

Abstract
Uterine fibroids (UFs) are benign smooth muscle neoplasms affecting up to 70% of reproductive age women. Treatment of symptomatic UFs places a significant economic burden on the US health-care system. Several specific genetic abnormalities have been described as etiologic factors of UFs, suggesting that a low DNA damage repair capacity may be involved in the formation of UF. In this study, we used human fibroid and adjacent myometrial tissues, as well as an in vitro cell culture model, to evaluate the expression of MutS homolog 2 (MSH2), which encodes a protein belongs to the mismatch repair system. In addition, we deciphered the mechanism by which polycomb repressive complex 2 protein, EZH2, deregulates MSH2 in UFs. The RNA expression analysis demonstrated the deregulation of MSH2 expression in UF tissues in comparison to its adjacent myometrium. Notably, protein levels of MSH2 were upregulated in 90% of fibroid tissues (9 of 10) as compared to matched adjacent myometrial tissues. Human fibroid primary cells treated with 3-deazaneplanocin A (DZNep), chemical inhibitor of EZH2, exhibited a significant increase in MSH2 expression (P < .05). Overexpression of EZH2 using an adenoviral vector approach significantly downregulated the expression of MSH2 (P < .05). Chromatin immunoprecipitation assay demonstrated that enrichment of H3K27me3 in promoter regions of MSH2 was significantly decreased in DZNep-treated fibroid cells as compared to vehicle control. These data suggest that EZH2-H3K27me3 regulatory mechanism dynamically changes the expression levels of DNA mismatch repair gene MSH2, through epigenetic mark H3K27me3. MSH2 may be considered as a marker for early detection of UFs.
Keywords: fibroid, DNA mismatch repair, EZH2, H3K27me3, MSH2, uterine fibroid
Introduction
Uterine fibroids (UFs) are benign, smooth muscle neoplasms affecting up to 70% of reproductive age women. Treatment of symptomatic UFs places a significant economic burden on the US health-care system.1,2 Etiology and pathogenesis of UFs are complex. Several genetic abnormalities related to the pathogenesis of UFs have been investigated, including deletions in 7q, trisomy of chromosome 12, and rearrangements in the HMGA2 gene, particularly, mutations in exons 1 and 2 of the MED12 gene are very common and can be detected in up to 85% of all sporadic UFs lesions.3–11 An inadequate repair of the acquired DNA damage is responsible for the undifferentiated cell proliferation and tumorigenesis.12,13 Endogenously and/or exogenously induced DNA damage commonly results in genomic instability leading to a variety of chromosomal aberrations.14 Increasing scientific evidence supports a link between low DNA repair capacity and an increased risk for neoplastic development.12,14–16 Direct repair of DNA damage, by endogenous repair enzymes, lessens the rate of mutagenesis and strengthens the immune response to tumor cells.14 Moreover, knowledge of the DNA repair system in normal and tumorigenic tissue may help predict and guide development of effective nonsurgical treatment for UFs.
The mismatch repair (MMR) system recognizes and repairs erroneous insertion, deletion, and misincorporation of bases that can arise during DNA replication and recombination. Several MutS homolog (MSH) proteins and other members including MSH1, MSH2, MSH3, MSH6, PCNA, and EXO1 belong to MMR system. MSH2, a component of the postreplicative DNA MMR system, is able to form 2 different heterodimers with MSH3 and MSH6, which binds to DNA mismatched segment, thereby initiating DNA repair.17 The MMR system, which includes MSH2 protein, is essential for maintaining the stability of the genome during repeated duplication.18
The polycomb repressive complex 2 (PRC2), the mammalian enzymatic complex, is involved in the gene-repressive high-degree methylation of histone H3 at lysine 27 (H3K27me3).19–22 Numerous studies have shown that EZH2 as a catalytic subunit of PRC2 plays a critical role in cancer initiation and progression, as well as in cancer stem cell biology.23–25 Furthermore, the molecular response to EZH2 alteration appears to be diverse and depends largely on the type of cancer.26–31 Notably, EZH2 has been shown to deregulate several DNA repair genes leading to the development of cancers.32–34
Although genetic abnormalities have been well described in human UFs, little is known about the DNA damage repair system related to epigenetic abnormalities in this common disease.2,35,36 Since a wide array of diverse genetic alterations and mutations have been identified in UFs, the purpose of this study was to determine the gene expression pattern of MSH2 in UFs and adjacent myometrium tissues and decipher the mechanism underlying deregulation of MSH2 expression.
Materials and Methods
Cell Line and Primary Cell Cultures
The immortalized human uterine fibroid cell line (HuLM), which expresses both estrogen and progesterone receptors, was a generous gift from Dr Darlene Dixon (National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina).37 The primary human UF cells were generated from UF tissue specimens. Isolation of the primary cell population, from tissues, was performed as previously described38; briefly, a portion (∼5 mm3) of the fresh UF tissue was washed in culture medium to remove blood, chopped into small pieces under sterile conditions, transferred into a 15-mL capped tube, and suspended in Hank balanced salt solution containing 1× antibiotic–antimycotic (Life Technologies, Grand Island, NY) and 300 U/mL collagenase type 4 (Worthington Biochemical Corp, Lakewood, New Jersey). The tissue suspension was then incubated at 37°C for at least 12 hours to obtain individual cells and any clumps of cells. Next, the suspension was passed through a 100-µm pore-sized sterile nylon filter and individual cells were plated out and incubated at 37°C, allowing the cells to attach to the 100-mm sterile tissue culture-treated plate containing smooth muscle cell basal medium (SmBM, catalog no. CC-3181, Lonza, Walkersville, MD) containing 5% fetal bovine serum (FBS) and supplemented with SmBM SingleQuots (catalog no. CC-4149). The SmBM singlequots contained human epidermal growth factor, insulin, human fibroblastic growth factor B, and gentamicin/amphotericin B.
Patients and Tumor Specimens
The study was approved by the institutional review board of Georgia Regents University. Fibroid tissues were consistently collected from peripheral parts of intramural fibroid lesions (≥5 cm in diameter) with care to avoid areas of apparent necrosis, bleeding, or degeneration. Adjacent myometrium was collected at least 2 cm away from the closest fibroid lesion. Patient records were reviewed for the demographic characteristics. Tissue samples were collected from the patients with no hormonal treatment for 3 months. All tissues used in this study were collected from African American women.
Cell Treatment
Human fibroid primary cells were cultured in media in the presence or absence of EZH2 inhibitor 3-deazaneplanocin A (DZNep; Cayman, Ann Arbor, MI) for 3 days as we have described previously.
Cell Viral Infection
Immortalized human uterine fibroid cells (HuLM) were cultured in 60-mm dishes; at 30% to 40% confluence, cells were transduced with Ad-EZH2 (adenovirus expressing EZH2 under CMV5 promoter; Vector Biolabs, Malvern, Pennsylvania) and Ad- Green Fluorescent Protein (GFP) at varying multiplicity of infections (0-100 plaque-forming unit/cell), as we have described previously.39,40 After 6 hours, transduction was stopped by changing the media to smooth muscle growth medium (SMGM-2; Lonza #CC3182) supplemented with growth factors and 5% FBS. Protein lysates were prepared on day 5 of transduction for measurement of EZH2 and H3K27me3 levels. RNA was isolated on day 3 of transduction, for measurement of MSH2 and p27 expression.
RNA Extraction, Complementary DNA Synthesis, and SYBR Green Real-Time Polymerase Chain Reaction
Human UFs and adjacent myometrial tissue (MyoF) samples were pulverized to a fine powder in liquid nitrogen using the Cellcrusher tissue pulverizer (Cell Crusher Limited, Cork, Ireland). RNA was isolated from pulverized UFs and MyoF tissues from patients, as well as from HuLM and fibroid primary cells, using TRIzol reagent (Life Technologies) and reverse transcribed into the first-strand complementary DNA (cDNA) using Superscript III cDNA transcription kit (Invitrogen) using standard techniques.41 All assays were carried out in 96-well format. Each sample was run in triplicate. Real-time fluorescence detection of polymerase chain reaction (PCR) products was performed using the following thermocycling conditions: 1 cycle of 95°C for 2 minutes, 40 cycles of 95°C for 5 seconds, and 60°C for 30 seconds. Sequences of the primers are shown in Table 1. 18S was used as an endogenous control for gene expression. For data analysis, the comparative method (ΔΔCt) was used to calculate relative quantities of the nucleic acid sequence.
Table 1.
Primer Sequences and Assays.
| Gene | Forward/Reverse | Primer Sequences | Assay | Species | Note |
|---|---|---|---|---|---|
| MSH2 | Forward | AAGAAGTGCTATCTGGAAAGAG | q-PCR | Human | |
| MSH2 | Reverse | ACATTTCAGTAAAGGGCATTTG | q-PCR | Human | |
| p27 | Forward | GGACTGCGGGACGATCCT | q-PCR | Human | |
| p27 | Reverse | TGACAAGCCACGCAGTAGATTT | q-PCR | Human | |
| 18S | Forward | CGAACGTCTGCCCTATCAACTT | q-PCR | Human | |
| 18S | Reverse | ACCCGTGGTCACCATGGTA | q-PCR | Human | |
| MSH2 | Forward | ATCCTCAGAGCCAAGAAGAG | ChIP | Human | Distal region |
| MSH2 | Reverse | CTGCCTGTTAGCCACATTATC | ChIP | Human | Distal region |
| MSH2 | Forward | GATGTTACTCCCATGCTTCC | ChIP | Human | Proximal region |
| MSH2 | Reverse | GAGCTCCTTTCTGTGTTTACT | ChIP | Human | Proximal region |
Abbreviations: ChIP, chromatin immunoprecipitation; q-PCR, quantitative PCR.
Nuclear and Cytoplasmic Extraction
Nuclear–cytoplasmic fractionation was conducted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, Waltham, Massachusetts) according to the manufacturer’s instructions.
Western Blot Analysis
Total proteins from fibroid primary cells treated with vehicle or DZNep were extracted by lysing and by sonication in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitor cocktails. Protein concentrations were determined using the Bradford protein assay kit (Bio-Rad, Hercules, California). Equal amounts of total proteins (20 μg) were subjected to sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membrane for 90 minutes at 100 V. Membranes were blocked for 1 hour at room temperature in Tris-buffered saline (TBS) containing 5% nonfat powdered milk and probed with primary antibody in TBS at dilution of 1:1000 to 20 000 overnight in accordance with the manufacturer’s instruction and our experience (Table 2). In all cases, a secondary antibody labeled with horseradish peroxidase (Santa Cruz, Dallas, Texas) was used at a dilution of 1:5000 for 1 hour at room temperature, and immunoreactive bands were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and recorded on photosensitive film. The relative intensities of immunoreactive bands were detected by Western blot analysis and quantified by densitometry using NIH ImageJ Software and normalized with β-actin levels.
Table 2.
List of Antibodies Used for WB and ChIP Assay Analysis.
| Antigen | Catalog No | Assay | Concentration | Dilution | Isotype | Supplier |
|---|---|---|---|---|---|---|
| H3K27me3 | 39155 | WB | 1 μg/μL | 1:2000 | Rabbit IgG | Active Motif |
| EZH2 | 39933 | WB | 1 μg/μL | 1:2000 | Rabbit IgG | Active Motif |
| MSH2 | ab52266 | WB | 1 μg/μL | 1;2000 | Mouse IgG | Abcam |
| β-actin | A5441 | WB | 1-4 μg/μL | 1:20000 | Mouse IgG | Sigma |
| Histone H3 | ab1791 | WB | 1 μg/μL | 1:10000 | Rabbit IgG | Abcam |
| H3K27me3 | 39155 | ChIP | 1 μg/μL | 1:100 | Rabbit IgG | Active Motif |
| Negative control | ab171870 | ChIP | 1 μg/μL | 1:100 | Rabbit IgG | Abcam |
Abbreviations: ChIP, chromatin immunoprecipitation; IgG, immunoglobulin G; WB, Western blot.
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assay was performed as we have previously described.42,43 Primary cells from UFs were grown in media, either in the presence or absence of DZNep for 3 days. Cells (1 × 107) were then incubated with 1% formaldehyde for 10 minutes to cross-link histones to DNA. After washing with cold phosphate-buffered saline, cell pellets were resuspended in a cell lysis buffer (10 mmol/L Tris, pH 8.0, 10 mmol/L NaCl, 0.2% NP40). Nuclei were resuspended in nuclei lysis buffer (50 mmol/L Tris pH 8.0, 10 mmol/L EDTA, 1% SDS) and sonicated for 25 minutes. The soluble chromatin fraction was collected, and 5 μL of antibody for H3K27me3 (Active Motif, Carlsbad, CA) or normal rabbit immunoglobulin G was added. After incubation, chromatin–antibody complexes were collected using A/G magnetic beads (Millipore, Billerica, MA). After washing, immunoprecipitated DNA was treated with proteinase K at 62°C for 2 hours. DNA was extracted with a QIAquick PCR Purification kit (Qiagen, Valencia, CA) and analyzed by SYBR green real-time PCR. Primer pairs used for ChIP assays are shown in Table 1.
Statistical Analysis
Statistical analysis of the data was performed using a single-factor analysis of variance and the standard 2-sample Student t test for normally distributed continuous variables. Statistical significance was determined using a 2-tailed distribution assumption and set at P < .05.
Results
The Expression Levels of MSH2 in Fibroid as Compared to Matched Myometrial Tissues
Since MSH2 has been shown to be involved in the development of varied types of cancer, we initially measured expression levels of MSH2 by quantitative PCR in 8 patients using fibroids and adjacent myometrial samples. As shown in Figure 1A, expression levels of MSH2 were deregulated in fibroid tissues as compared to adjacent myometrial tissues. The RNA expression of MSH2 was upregulated in 62.5% (5 of 8) of UF lesions as compared to matched adjacent myometrial tissues. In addition, 37.5% (3 of 8) of UF lesions exhibit reduced expression as compared to myometrium samples.
Figure 1.

The expression levels of DNA mismatch repair gene MSH2 in human fibroid and adjacent myometrial tissues. A, The RNA expression of MSH2 was determined in fibroid tumors as compared to matched adjacent myometrial samples by real-time polymerase chain reaction (n = 8). 18S was used as an endogenous control. B, The protein lysates were prepared from fibroids (F; n = 10) and matched myometrium tissues (M). The protein expression of MSH2 was determined by Western blot analysis. MSH2 protein bands were quantified and normalized to β-actin, and relative values were used to generate data graphs (bottom panel). Each short horizontal line indicates a pair of M and F from same patient.
Next, we performed Western blot analysis to determine the expression levels of MSH2 in UFs as compared to adjacent MyoF myometrial tissues (n = 10). As shown in Figure 1B, protein levels of MSH2 were upregulated in 90% of UF lesions (9 of 10 patients) as compared to matched adjacent myometrial tissues.
Inhibition of EZH2 Increased the Expression of MSH2 in Human Primary Fibroid Cells
EZH2 has been shown to regulate several DNA repair genes.32–34 In this regard, we determine the mechanism underlying the deregulation of MSH2 expression in human fibroid lesions. We first treated human primary fibroid cells (PFCs) with EZH2 inhibitor (DZNep). As shown in Figure 2A, DZNep treatment decreased expression levels of H3K27me3 in a dose-dependent manner, suggesting that the expression of the epigenetic mark H3K27me3 is highly dependent on EZH2 activity. To determine whether MSH2 expression was regulated by EZH2, we measured the expression levels of MSH2 in the PFCs treated with DZNep. As shown in Figure 2B, MSH2 protein expression is significantly upregulated in PFCs treated with DZNep in a dose-dependent manner. Accordingly, MSH2 RNA expression was also upregulated in the PFCs treated with DZNep (Figure 2C). Since PRC2 complex also contains SUZ12 and EED, we also measured the expression levels of these genes. As shown in Figure 2D and E, treatment with DZNep slightly upregulated EED, but not SUZ12. It has been reported that EZH2 is associated with cell proliferation,19 therefore, we determined the cell cycle regulatory gene p27. As shown in Figure 2F, inhibition of EZH2 significantly upregulated p27 expression in a dose-dependent manner.
Figure 2.

Inhibition of EZH2 increases the expression of MSH2 in a dose-dependent manner in human fibroid cells. A, Inhibition of EZH2 by 3-deazaneplanocin A (DZNep) decreased levels of H3K27me3 by Western blot analysis. Primary fibroid cells (PFCs) were plated in 60-mm dish. Cells were treated with varying concentrations of EZH2 inhibitor DZNep for 3 days. Dimethyl sulfoxide (DMSO) was used as a vehicle control. B, The protein lysates were prepared from PFCs treated with vehicle or 1 μmol/L DZNep for 3 days. The protein levels of MSH2 were determined by Western blot analysis. In addition, cells were treated with varying concentrations of EZH2 inhibitor DZNep for 3 days. Cells were collected and subjected to RNA extraction and complementary DNA (cDNA) synthesis. Quantitative polymerase chain reaction was performed to measure the expression levels of MSH2 (C), SUZ12 (D), EED (E), and P27 (F). 18S was used as an endogenous control.
Overexpression of EZH2 Enhances HuLM Cell Proliferation
To determine the effect of EZH2 on cell proliferation, 3-(4,5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assay was performed. As shown in Figure 3, although there is no significant difference in the rate of cell proliferation between EZH2 and GFP control group after infection for 3 days, overexpression of EZH2 significantly enhanced the HuLM cell proliferation by 44.4% ± 2.7% as compared to GFP control after 6 days of infection (P < .05).
Figure 3.
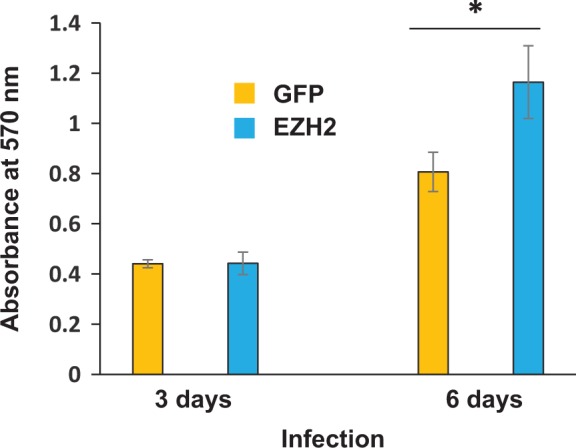
Overexpression of EZH2 increases HuLM cell proliferation. HuLM cells were plated in 96-well dish. Cellular confluence was approximately 30%. Equal amounts of virus (20 plaque-forming unit [pfu]/cell) containing GFP or EZH2 gene were added to the medium, respectively. Eight hours later, the viral supernatant was removed and fresh medium was added. After 3 days and 6 days of infection, MTT assay was performed to determine absorbance at 570 nm. *P < .05 compared with the control.
Overexpression of EZH2 Upregulated H3K27me3 Levels in HuLM Cells Associated With Downregulation of MSH2
We next determined whether an overexpression of EZH2 would result in upregulation of H3K27me3 in human fibroid cells. As shown in Figure 4A, overexpression of EZH2 markedly elevated the level of epigenetic mark H3K27me3 in HuLM cells. The expression of EZH2 was primarily in the nucleus (Figure 4B), suggesting that ectopic induction of EZH2 may directly affect gene expression. Quantitative RNA expression analysis exhibited significant downregulation of MSH2 in EZH2-overexpressed HuLM cells as compared to the GFP control, P < .05 (Figure 4C). Additionally, overexpression of EZH2 significantly downregulated cell cycle regulatory gene p27 in HuLM cells as compared to the control group, P < .05 (Figure 4D). To further determine the EZH2-mediated downregulation of MSH2 expression and localization at protein level, we prepared protein lysates from total cells, nucleus, and cytoplasm of HuLM cells infected with Ad-EZH2 and Ad-GFP, respectively, and measure the MSH2 protein levels by Western blot analysis. As shown in Figure 4E, MSH2 protein levels were markedly downregulated in the HuLM cells infected with Ad-EZH2 virus as compared to Ad-GFP virus (Figure 4E, top panel). The major decrease in EZH2 protein expression comes from reduced levels of EZH2 protein in the nucleus (Figure 4E, middle panel), although slight decrease in the MSH2 expression in the cytoplasm from the cells infected with Ad-EZH2 can be seen (Figure 4E. low panel).
Figure 4.

Overexpression of EZH2 using adenoviral vector decreases MSH2 expression in HuLM cells. A, EZH2 was overexpressed in HuLM cells infected with adenovirus containing EZH2 as compared to cells infected with adenovirus containing GFP. Levels of epigenetic mark H3K27me3 were also increased in HuLM cells infected with virus containing EZH2. B, EZH2 was found in both cytoplasm and nucleus; however, higher expression of EZH2 was observed in the nucleus as compared to the cytoplasm. C, MSH2 RNA expression was downregulated in HuLM cells overexpressed with EZH2 as compared to GFP control. D, Cell cycle regulatory gene p27 is downregulated in EZH2-overexpressed cells as compared to GFP control cells. *P < .05 compared with the control. Total cell lysates (E), nucleus (F), and cytoplasm (G) from HuLM cells infected with Ad-EZH2 or Ad-GFP were analyzed by Western blot using Anti-MSH2 antibody. Western blot with anti–β-actin antibody was used as the loading control. MSH2 protein bands were quantified and normalized to β-actin, and relative values were used to generate data graphs (right panels).
MSH2 is an Epigenetic Target of EZH2
To determine whether MSH2 is directly regulated by EZH2, the enrichment of the bivalent mark of H3K27me3 in the promoter regions of MSH2 was examined by ChIP assay in human fibroid primary cells. The promoter mapping in upper regulatory region of the MSH2 gene has been previously described44 and also in Figure 5A. Following DZNep treatment at 0.25 μmol/L, the enrichment of H3K27me3 in the distal promoter region of MSH2 was unaffected, as shown in Figure 5B. Following DZNep treatment at 1 μmol/L, enrichment of H3K27me3 in the distal promoter region of MSH2 was significantly decreased in response to DZNep treatment (Figure 5B, P < .05). Enrichment of H3K27me3 in the proximal promoter region of MSH2 was also investigated, as shown in Figure 5C. Decreased enrichment of H3K27me3 in the proximal promoter region of MSH2 was detected after treatment with 0.25 μmol/L DZNep as compared to the vehicle control, although a significant difference was not reached. At 1 μmol/L DZNep treatment, no binding of H3K27me3 in the proximal promoter region of MSH was observed as shown in Figure 5C (P < .05). These data suggest that disruption of PRC2 by DZNep upregulates expression of MSH2 through lowering or even eliminating the level of epigenetic mark H3K27me3 presence at both the distal and proximal MSH2 promoter regions.
Figure 5.

DZNep treatment restores expression levels of MSH2 through epigenetic mark H3K27me3. A, Location of regions analyzed by chromatin immunoprecipitation (ChIP)/polymerase chain reaction (PCR) along human MSH2 promoter. The position of the transcriptional start site is designated as +1. Short horizontal lines indicate regions analyzed by ChIP/PCR. B, ChIP/quantitative PCR (q-PCR) was performed with anti-H3K27me3 antibody in the distal promoter region of MSH2 in human fibroid primary cells in the presence or absence of DZNep. C, ChIP/q-PCR was performed with anti-H3K27me3 antibody in the proximal promoter region of MSH2 in human fibroid primary cells in the presence or absence of DZNep. *P < .05 compared with the control.
Discussion
Deficient DNA MMR results in a strong mutator phenotype, known as microsatellite instability.45 Accumulating evidence shows that MSH2 aberrations are involved in many biological events associated with a mutator phenotype and cancer susceptibility.46–53 Yoo et al54 reported that deficiency of MSH2 is associated with clear cell renal carcinoma. Pritchard et al demonstrated that mutations in MSH2/MSH6 complex or MSH6 structural rearrangements are frequently encountered in advanced prostate cancer.55 Somatic rather than germline mutation of MMR genes can be found in colon and endometrial cancers.56,57 Immunohistochemical staining of MMR proteins including MSH2, MLH1, MSH6, and PMS2 demonstrates loss of MMR proteins in some uterine sarcomas and carcinosarcomas.58
A growing amount of evidence consistently supports the concept that PcG proteins play a role in cell cycle progression.19,59 For instance, overexpression of EZH2 enhances proliferation of B-cell lymphoma cell line.60 Furthermore, EZH2 is highly expressed in a wide range of cancers, including breast, prostate, bladder, colon, lung, pancreatic cancers, sarcoma, and lymphomas.61–64 Overexpression of EZH2 is frequently correlated with advanced stages of human cancer and with poor prognosis.27,65 In this study, we reported that overexpression of EZH2 increased the HuLM cell proliferation associated with decreased expression of cell cycle regulatory gene p27, suggesting that EZH2 is capable of altering fibroid cell phenotype. Our study is consistent with previous findings showing similar impact of EZH2 on cell proliferation of other types of tumors.66,67 For example, deactivation of EZH2 by DZNep treatment resulted in decreased proliferation of cholangiocarcinoma and non-small cell lung cancer cells and induced G1-phase cell cycle arrest.66,67 Additionally, disruption of EZH2 by DZNep inhibits cell proliferation, tumorigenicity, and tumor progression in prostate cancer.68 Specific inhibition of EZH2 by short hairpin RNA efficiently inhibits the growth of numerous cancer cell types.69,70
The mechanism by which EZH2 promotes tumor cell proliferation has been investigated in several types of tumors.28,62 EZH2 increases cell proliferation through cell cycle regulatory genes.27 For instance, G1 cell cycle arrest, induced by inhibition of EZH2, in non-small cell lung cancer cells is associated with p27 accumulation,67 which is consistent with our finding (Figure 4D); we showed that overexpression of EZH2 in fibroid cells led to increased cell proliferation, which was associated with decreased expression of p27. In addition, EZH2-dependence suppression of a cellular senescence phenotype in melanoma cells occurs through the inhibition of p21 expression.71 EZH2 depletion inhibited the proliferation and arrested G1/S phase of nasopharyngeal carcinoma cells with associated increase in the expression of p16.72 Recent studies also demonstrate that several microRNAs regulate many types of cancer cell proliferation or phenotype by targeting EZH2.73–76 These studies strongly suggest that the aberrant expression of polycomb protein EZH2 is involved in abnormal cell proliferation and the pathogenesis of tumor initiation and progression.
Independent clonal origin of multiple UFs was determined by microsatellite analysis,77,78 suggesting that dysfunctional DNA MMR system may result in lower DNA repair capacity leading to UF development. In this study, we found that RNA expression levels of MSH2 were deregulated in a subset of fibroid tumors as compared to adjacent myometrial tissues. Notably, protein levels of MSH2 were upregulated in 90% (9/10) of fibroid tissues as compared to matched adjacent myometrial tissues. These findings are in accordance with other studies showing increased expression of MSH2 in cancers such as gastric cancer.79,80 The increased expression of MSH2 in fibroid tumors as compared to adjacent myometrium samples may be due to several possible mechanisms. It is possible that increased expression of MSH2 could be a cellular adaption for DNA lesion repair. Another explanation may be that the increased expression of MSH2 could represent a possible response to the rapidly growing number of replication errors in the fibroid tissue with an increased rate of cell divisions.79,80 Moreover, MSH2 levels can be reduced by ubiquitin-proteasome pathway.81 Since PRC has been shown to regulate several DNA repair genes, we performed experiment to determine whether EZH2 regulates MSH2 expression. We first treated PFCs with EZH2 inhibitor (DZNep) and determine whether inhibition of EZH2 alters MSH2 expression. As shown in Figure 2, DZNep treatment showed a robust increase in the expression of MSH2 in a dose-dependent manner associated with decreased levels of H3K27me3. In addition, overexpression of EZH2 decreased the MSH2 protein levels mainly in the nucleus (Figure 4). Previous studies82 have shown that DZNep treatment in cancer cells resulted in dramatic decreases in the protein levels of 3 PRC2 components including SUZ12, EZH2, and EED. In this study, we determined the RNA levels of SUZ12 and EED following treatment with DZNep. Although expression level of EED is slightly increased in response to DZNep treatment, there is no difference in SUZ12 RNA levels between DZNep- and vehicle-treated cells. Our data are in line with a previous observation that DZNep treatment does not decrease RNA levels of PRC2 components but rather may deplete the protein levels of PRC2 components through the protein degradation pathway.82
It is well recognized that polycomb group proteins (PcG), which alter chromatin structure such that epigenetic silencing of genes take place, bind to specific regions of gene promoters and direct posttranslational modifications at certain histone sites, thereby silencing gene expression.83 The genome-wide mapping of PcG target genes revealed more than 2000 sites in the mouse embryonic stem cell genome. These loci are associated with increased levels of H3K27me3 repressive marks, suggesting that polycomb repression affects numerous genes encoding key developmental regulators and signaling proteins.84 In humans, many genes regulated by canonical or noncanonical EZH2 activity have been identified.85–87 Among them, EZH2-mediated double-strand breaks have been discovered, which are related to dysfunctional DNA damage repair system.88 Overexpression of EZH2 in breast epithelial cells results in a decrease in messenger RNA and protein levels of the RAD51 paralog genes.88 In addition, EZH2 overexpression led to a significant decrease in the number of RAD51 repair nuclear foci after induction of double-strand breaks.34 Studies by Chang et al identify a mechanism by which EZH2-mediated downregulation of DNA damage-repair leads to accumulation of recurrent RAF1 gene amplification in breast tumor-imitating cells (BTICs), which in turn activates downstream signaling to promote BTIC expansion in aggressive breast cancer.32 In our study, we first demonstrated that MSH2, an MMR gene, is a novel target of EZH2 in fibroid cells. We demonstrated that overexpression of EZH2 by viral transduction decreased the expression of MSH2 expression. EZH2 expression was found to be mainly located in the nucleus. Disruption of EZH2 by DZNep treatment increased MSH2 expression in fibroid cells. Moreover, we found that EZH2 is involved in the downregulation of DNA MMR gene MSH2 through H3K27me3 in fibroids. Enrichment of H3K27me3 in both the distal and proximal promoter regions of MSH2 is markedly decreased in fibroid primary cells following treatment with EZH2 inhibitor, which is concurrently associated with increased expression of MSH2. The decreased binding levels of H3K27me3 in the promoter region of MSH2 in response to DZNep treatment suggests the important role of canonical EZH2 activity in regulating DNA MMR gene MSH2 in human UFs. Although through gain and loss function of EZH2 study, we demonstrated that EZH2 regulated MSH2 RNA expression leading to altering its protein levels; no clear correlation between RNA and protein expression was observed in fibroid tissues and myometrium, suggesting that translational regulation or protein stability pathway may be involved in regulation of MSH2 protein levels.
In conclusion, our studies provide the first evidence showing that expression of DNA MMR gene MSH2 is deregulated in fibroid tumors as compared with matched adjacent myometrial tissues. Importantly, polycomb protein EZH2 regulates MSH2 through epigenetic mark H3K27me3 in the promoter regions. Further research aimed at better understanding of the role of MSH2 and other MMR proteins in UF development is warranted. MSH2 could be considered as a potential marker for early detection of UFs. This in turn could lead to novel medical treatments for women impacted by symptomatic UFs.
Acknowledgements
The authors thank Walidah Walker, MPH, for editing this manuscript.
Authors’ Note: Q.Y. and A.A. contributed to conception and design of research. Q.Y., A.L., and L.E. conducted the experiments. Q.Y. and A.L. analyzed the results. L.G. and J.L. provided patient samples. Q.Y. interpreted results of experiments and drafted the manuscript. Q.Y., N.I., L.G., M.D., and A.A. revised the manuscript. All authors approved the revised version of the manuscript.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by an Augusta University Startup package, the National Institutes of Health Grant HD04622811 (to A.A.), and the Augusta University Intramural Grants Program (Q.Y.).
References
- 1. Bulun SE. Uterine fibroids. N Engl J Med. 2013;369(14):1344–1355. [DOI] [PubMed] [Google Scholar]
- 2. Al-Hendy A, Salama S. Gene therapy and uterine leiomyoma: a review. Hum Reprod Update. 2006;12(4):385–400. [DOI] [PubMed] [Google Scholar]
- 3. Heinonen HR, Sarvilinna NS, Sjoberg J, et al. MED12 mutation frequency in unselected sporadic uterine leiomyomas. Fertil Steril. 2014;102(4):1137–1142. [DOI] [PubMed] [Google Scholar]
- 4. Rieker RJ, Agaimy A, Moskalev EA, et al. Mutation status of the mediator complex subunit 12 (MED12) in uterine leiomyomas and concurrent/metachronous multifocal peritoneal smooth muscle nodules (leiomyomatosis peritonealis disseminata). Pathology. 2013;45(4):388–392. [DOI] [PubMed] [Google Scholar]
- 5. Halder SK, Laknaur A, Miller J, Layman LC, Diamond M, Al-Hendy A. Novel MED12 gene somatic mutations in women from the Southern United States with symptomatic uterine fibroids. Mol Genet Genomics. 2015;290(2):505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bertsch E, Qiang W, Zhang Q, et al. MED12 and HMGA2 mutations: two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol. 2014;27(8):1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hunter DS, Klotzbucher M, Kugoh H, et al. Aberrant expression of HMGA2 in uterine leiomyoma associated with loss of TSC2 tumor suppressor gene function. Cancer Res. 2002;62(13):3766–3772. [PubMed] [Google Scholar]
- 8. Ingraham SE, Lynch RA, Surti U, et al. Identification and characterization of novel human transcripts embedded within HMGA2 in t(12;14)(q15;q24.1) uterine leiomyoma. Mutat Res. 2006;602(1-2):43–53. [DOI] [PubMed] [Google Scholar]
- 9. El-Shennawy GA, Elbialy AA, Isamil AE, El Behery MM. Is genetic polymorphism of ER-alpha, CYP1A1, and CYP1B1 a risk factor for uterine leiomyoma? Arch Gynecol Obstet. 2011;283(6):1313–1318. [DOI] [PubMed] [Google Scholar]
- 10. Vikhliaeva EM. Molecular-genetic determinants of the neoplastic process and state-of-the-art treatment of patients with uterine leiomyoma [in Russian]. Vopr Onkol. 2001;47(2):200–204. [PubMed] [Google Scholar]
- 11. Yang Y, Zhai XD, Gao LB, Li SL, Wang Z, Chen GD. Genetic polymorphisms of DNA repair gene XRCC1 and risk of uterine leiomyoma. Mol Cell Biochem. 2010;338(1-2):143–147. [DOI] [PubMed] [Google Scholar]
- 12. Ramos JM, Ruiz A, Colen R, Lopez ID, Grossman L, Matta JL. DNA repair and breast carcinoma susceptibility in women. Cancer. 2004;100(7):1352–1357. [DOI] [PubMed] [Google Scholar]
- 13. Ricks-Santi LJ, Sucheston LE, Yang Y, et al. Association of Rad51 polymorphism with DNA repair in BRCA1 mutation carriers and sporadic breast cancer risk. BMC Cancer. 2011;11:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. [DOI] [PubMed] [Google Scholar]
- 15. Benhamou S, Sarasin A. Variability in nucleotide excision repair and cancer risk: a review. Mutat Res. 2000;462(2-3):149–158. [DOI] [PubMed] [Google Scholar]
- 16. Matta JL, Villa JL, Ramos JM, et al. DNA repair and nonmelanoma skin cancer in Puerto Rican populations. J Am Acad Dermatol. 2003;49(3):433–439. [DOI] [PubMed] [Google Scholar]
- 17. Belcheva A, Kolaj B, Martin A. Missing mismatch repair: a key to T cell immortality. Leuk Lymphoma. 2010;51(10):1777–1778. [DOI] [PubMed] [Google Scholar]
- 18. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7(3):153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bachmann IM, Halvorsen OJ, Collett K, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24(2):268–273. [DOI] [PubMed] [Google Scholar]
- 20. Kleer CG, Cao Q, Varambally S, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100(20):11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Majewski IJ, Blewitt ME, de Graaf CA, et al. Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biol. 2008;6(4):e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stefansson OA, Esteller M. EZH2-mediated epigenetic repression of DNA repair in promoting breast tumor initiating cells. Breast Cancer Res. 2011;13(3):309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoo KH, Hennighausen L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Biol Sci. 2012;8(1):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu H, Simons DL, Segall I, et al. PRC2/EED-EZH2 complex is up-regulated in breast cancer lymph node metastasis compared to primary tumor and correlates with tumor proliferation in situ. PloS One. 2012;7(12):e51239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamaguchi H, Hung MC. Regulation and role of EZH2 in cancer. Cancer Res Treat. 2014;46(3):209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106(2):243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen YH, Hung MC, Li LY. EZH2: a pivotal regulator in controlling cell differentiation. Am J Transl Res. 2012;4(4):364–375. [PMC free article] [PubMed] [Google Scholar]
- 29. Holm K, Grabau D, Lovgren K, et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol Oncol. 2012;6(5):494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan JZ, Yan Y, Wang XX, Jiang Y, Xu HE. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin. 2014;35(2):161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kondo Y. Targeting histone methyltransferase EZH2 as cancer treatment. J Biochem. 2014;156(5):249–257. [DOI] [PubMed] [Google Scholar]
- 32. Chang CJ, Yang JY, Xia W, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19(1):86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Puppe J, Drost R, Liu X, et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to polycomb repressive complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009;11(4):R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeidler M, Varambally S, Cao Q, et al. The polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia. 2005;7(11):1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Akinyemi BO, Adewoye BR, Fakoya TA. Uterine fibroid: a review. Niger J Med. 2004;13(4):318–329. [PubMed] [Google Scholar]
- 36. Yang Q, Mas A, Diamond MP, Al-Hendy A. The mechanism and function of epigenetics in uterine leiomyoma development. Reprod Sci. 2016;23(2):163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carney SA, Tahara H, Swartz CD, et al. Immortalization of human uterine leiomyoma and myometrial cell lines after induction of telomerase activity: molecular and phenotypic characteristics. Lab Invest. 2002;82(6):719–728. [DOI] [PubMed] [Google Scholar]
- 38. Halder SK, Osteen KG, Al-Hendy A. Vitamin D3 inhibits expression and activities of matrix metalloproteinase-2 and -9 in human uterine fibroid cells. Hum Reprod. 2013;28(9):2407–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nair S, Curiel DT, Rajaratnam V, Thota C, Al-Hendy A. Targeting adenoviral vectors for enhanced gene therapy of uterine leiomyomas. Hum Reprod. 2013;28(9):2398–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nair S, Saed GM, Atta HM, et al. Towards gene therapy of postoperative adhesions: fiber and transcriptional modifications enhance adenovirus targeting towards human adhesion cells. Gynecol Obstet Invest. 2013;76(2):119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang Q, Sun M, Ramchandran R, Raj JU. IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: role of epigenetic regulation. Vascular Pharmacol. 2015;73:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Q, Tian Y, Ostler KR, et al. Epigenetic alterations differ in phenotypically distinct human neuroblastoma cell lines. BMC Cancer. 2010;10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang Q, Dahl MJ, Albertine KH, Ramchandran R, Sun M, Raj JU. Role of histone deacetylases in regulation of phenotype of ovine newborn pulmonary arterial smooth muscle cells. Cell Prolif. 2013;46(6):654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iwahashi Y, Ito E, Yanagisawa Y, et al. Promoter analysis of the human mismatch repair gene hMSH2. Gene. 1998;213(1-2):141–147. [DOI] [PubMed] [Google Scholar]
- 45. Yamamoto H, Imai K. Microsatellite instability: an update. Arch Toxicol. 2015;89(6):899–921. [DOI] [PubMed] [Google Scholar]
- 46. Campbell MR, Wang Y, Andrew SE, Liu Y. MsH2 deficiency leads to chromosomal abnormalities, centrosome amplification, and telomere capping defect. Oncogene. 2006;25(17):2531–2536. [DOI] [PubMed] [Google Scholar]
- 47. Campbell MR, Nation PN, Andrew SE. A lack of DNA mismatch repair on an athymic murine background predisposes to hematologic malignancy. Cancer Res. 2005;65(7):2626–2635. [DOI] [PubMed] [Google Scholar]
- 48. Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene MsH2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12(3):273–281. [DOI] [PubMed] [Google Scholar]
- 49. Lal G, Ash C, Hay K, et al. Suppression of intestinal polyps in MsH2-deficient and non-MsH2-deficient multiple intestinal neoplasia mice by a specific cyclooxygenase-2 inhibitor and by a dual cyclooxygenase-1/2 inhibitor. Cancer Res. 2001;61(16):6131–6136. [PubMed] [Google Scholar]
- 50. Bridge G, Rashid S, Martin SA. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers. 2014;6(3):1597–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Russo MT, De Luca G, Casorelli I, et al. Role of MUTYH and MSH2 in the control of oxidative DNA damage, genetic instability, and tumorigenesis. Cancer Res. 2009;69(10):4372–4379. [DOI] [PubMed] [Google Scholar]
- 52. Meira LB, Cheo DL, Reis AM, et al. Mice defective in the mismatch repair gene MsH2 show increased predisposition to UVB radiation-induced skin cancer. DNA Repair. 2002;1(11):929–934. [DOI] [PubMed] [Google Scholar]
- 53. Reitmair AH, Schmits R, Ewel A, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11(1):64–70. [DOI] [PubMed] [Google Scholar]
- 54. Yoo KH, Won KY, Lim SJ, Park YK, Chang SG. Deficiency of MSH2 expression is associated with clear cell renal cell carcinoma. Oncol Lett. 2014;8(5):2135–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pritchard CC, Morrissey C, Kumar A, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014;5:4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308–1316.e1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Amant F, Dorfling CM, Dreyer L, Vergote I, Lindeque BG, Van Rensburg EJ. Microsatellite instability in uterine sarcomas. Int J Gynecol Cancer. 2001;11(3):218–223. [DOI] [PubMed] [Google Scholar]
- 58. Hoang LN, Ali RH, Lau S, Gilks CB, Lee CH. Immunohistochemical survey of mismatch repair protein expression in uterine sarcomas and carcinosarcomas. Int J Gynecol Pathol. 2014;33(5):483–491. [DOI] [PubMed] [Google Scholar]
- 59. Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. [DOI] [PubMed] [Google Scholar]
- 60. Visser HP, Gunster MJ, Kluin-Nelemans HC, et al. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112(4):950–958. [DOI] [PubMed] [Google Scholar]
- 61. Cao W, Younis RH, Li J, et al. EZH2 promotes malignant phenotypes and is a predictor of oral cancer development in patients with oral leukoplakia. Cancer Prev Res (Phila). 2011;4(11):1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17(9):2613–2618. [DOI] [PubMed] [Google Scholar]
- 63. Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647(1-2):21–29. [DOI] [PubMed] [Google Scholar]
- 64. Wu Z, Lee ST, Qiao Y, et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18(11):1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7(3):299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nakagawa S, Sakamoto Y, Okabe H, et al. Epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A inhibits the growth of cholangiocarcinoma cells. Oncol Rep. 2014;31(2):983–988. [DOI] [PubMed] [Google Scholar]
- 67. Kikuchi J, Takashina T, Kinoshita I, et al. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer. 2012;78(2):138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Crea F, Hurt EM, Mathews LA, et al. Pharmacologic disruption of polycomb repressive complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer. 2011;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40(6):741–750. [DOI] [PubMed] [Google Scholar]
- 70. Bracken AP, Kleine-Kohlbrecher D, Dietrich N, et al. The polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21(5):525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fan T, Jiang S, Chung N, et al. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res. 2011;9(4):418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhong J, Min L, Huang H, et al. EZH2 regulates the expression of p16 in the nasopharyngeal cancer cells. Technol Cancer Res Treat. 2013;12(3):269–274. [DOI] [PubMed] [Google Scholar]
- 73. Lin L, Zheng Y, Tu Y, et al. MicroRNA-144 suppresses tumorigenesis and tumor progression of astrocytoma by targeting EZH2. Hum Pathol. 2015;46(7):971–980. [DOI] [PubMed] [Google Scholar]
- 74. Chen DL, Zhang DS, Lu YX, et al. microRNA-217 inhibits tumor progression and metastasis by downregulating EZH2 and predicts favorable prognosis in gastric cancer. Oncotarget. 2015;6(13):10868–10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu F, He Y, Shu R, Wang S. MicroRNA-1297 regulates hepatocellular carcinoma cell proliferation and apoptosis by targeting EZH2. Int J Clin Exp Pathol. 2015;8(5):4972–4980. [PMC free article] [PubMed] [Google Scholar]
- 76. Sun J, Zheng G, Gu Z, Guo Z. MiR-137 inhibits proliferation and angiogenesis of human glioblastoma cells by targeting EZH2. J Neurooncol. 2015;122(3):481–489. [DOI] [PubMed] [Google Scholar]
- 77. Canevari RA, Pontes A, Rosa FE, Rainho CA, Rogatto SR. Independent clonal origin of multiple uterine leiomyomas that was determined by X chromosome inactivation and microsatellite analysis. Am J Obstet Gynecol. 2005;193(4):1395–1403. [DOI] [PubMed] [Google Scholar]
- 78. French D, Cermele C, Lombardi AM, et al. Microsatellite alterations in uterine leiomyomas. Anticancer Res. 1998;18(1A):349–351. [PubMed] [Google Scholar]
- 79. Li M, Liu L, Wang Z, et al. Overexpression of hMSH2 and hMLH1 protein in certain gastric cancers and their surrounding mucosae. Oncol Rep. 2008;19(2):401–406. [PubMed] [Google Scholar]
- 80. Dracea A, Angelescu C, Danciulescu M, Ciurea M, Ioana M, Burada F. Mismatch repair gene expression in gastroesophageal cancers. Turk J Gastroenterol. 2015;26(5):373–377. [DOI] [PubMed] [Google Scholar]
- 81. Zhang M, Xiang S, Joo HY, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Molecular Cell. 2014;55(1):31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21(9):1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Muller J, Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev. 2009;19(2):150–158. [DOI] [PubMed] [Google Scholar]
- 84. Volkel P, Dupret B, Le Bourhis X, Angrand PO. Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res. 2015;7(2):175–193. [PMC free article] [PubMed] [Google Scholar]
- 85. Shi B, Liang J, Yang X, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27(14):5105–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lee ST, Li Z, Wu Z, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Molecular Cell. 2011;43(5):798–810. [DOI] [PubMed] [Google Scholar]
- 87. Gao SB, Zheng QF, Xu B, et al. EZH2 represses target genes through H3K27-dependent and H3K27-independent mechanisms in hepatocellular carcinoma. Mol Cancer Res. 2014;12(10):1388–1397. [DOI] [PubMed] [Google Scholar]
- 88. Zeidler M, Kleer CG. The Polycomb group protein Enhancer of Zeste 2: its links to DNA repair and breast cancer. J Mol Histol. 2006;37(5-7):219–223. [DOI] [PubMed] [Google Scholar]