of cage mates increased (Figure 1C). For example, a male offspring with 4 male cage mates was predicted to have 4.8% hyperplastic ducts if he was exposed to maternal DIO, whereas a similarly housed mouse with a control mother would be predicted to have 2.0% hyperplastic ducts. Thus, Cage density was a predictor of prostate hyperplasia in mice.
The variable Body weight did not show evidence of an effect on prostate hyperplasia in the GLM (−0.0202, P = .5513). This may be due, in part, to the fact that mean body weights for control- and DIO-exposed offspring were 32.2 and 31.7 g, respectively, indicating that DIO exposure did not alter adult offspring body size (P = .767). Despite this, including the explanatory covariate Body weight was critical to maintain the fit of the prostate hyperplasia model, suggesting that Body weight interacted with other covariates. Graphing the distribution of body weights for the cohort demonstrated a bimodal distribution at a cutoff point of 34.0 g (group 1 = 25.0-34.0 g and group 2 = 35.0-45.0 g; Figure 2). The majority (39 of 54) of the mice (group 1) were within normal body weight ranges for males (from 24.0 to 35.0 g3). Notably, all of the heavier mice were from cages with more cage mates (Figure 2, mean Cage density per weight range: group 1 = 2.96 and group 2 = 4.43), suggesting that Cage density influenced Body weight. Simple bivariate linear regression analysis of Body weight and Cage density indicated that these covariates associated (Table 5). Taken together, these data suggested that Cage density influenced both the body weight and prostate health of the male subjects.
Figure 2.
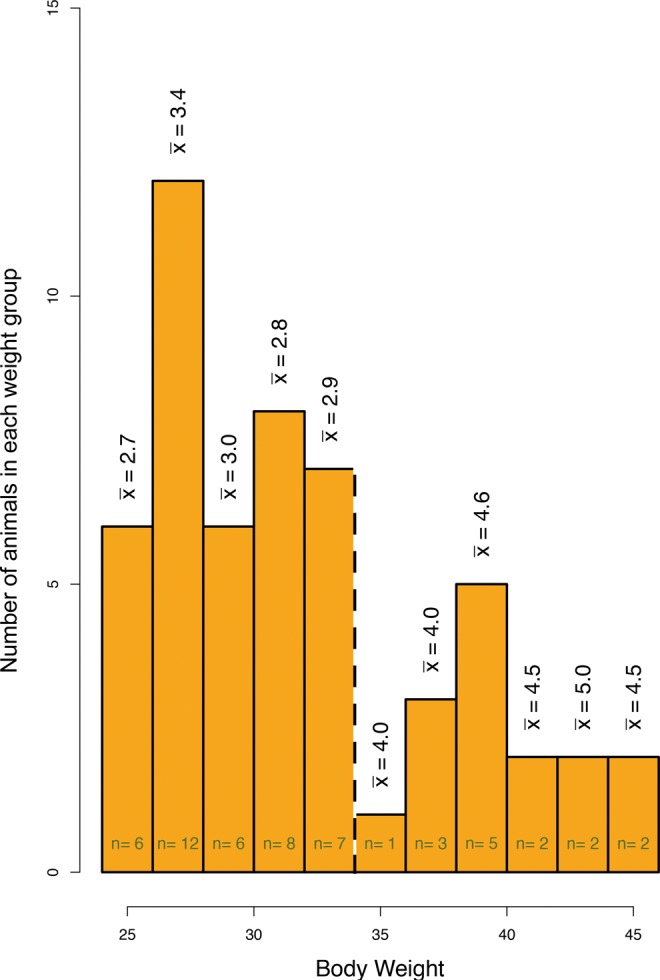
Heavier offspring came from cages with more cage mates. The dashed line indicates the cutoff of the bimodal distribution of body weights. The vertical values denote the mean number of male pups in a cage for that weight cohort.
Table 5.
A Linear Regression Assessing Correlation Between Cage Density and the Body Weight Outcome.a
| Simple Linear Regression of Cage Density and Body Weight | ||||
|---|---|---|---|---|
| Estimated Effect Size | Std Error | t Value | Pr(>|t|) | |
| (Intercept) | 29.3401 | 1.1890 | 24.6710 | 0.0000 |
| Cage density | 3.8861 | 1.5212 | 2.5542 | 0.0136 |
aResidual standard error: 5.45 on 52 degrees of freedom; multiple R 2: .1115; adjusted R 2: .09438; F-statistic: 6.524 on 1 and 52 df.
Glucocorticoid Receptor Expression is Elevated in Overcrowded Male Cages
Evaluation of the GLM indicated that Cage density positively predicted both prostate hyperplasia and Body weight in group-housed animals. One potential explanation was that male animals housed in crowded cages experienced alterations in stress homeostasis, which is regulated by adrenal secretion of glucocorticoids that act on GRs to stimulate physiologic responses to stressors.25 The role of GR in prostate tissue homeostasis and cancer is not well understood, but from studies in human prostate cancer cell lines GR is abundant and overlaps with androgen receptor to regulate many genes.26,27 For these reasons, we hypothesized that GR could be altered in the prostates of overcrowded animals. Immunofluorescence labeling of GRα was performed on the remaining paraffin-embedded prostate sections from the same cohort modeled in Table 4. We quantitated immunofluorescence (Figure 3A and B) as gray values and normalized to nuclear dye (gray value/TOPRO). The ordinally ranked values demonstrated a linear positive relationship with Cage density (Figure 3C; r = .526, r 2 = .277). When the data were dichotomized (animals housed with either ≤3 or ≥4 cage mates), GR protein abundance was significantly higher in animals from more crowded cages (Figure 3D; P < .0001). Poisson GLM modeling was used to determine whether other explanatory covariates contributed to the outcome of increased GR protein (Table 6). Only Cage density had a positive association with GR, whereas Age cohort had a statistically significant, but negative, association. No other potential explanatory variables associated, indicating that, of the variables assessed, only number of cage mates positively predicted expression of GR in offspring prostates.
Figure 3.

Expression of gluococorticoid receptor in prostates correlates with the number of cage mates. Glucocorticoid receptor (GR; red) and TOPRO (blue) staining of a prostate section from a mouse maintained in a cage with (A) 3 or fewer or (B) 4 or more cage mates. (C) Normalized gray value analysis ranked ordinally by cage density (r = .526, r 2 = .277; solid line: best-fit linear regression; dashed lines: 95% confidence interval [CI]). (D) The same data as in (C), dichotomized as ≥4 or ≤3 cage mates. ***P ≤ .0001.
Table 6.
A Multivariate Analysis of Exposures That Could Contribute to Glucocorticoid Receptor-α Protein Abundance Outcome in Offspring.
| Multivariate GLM of Offspring Glucocorticoid Receptor Expression | ||||
|---|---|---|---|---|
| Estimated Effect Size | Std Error | t Value | Pr(>|t|) | |
| (Intercept) | 0.8125 | 0.5334 | 1.5234 | 0.1409 |
| Age cohort | −0.8755 | 0.3540 | −2.4729 | 0.0134 |
| Maternal diet | −0.1088 | 0.1622 | −0.6707 | 0.5024 |
| Days old when analyzed | 0.0012 | 0.0014 | 0.9160 | 0.3620 |
| Body weight | 0.0350 | 0.0246 | 1.4217 | 0.1729 |
| Cage density | 0.5551 | 0.2126 | 2.6114 | 0.0092 |
| Log (maternal exposure length) | −0.0010 | 0.0023 | −0.4170 | 0.6782 |
| Prostate hyperplasia | 0.0222 | 0.0460 | 0.4826 | 0.6294 |
Abbreviation: GLM, generalized linear model
Discussion
Here, we used GLM to identify environmental exposures that may contribute to prostate hyperplasia. Three findings emerged from the GLM. First, prostate hyperplasia counts were positively associated with age of the subject. Second, the length of time the mothers were exposed to DIO did not influence prostate hyperplasia in offspring, suggesting that obesity per se, and not worsening obesity, of the mother was the key factor for that particular environmental effect. Third, Cage density associated with increased prostate hyperplasia and correlated with increased GR expression. Discovery of the Cage density effect through this statistical modeling approach illustrated the utility of developing a multivariate regression model to evaluate multiple explanatory variables in small n experiments. Together, these explanatory variables (maternal DIO, age of the subject, and cage density) all mediate risk of prostate hyperplasia in mice (Figure 4).
Figure 4.

A schematic model of factors that contribute to prostate hyperplasia in mice over the lifespan. Age of subjects (green) and maternal diet-induced obesity (DIO) exposure (red) positively correlated with hyperplasia counts. Additionally, increased number of male cage mates from weaning to sacrifice correlated with increased hyperplasia counts (solid blue). Litter size from preconception (in utero) through weaning (dotted blue) could also influence hyperplasia counts.
Aging and Maternal DIO May Coregulate Prostate Hyperplasia
The maternal DIO model stimulates prostate hyperplasia in C57BL/6J mice, a species that does not spontaneously develop prostate cancer.2 Thus, it is unknown whether DIO-stimulated prostate hyperplasia is a risk factor for frank prostatic carcinoma in situ. However, the current analysis indicated that an animal’s age cohort had a statistically significant positive association with the prostate hyperplasia phenotype. Given that aging is widely accepted as the most significant risk factor for prostate hyperplasia and cancer in humans, this finding lends credence to our model as one that recapitulates human prostate phenotypes.1,28 The current findings further suggest that maternal obesity exacerbated aging-related hyperplasia. Aging influences prostate hyperplasia in humans by altering the autonomous nervous system, neuroendocrine cell function, sex steroid hormone metabolism, epithelial/stromal cell interactions, and inflammation.29 The resulting signaling changes may coordinate with signaling changes brought about by maternal DIO exposure to promote prostate hyperplasia. Future studies will seek to identify molecular mechanisms that are influenced by maternal obesity versus aging.
Increasing Obesity of the Mother May Not Exacerbate Prostate Hyperplasia Outcomes
To our knowledge, our previous study was the first to describe a maternal DIO model of prostate hyperplasia, so the length of time that mothers needed to be exposed to DIO to influence offspring prostate health was unknown. We chose 1 month before mating through gestation and lactation as a starting point. The shortest amount of time between the start of maternal DIO exposure and birth of a subject was 57 days (which may have been sufficient to affect the oocytes), and the longest was 209 days. Despite this large range, the GLM showed that increased DIO exposure length did not exacerbate the effect; any length of DIO exposure was sufficient to stimulate prostate hyperplasia in offspring. Thus, although maternal obesity and/or exposure to a high-sugar/high-fat diet during gestation was critical for prostate hyperplasia, secondary effects due to increasing maternal obesity, and associated comorbidities may be irrelevant to the effect. Several possible time windows of exposure to high sugar/high fat and DIO could alter the oocyte, embryo, or young offspring, leading to the prostate hyperplasia response. First, the offspring may be indirectly exposed to the high-sugar/high-fat diet during gestation, which may directly alter oocyte and embryo gene expression patterning. Second, maternal DIO could indirectly influence the oocyte or the intrauterine environment of the offspring. Third, offspring consumption of high-sugar/high-fat diet after birth but prior to weaning may influence early postnatal prostate development. Forth, sires mated to high-sugar/high-fat dams are also briefly exposed to adverse dietary conditions during mating. Early life patterning has been demonstrated to influence diseases ranging from asthma/atopic conditions to mammary tumor development, often through metabolic and epigenetic misregulation,23,30–37 making dissection of the relevant time window of exposure an important next step. Future experiments, switching mothers’ diets between high sugar/high fat and control chow before, during and after conception and gestation will dissect the minimum exposure window to pattern prostate hyperplasia.
Cage Density is a Novel Potential Risk Factor for Prostate Hyperplasia
We observed that cage density is positively associated with prostate hyperplasia. Previous studies have indicated that stress homeostasis is influenced by cage density of rodents. Female rodents subjected to isolation stress exhibit increased incidence of mammary tumors.38 We have modeled cage density stress during adult life in males. This stress is possibly imparted by nonreproductive aggression experienced between cage mates that may correlate with density. Glucocorticoids have been implicated in mediating nonreproductive-based aggression in rodents39,40 and have been demonstrated to regulate similar gene expression profiles as androgen receptor, the canonical regulator of prostate proliferation, in prostate.27 We hypothesized that glucocorticoid signaling might mediate interactions between cage density and prostate hyperplasia. As an exploratory study, we were limited to retrospective examination of paraffin-embedded prostate tissues. Despite this limitation, the data from this preliminary study show that expression level of GRα is positively associated with cage density in offspring prostates. Continued work is needed in another animal cohort to test cortisol levels in mice housed at different cage densities, to quantify GRα with other standard techniques, and to test receptor activity at the nucleus in DIO exposure models to confirm these findings. Additionally, a limitation of this study is that litter size in utero might contribute to the hyperplasia effect (depicted in Figure 4) and parsing out this contribution should be the topic of future study.
Because established prostate cancer risk factors are few and cannot be modified, these findings provide an interesting avenue for future studies. Population dynamics studies have demonstrated that animal health exhibits plasticity in response to exploitation of resources and the physical restraint of crowding.41 Additionally, animals housed in crowded conditions may undergo hormone signaling that can influence cage mates.25–27 For example, pheromones can be used to signal between organisms (from Caenorhabditis elegans to rodents) via chemosensory receptors.42–45 Androgen signaling directly regulates prostate proliferation and, in hamsters, the number of cells expressing androgen receptor is altered by chemosensory/pheromone input in the amygdala.46,47 Furthermore, androgen levels in rodents are influenced by the presence of other males and can result in fighting behaviors and cortisol hormone signaling.48,49 In our study, the number and aggressiveness of the males in close proximity to a test subject could have influenced chemosensory regulation and crowding-related signals, possibly influencing prostate tissue patterning. Therefore, controlled experiments testing the effect of group housing on prostate hyperplasia and hormone signaling are exciting avenues of future study.
No Evidence of a Body Weight Effect on Prostate Hyperplasia
This analysis indicated that body weight of the subject did not correlate with maternal DIO or prostate hyperplasia. In humans, only advanced prostate cancer is consistently associated with body weight,50 and obesity may even be modestly protective in adolescent males.4 Nonetheless, the body weight variable was required to maintain the fit of the overall GLM and could not be completely removed. By contrast, weights of the urogenital sinus region and DLP lobes were each irrelevant to the model (data not shown). These results suggested that the effect of body weight, as a whole, is more nuanced than the measured variables examined here. Figure 2 and Table 5 demonstrated that body weight was bimodal and that Cage density was an underlying predictor of body weight. It is possible that crowded cages could lead to competitive behaviors, increased corticosteroid hormone signaling, subsequent overeating, and increased body weight. This multifactorial interplay is similar to the etiology of human obesity.51 Future studies will investigate these relationships.
Acknowledgment
We thank Dr Deborah Frank for careful editing of this manuscript.
Authors’ Note: ECB, JG, and KHM designed the study and ECB performed the data collection and analyzed the findings with LEL and KHM. JG performed the statistical analysis. ECB and JG wrote the manuscript.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ECB, JG, and KHM were supported by the Washington University TREC Program and National Cancer Institute (NIH U54 CA 155496), ECB was supported by the Washington University Department of Pathology Training and National Institute of Diabetes and Digestive and Kidney Diseases (NIH 5T32 DK 7296-33), and LEL was supported by a Department of Defense Prostate Cancer Postdoctoral Training Award and Congressionally Directed Medical Research Programs (DoD W81XWH-12-1-0119).
References
- 1. Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. (eds). SEER Cancer Statistics Review, 1975–2011. Bethesda, MD: National Cancer Institute; 2014. Web site http://seer.cancer.gov/csr/1975_2011/. Accessed 15 March, 2015. [Google Scholar]
- 2. Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14(19):2410–2434. [DOI] [PubMed] [Google Scholar]
- 3. Benesh EC, Humphrey PA, Wang Q, Moley KH. Maternal high-fat diet induces hyperproliferation and alters Pten/Akt signaling in prostates of offspring. Sci Rep. 2013;3:3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sutcliffe S, Colditz GA. Prostate cancer: is it time to expand the research focus to early-life exposures? Nat Rev Cancer. 2013;13(3):208–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gapp K, Jawaid A, Sarkies P, et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci. 2014;17(5):667–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nelder JA, Lee Y. Likelihood, quasi-likelihood and pseudolikelihood—some comparisons. J Roy Stat Soc B Met. 1992;54(1):273–284. [Google Scholar]
- 7. Surwit RS, Wang S, Petro AE, et al. Diet-induced changes in uncoupling proteins in obesity-prone and obesity-resistant strains of mice. Proc Natl Acad Sci U S A. 1998;95(7):4061–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu ZH, Wright JD, Belt B, Cardiff RD, Arbeit JM. Hypoxia-inducible factor-1 facilitates cervical cancer progression in human papillomavirus type 16 transgenic mice. Am J Pathol. 2007;171(2):667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suwa T, Nyska A, Haseman JK, Mahler JF, Maronpot RR. Spontaneous lesions in control B6C3F1 mice and recommended sectioning of male accessory sex organs. Toxicol Pathol. 2002;30(2):228–234. [DOI] [PubMed] [Google Scholar]
- 10. Sugimura Y, Cunha GR, Donjacour AA. Morphogenesis of ductal networks in the mouse prostate. Biol Reprod. 1986;34(5):961–971. [DOI] [PubMed] [Google Scholar]
- 11. Sugimura Y, Cunha GR, Donjacour AA, Bigsby RM, Brody JR. Whole-mount autoradiography study of DNA synthetic activity during postnatal development and androgen-induced regeneration in the mouse prostate. Biol Reprod. 1986;34(5):985–995. [DOI] [PubMed] [Google Scholar]
- 12. Garabedian EM, Humphrey PA, Gordon JI. A transgenic mouse model of metastatic prostate cancer originating from neuroendocrine cells. Proc Natl Acad Sci U S A. 1998;95(26):15382–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Humphrey PA; American Society for Clinical Pathology. Prostate Pathology. Chicago: American Society for Clinical Pathology; 2003. [Google Scholar]
- 14. Gill J. Generalized Linear Models: A Unified Approach. Thousand Oaks, CA: Sage; 2000. [Google Scholar]
- 15. Little RJA, Rubin DB. Statistical Analysis with Missing Data. 2nd ed Hoboken, NJ: John W. Wiley and Sons; 2002. [Google Scholar]
- 16. Rubin DB. Multiple Imputation for Nonresponse in Surveys. New York, NY: John Wiley & Sons; 1987. [Google Scholar]
- 17. Cranmer SJG, Gill J. We have to be discrete about this: a non-parametric imputation technique for missing categorical data. Br J Polit Sci. 2012;43(2):425–449. [Google Scholar]
- 18. Rubin DB. Multiple Imputation after 18+ Years. J Am Stat Assoc. 1996;91(434):473–489. [Google Scholar]
- 19. Schafer JL. Analysis of Incomplete Multivariate Data. London: Chapman & Hall; 1997. [Google Scholar]
- 20. Esakky P, Hansen DA, Drury AM, Moley KH. Molecular analysis of cell type-specific gene expression profile during mouse spermatogenesis by laser microdissection and qRT-PCR. Reprod Sci. 2013;20(3):238–252. [DOI] [PubMed] [Google Scholar]
- 21. Allott EH, Masko EM, Freedland SJ. Obesity and prostate cancer: weighing the evidence. Eur Urol. 2013;63(5):800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luzzo KM, Wang Q, Purcell SH, et al. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7(11):e49217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sullivan EL, Nousen EK, Chamlou KA. Maternal high fat diet consumption during the perinatal period programs offspring behavior. Physiol Behav. 2014;123:236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jungheim ES, Travieso JL, Carson KR, Moley KH. Obesity and reproductive function. Obstet Gynecol Clin North Am. 2012;39(4):479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kino T. Glucocorticoid receptor In: De Groot LJ, Beck-Peccoz P, Chrousos G, et al., eds. Endotext. South Dartmouth, MA: MDText.com, Inc; 2000. [Google Scholar]
- 26. Sahu B, Laakso M, Pihlajamaa P, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73(5):1570–1580. [DOI] [PubMed] [Google Scholar]
- 27. Yemelyanov A, Bhalla P, Yang X, et al. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle. 2012;11(2):395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kitagishi Y, Matsuda S. Redox regulation of tumor suppressor PTEN in cancer and aging [review]. Int J Mol Med. 2013;31(3):511–515. [DOI] [PubMed] [Google Scholar]
- 29. Untergasser G, Madersbacher S, Berger P. Benign prostatic hyperplasia: age-related tissue-remodeling. Exp Gerontol. 2005;40(3):121–128. [DOI] [PubMed] [Google Scholar]
- 30. Dong M, Zheng Q, Ford SP, Nathanielsz PW, Ren J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J Mol Cell Cardiol. 2013;55:111–116. [DOI] [PubMed] [Google Scholar]
- 31. Harpsoe MC, Basit S, Bager P, et al. Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: a study within the Danish National Birth Cohort. J Allergy Clin Immunol. 2013;131(4):1033–1040. [DOI] [PubMed] [Google Scholar]
- 32. Li M, Sloboda DM, Vickers MH. Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp Diabetes Res. 2011;2011:592408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lowe A, Braback L, Ekeus C, Hjern A, Forsberg B. Maternal obesity during pregnancy as a risk for early-life asthma. J Allergy Clin Immunol. 2011;128(5):1107–1109. e1101–e1102. [DOI] [PubMed] [Google Scholar]
- 34. Yessoufou A, Moutairou K. Maternal diabetes in pregnancy: early and long-term outcomes on the offspring and the concept of “metabolic memory”. Exp Diabetes Res. 2011;2011:218598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang S, Rattanatray L, McMillen IC, Suter CM, Morrison JL. Periconceptional nutrition and the early programming of a life of obesity or adversity. Prog Biophys Mol Biol. 2011;106(1):307–314. [DOI] [PubMed] [Google Scholar]
- 36. de Assis S, Warri A, Cruz MI, et al. High-fat or ethinyl-oestradiol intake during pregnancy increases mammary cancer risk in several generations of offspring. Nat Commun. 2012;3:1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. La Merrill M, Harper R, Birnbaum LS, Cardiff RD, Threadgill DW. Maternal dioxin exposure combined with a diet high in fat increases mammary cancer incidence in mice. Environ Health Perspect. 2010;118(5):596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McClintock MK, Conzen SD, Gehlert S, Masi C, Olopade F. Mammary cancer and social interactions: identifying multiple environments that regulate gene expression throughout the life span. J Gerontol B Psychol Sci Soc Sci. 2005;60(spec no 1):32–41. [DOI] [PubMed] [Google Scholar]
- 39. Haller J, Halasz J, Mikics E, Kruk MR. Chronic glucocorticoid deficiency-induced abnormal aggression, autonomic hypoarousal, and social deficit in rats. J Neuroendocrinol. 2004;16(6):550–557. [DOI] [PubMed] [Google Scholar]
- 40. Harding CF, Leshner AI. The effects of adrenalectomy on the aggressiveness of differently housed mice. Physiol Behav. 1972;8(3):437–440. [DOI] [PubMed] [Google Scholar]
- 41. Gergs A, Preuss TG, Palmqvist A. Double trouble at high density: cross-level test of resource-related adaptive plasticity and crowding-related fitness. PLoS One. 2014;9(3):e91503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roy A, Derakhshan F, Wilson RJ. Stress peptide PACAP engages multiple signaling pathways within the carotid body to initiate excitatory responses in respiratory and sympathetic chemosensory afferents. Am J Physiol Regul Integr Comp Physiol. 2013;304(12):R1070–R1084. [DOI] [PubMed] [Google Scholar]
- 43. Ludewig AH, Izrayelit Y, Park D, et al. Pheromone sensing regulates Caenorhabditis elegans lifespan and stress resistance via the deacetylase SIR-2.1. Proc Natl Acad Sci USA. 2013;110(14):5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Herrero P. Fruit fly behavior in response to chemosensory signals. Peptides. 2012;38(2):228–237. [DOI] [PubMed] [Google Scholar]
- 45. Liberles SD, Buck LB. A second class of chemosensory receptors in the olfactory epithelium. Nature. 2006;442(7103):645–650. [DOI] [PubMed] [Google Scholar]
- 46. da Silva HB, Amaral EP, Nolasco EL, et al. Dissecting major signaling pathways throughout the development of prostate cancer. Prostate Cancer. 2013;2013:920612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blake CB, Meredith M. Change in number and activation of androgen receptor-immunoreactive cells in the medial amygdala in response to chemosensory input. Neuroscience. 2011;190:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Christian JJ, Lemunyan CD. Adverse effects of crowding on lactation and reproduction of mice and two generations of their progeny. Endocrinology. 1958;63(5):517–529. [DOI] [PubMed] [Google Scholar]
- 49. Reber SO, Obermeier F, Straub RH, Falk W, Neumann ID. Chronic intermittent psychosocial stress (social defeat/overcrowding) in mice increases the severity of an acute DSS-induced colitis and impairs regeneration. Endocrinology. 2006;147(10):4968–4976. [DOI] [PubMed] [Google Scholar]
- 50. Holmberg L. Obesity, nutrition, and prostate cancer: insights and issues. Eur Urol. 2013;63(5):821–822. [DOI] [PubMed] [Google Scholar]
- 51. Sinha R, Jastreboff AM. Stress as a common risk factor for obesity and addiction. Biol Psychiatry. 2013;73(9):827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]