

Abstract
Endometriosis is an estrogen (ER)-dependent gynecological disease caused by the growth of endometrial tissue at extrauterine sites. Current endocrine therapies address the estrogenic aspect of disease and offer some relief from pain but are associated with significant side effects. Immune dysfunction is also widely believed to be an underlying contributor to the pathogenesis of this disease. This study evaluated an inhibitor of c-Jun N-terminal kinase, bentamapimod (AS602801), which interrupts immune pathways, in 2 rodent endometriosis models. Treatment of nude mice bearing xenografts biopsied from women with endometriosis (BWE) with 30 mg/kg AS602801 caused 29% regression of lesion. Medroxyprogesterone acetate (MPA) or progesterone (PR) alone did not cause regression of BWE lesions, but combining 10 mg/kg AS602801 with MPA caused 38% lesion regression. In human endometrial organ cultures (from healthy women), treatment with AS602801 or MPA reduced matrix metalloproteinase-3 (MMP-3) release into culture medium. In organ cultures established with BWE, PR or MPA failed to inhibit MMP-3 secretion, whereas AS602801 alone or MPA + AS602801 suppressed MMP-3 production. In an autologous rat endometriosis model, AS602801 caused 48% regression of lesions compared to GnRH antagonist Antide (84%). AS602801 reduced inflammatory cytokines in endometriotic lesions, while levels of cytokines in ipsilateral horns were unaffected. Furthermore, AS602801 enhanced natural killer cell activity, without apparent negative effects on uterus. These results indicate that bentamapimod induced regression of endometriotic lesions in endometriosis rodent animal models without suppressing ER action. c-Jun N-terminal kinase inhibition mediated a comprehensive reduction in cytokine secretion and moreover was able to overcome PR resistance.
Keywords: endometriosis, JNK inhibitor, MMP, progesterone, cytokine, bentamapimod, MPA.
Introduction
Endometriosis is characterized by the presence of endometrial tissue outside the uterine cavity that results in lesions containing glandular epithelium and stromal cells, macrophages, natural killer (NK) cells, and T lymphocytes.1-3 Although most women experience retrograde menstruation causing introduction of endometrial fragments into the peritoneal cavity, endometriosis only develops in a proportion (5%-10%) of women. It is currently believed that external factors influence the response to this ectopic tissue in susceptible women, and the response does not effectively eliminate the adhered tissue from the peritoneal surface. Current hypotheses propose that over time (≥9 months), lesions progress from a hormone-dependent lesion to an endocrine-independent lesion,4,5 and a subpopulation of cells undergo epithelial–mesenchymal transdifferentiation6,7 and over time develop perturbed gene expression patterns in response to progesterone (PR) or cyclic adenosine monophosphate.8 As a result of endocrine resistance and moderate cellular differentiation, lesions become resistant to conventional steroidal-based treatments.
Currently approved treatments of endometriosis consist of hormonal therapy and/or nonsteroidal anti-inflammatory drugs (NSAIDs) and surgery.9,10 Among the hormonal therapies are combined oral contraceptives (combinations of estrogen [ER] and PR), PR only (norethindrone), ER antagonists (danazol), and GnRH agonists (Lupron).9 Approved endocrine-based therapies all share as a common mechanism the disruption of ER production or action that also result in side effects associated with menopausal symptoms or increased androgenic effects. Approved anti-inflammatory drugs (NSAIDs; ibuprofen, naproxen, ketoprofen, and mefenamic acid) are used to reduce prostanoid synthesis and are prescribed in anticipation of pain associated with disease.11
There is considerable evidence to suggest that the immune system is involved in the pathogenesis of endometriosis.12,13 The presence of ectopic endometrium in the peritoneum elicits classical inflammatory responses, including activation of peritoneal macrophages and increased cytokine production.14 Elevated tumor necrosis factor α (TNF-α) levels in peritoneal fluid have been associated with upregulation of this cytokine production in peritoneal macrophages and peripheral monocytes and increased endometrial cell proliferation and adhesion.15,16
Recently, several approaches for suppressing circulating levels of TNF-α have been shown to cause regression of disease in a baboon model. Recombinant human TNF-α binding protein (TBP; onercept) was shown to reduce the size and severity of endometriotic lesions in a primate model of endometriosis.17 Etanercept18 and infliximab19 have also been shown to cause regression of lesions in baboons with spontaneous or induced endometriosis. However, further development of molecules that neutralize circulating TNF-α for the treatment of endometriosis has proceeded cautiously due to the risk-to-benefit ratio for these agents in this disease. Therapeutic agents that neutralize circulating TNF-α levels have been demonstrated to increase the risk of infections in immune-compromised patients.20
Stimulation of proinflammatory TNF-α receptor in endometriotic cell types leads to activation of kinase cascades. In endometriotic stromal cells, TNF-α stimulates ER receptor transcription via an extracellular signal-regulated kinase pathway, including c-Jun N-terminal kinase (JNK), and p38 protein kinases.21 c-Jun N-terminal kinase inhibitors decreased generation of TNF-α following LPS stimulation, decreased inflammation score in collagen-induced arthritis,22 and reduced proliferation of immune cells causing autoimmune inflammation in multiple sclerosis.23 Furthermore, several intracellular kinases linked with JNK pathways were found elevated in 3 independent studies using microarray analysis of human lesions.24-27 Levels of c-Jun were found 1.5-fold higher in endometrium from women with endometriosis compared to unaffected women.28 In the eutopic endometrium of patients, presence of phospho-JNK is elevated in endometrial endothelial cells.29 In baboons, MKK7, MAPK9, as well as c-fos and JunD were also increased soon after establishment of disease and persisted over the course of disease.5
In the endometriotic epithelial cell line 12Z,30 TNF-α was shown to cause an increase in the expression of several matrix metalloproteinases (MMP), growth factors, and growth factor receptors.31 The JNK inhibitor (SP600125) caused the most significant inhibition of 12Z cell motility without affecting cell viability.31 So, there is a reasonable expectation that JNK inhibitors may interrupt immune cell or endometrial cell response to proinflammatory cytokine-mediated processes and effectively reduce the number of endometriotic lesions and associated pain. Bentamapimod (also coded as AS602801 or PGL5001; Figure 1) was developed during the course of drug discovery efforts for neurological diseases22 including cerebral ischemia32 and multiple sclerosis.23 Based on its demonstrated efficacy to reduce motor neuron loss following induced ischemia and inflammation and that a recognized mechanism was through modulation of T-cell subtypes in animal models of multiple sclerosis, the efficacy of bentamapimod was evaluated in models of endometriosis, a disease characterized by pain associated with sensory nerve recruitment, inflammation, and T-cell involvement in the lesion.33 Our primary goal of experiments employing bentamapimod and medroxyprogesterone acetate (MPA) was to demonstrate that bentamapimod had unique ability to induce regression of endometriosis and that MPA (an approved progestin for contraception) did not interfere with the action of bentamapimod in preclinical models. A secondary goal was to determine if bentamapimod and MPA complemented each other when delivered as a combination therapy. Our results show that bentamapimod caused regression of endometriosis in a nude mouse xenograft model of human disease and in an autologous rat model of endometriosis. Furthermore, our results suggest that bentamapimod treatment restores PR receptor function in endometriotic lesions established from biopsies obtained from women with the disease, and our results demonstrate that bentamapimod inhibition does not interrupt ER action on uterine size. A parallel manuscript with bentamapimod describes that it causes regression of lesions in baboons with experimentally induced endometriosis (Hussein et al, unpublished data).
Figure 1.

Chemical structure of bentamapimod/AS602801-HCl/PGL5001-HCl; MW 595.
Materials and Methods
Acquisition of Human Tissues
Biopsies of endometrial tissue from normal volunteers (BNV, n = 4) were acquired by Pipelle (Unimar, Inc, Wilton, Connecticut) during the proliferative phase (days 9-12) of the menstrual cycle from a donor population (age 18-45 years) exhibiting normal menstrual cycles and no history of endometriosis. Endometrial tissues from women with surgically confirmed endometriosis (BWE, n = 4) were also obtained by biopsy during the proliferative phase. An endometrial thickness ≥9 mm (confirmed by vaginal ultrasound) and a serum PR level ≤1.5 ng/mL were required for inclusion in this study. Individuals with a recent (≤3 months) history of hormone therapy (ie, oral contraceptives) were excluded. Biopsies were washed in prewarmed phenol-red free Dulbecco Modified Eagles Medium/Ham F-12 Medium (DME/F-12; Sigma, St. Louis, MO) to remove residual blood and mucous prior to culturing. Informed consent was obtained prior to biopsy, and the use of human tissues was approved by Vanderbilt University’s Institutional Review Board and Committee for the Protection of Human Subjects.
Nude Mouse Model of Endometriosis
The model of endometriosis was performed as previously described.34 Briefly, 5-week-old athymic (ncr/nude) ovariectomized mice (Harlan, Indinapolis, Indiana) were anesthetized with isoflurane (Henry Schein, Dublin, OH) and subcutaneously implanted with a silastic capsule containing 8 μg estradiol. Twenty-four hours later, mice received subcutaneous or intraperitoneal injection with a phosphate-buffered saline (PBS) suspension of 8 to 10 human endometrial tissue fragments/mouse (biopsies obtained from volunteers or patients) on the ventral midline just below the umbilicus. For 24 hours immediately preceding injection, tissue fragments were established as organ cultures treated with 1 nmol/L estradiol, PR (Sigma, St. Louis, MO), or MPA (Sigma, St. Louis, MO). Oral administration of AS602801 (EMD Serono, Rockland, MA) was initiated 10 to 12 days following the injection of tissue. Progesterone was provided via a slow-release silastic capsule containing 25 μg PR, and MPA was given by twice-weekly injections (200 mg/kg) along the right flank using a tuberculin syringe. AS602801 was administered by gavage at a dose of 10 mg/kg and 30 mg/kg/animal for 30 days. Following the completion of treatment, mice were again anesthetized and sacrificed by cervical dislocation for direct examination of lesion size and number. Uteri were measured and weighed, and excised lesions rapidly frozen for further analysis. Experiments described here were approved by the Vanderbilt University Instructional Animal Care and Use Committee in accordance with the Animal Welfare Act.
Proliferative Organ Culture Model
Endometrial biopsies were dissected into small cubes (∼1 × 1 mm3), and 8 to 10 pieces of tissue per treatment group were suspended in tissue culture inserts. Organ cultures were maintained a total of 72 hours at 37°C in DME/F-12 media with serum supplements as described previously.35 Tissue treatments included 17β-estradiol (E2), E2 plus PR, or E2 plus MPA with and without AS602801. Cultures were maintained for 24 hours in E2 only until the initiation of experimental conditions. AS602801 was used at a concentration of 5 or 15 μmol/L, and MPA was used at a concentration of 50, 100, or 250 μmol/L. Following termination of the experiment, media were collected and stored at −80°C until further analysis.
Western Blots
Media were collected from human endometrial organ cultures and secreted proteins quantified using the Coomassie Plus Protein Assay (Pierce, Grand Island, NY) and 20 μg total protein subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membrane and blocked in PBS with 10% nonfat milk and 0.05% Tween-20. Blots were incubated overnight at 4°C in PBS/milk/Tween with a primary antibody recognizing human MMP-3 (mouse monoclonal; Oncogene Research Products, San Diego, CA), a marker for estrogenic effects in endometrial tissue. The blots were subsequently washed and incubated with secondary antibody (horseradish peroxidase-conjugated anti-mouse; Amersham, Pittsburgh, PA) for 1 hour. Proteins were visualized by chemiluminescence (Amersham, Pittsburgh, PA) and autoradiography. As a negative control, identical blots were incubated without a primary antibody.
Quantitative Real-Time Polymerase Chain Reaction
RNA was isolated from lesions retrieved from nude mice treated with vehicle or 10 mg/kg AS602801 using TRIzol (Invitrogen Life Technologies) according to the manufacturer’s instructions. RNA was treated with DNase I (Qiagen, Valencia, CA) and followed with RNeasy kit (Qiagen, Valencia, CA) before complementary DNA (cDNA) synthesis. One microgram of total RNA was reverse transcribed using oligo-dT priming and the Superscript III First-strand cDNA synthesis kit (Invitrogen, Carlsbad, CA). The Reverse transcriptase (RT)-minus samples served as a control to exclude amplification from contaminating undigested genomic DNA. Complementary DNA corresponding to 100 ng of input RNA was amplified in duplicate with the TaqMan Universal PCR Master Mix on a custom TaqMan Low Density Array (Applied Biosystems, Foster City, California) as described earlier.30 Real-time polymerase chain reaction was conducted with an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Differential target gene expression was calculated according to the 2-ddCT method, using hypoxanthine phosphoribosyltransferase 1 (HPRT1) as an endogenous control. Interarray reproducibility was proven by repeated measurements of control cDNA samples on 3 different arrays.
Autologous Experimentally Induced Endometriosis in Rats
Endometriosis was induced in rats as described earlier.36 In brief, endometriosis was induced in rats by transplanting an autologous fragment of endometrial tissue onto the inner surface of the abdominal wall. Under pentobarbital (45 mg/kg) anesthesia, a distal segment 1 cm in length was resected from the right uterine horn and placed in PBS at 37°C. The other uterine horn was not manipulated. The uterine segment was opened by a longitudinal incision, and a 5 × 5 mm section was transplanted without removing the myometrium onto the inner surface of the abdominal wall using 7/0 nonabsorbable silk suture at 4 corners. Another group of rats similarly had a fragment of 1 uterine horn removed, but a 5 × 5 mm square of fat surrounding the uterus was transplanted (sham operated). One further group of rats, not undergoing any surgical procedure, was kept as a normal control group. Three weeks later, the animals were submitted to a second laparotomy (pretreatment laparotomy) to evaluate the size and viability of the ectopic endometrial tissue. The surface area of the engrafted tissue was measured using a caliper and recorded. The animals showing viable engraftments were stratified based on their size and assigned to the designated treatment groups. Treatment started after a 1-week recovery period. The control group received the vehicle only. AS602801 was administered orally twice a day (BID) at doses of 10, 30, and 60 mg/kg for 9 days, and Antide (gonadotropin-releasing hormone agonist agonist) was administered once by injection, similar to the study by D’Antonio et al.36 At the designated sacrifice time point (2 days after the last treatment, ie, 36 days after surgical engraftment), the animals were anesthetised and the surface area of endometriosis-like foci was measured for each animal.
The percentage variation versus pretreatment laparotomy value was calculated by the formula:
where xo is the size at the time of pretreatment laparotomy and x the size at the time of sacrifice. The mean value of percentage variation in each group was then computed. Data of percentage variation of engraftment size for experimental groups were analyzed by one-way analysis of variance, followed by Tukey test in order to evaluate the differences among treatment groups. Statistical analysis was performed using S-Plus 2000 (MathSoft, Inc, Seattle, Washington).
The endometriotic implants were finally removed and stored in 10% formalin for histological evaluation. The endometriotic foci and contralateral uterine horns were collected for measuring cytokine concentrations. The endometriotic-like foci and spleen were removed for histology and for NK cell activity measurement, respectively.
Immunohistochemistry of Rat Tissues
Paraformaldehyde-fixed and paraffin-embedded endometriotic foci were sectioned at about 4 to 5 μm of thickness and deparaffinized/rehydrated for immunoperoxidase staining using a Vectastain ABC kit (Vector Laboratories, Burlingame, California). Briefly, antigen unmasking was performed by incubation in 10 mmol/L sodium citrate buffer (pH 6.0), and endogenous peroxidase was quenched with 1% H2O2 for 10 minutes. Nonspecific immunoglobulin binding sites were blocked by incubating for 1 hour with normal goat serum, then sections were incubated with the primary antibodies anti-CD45 (30-F11; BD Pharmingen, San Diego, California), anti–phospho-c-Jun (Ser 73), and anti–total c-Jun (Cell Signaling Technology, Beverly, Massachusetts) overnight at 4°C. Sections were successively incubated for 30 minutes with a biotinylated secondary antibody solution followed by a 30-minute incubation with ABC reagent (Vectastain Elite ABC kit; Vector Laboratories). Immunoglobulin complexes were visualized by incubation with 3,3′-diaminobenzidine, then washed, counterstained with hematoxylin, cleared, dehydrated, mounted, and examined by light microscopy. Ten fields were observed for each sample. As negative control for the immunohistochemical staining, tissue sections were treated with normal serum instead of primary antibodies. For apoptotic cell detection, serial sections were stained with TUNEL reagents (In Situ Cell Death Detection, POD; Roche, Mannheim, Germany), according to the manufacturer’s instructions.
Determination of Cytokine Levels
Endometriotic foci, ipsilateral, and contralateral uterine horns were snap frozen in liquid nitrogen and homogenized in cold PBS in the presence of protease inhibitor cocktail (Sigma, St Louis, Missouri). After sonication, lysates were centrifuged, protein concentration determined, and 100 μg of proteins used for cytokine level determination. Interferon (IFN)-γ, TNF-α, interleukin (IL)-12p70, IL-10, IL-6, IL-5, IL-4, IL-2, and monocyte chemoattractant protein-1 (MCP-1) have been evaluated by flow cytometry with the Cytometric Bead Assay, according to the manufacturer’s instructions (CBA inflammation and Th1/Th2 kit; BD Pharmingen, San Diego, CA).
Natural Killer Cell Cytotoxicity Assay
Spleen was removed aseptically from sham-operated, control, and treated rats. Spleen cells were gently teased apart in 50 mL PBS. Cytotoxicity of NK cells in splenocyte was determined using 51Cr-release assay as described earlier36 with YAC-1, a murine lymphoma cell line that is sensitive to NK-cell cytotoxicity as the effector cell system. In brief, YAC-1 cells labeled with [51Cr]-sodium chromate were incubated with splenocytes at effector/target cell ratios of 40:1, 20:1, and 10:1. After 4-hour incubation at 37°C, 20 μL of the supernatant was transferred to a glass fiber filter, and the associated radioactivity was measured by a β-counter (LKB Pharmacia Wallac, Turku, Finland).
Results
The activity of bentamapimod in xenograft endometriosis experiments is summarized in Figure 2. All animals bearing xenografts were treated with estradiol. Medroxyprogesterone acetate has been approved by the FDA for the management of pain in women with endometriosis, but MPA was unable to cause regression of lesions relative to estradiol-treated animals in experiments using BNV (Figure 2A). Addition of bentamapimod (30 mg/kg) to estradiol + MPA-treated animals reduced the number of lesions 21%, but this difference was not significant compared to estradiol-treated mice (Figure 2A). On the other hand, PR caused 60% regression of BNV lesions as expected (Supplemental Figure 1; P < .05). Medroxyprogesterone acetate alone or in the presence of bentamapimod reduced uterine weight of the mice (Supplemental Figure 2; P < .05) consistent with its classic progestational effect on ER-primed mouse uterus but was without effect on the number of lesions. The effect of bentamapimod on lesion regression was next evaluated in the nude mouse model using BWE (laparoscopic diagnosis of endometriosis). On its own, bentamapimod had a weak effect on lesion regression (29% reduction at 30 mpk compared to E2 + PR-treated animals, P < .05; not significant compared to E2 only, P > .1) that was not evident at lower doses (10 mg/kg; Figure 2B). The modest response in BWE with the combination therapy of bentamapimod + MPA (10 mpk + MPA; 38%) was superior to the response obtained with PR (P < .05) but was not different from E2 alone. As expected, MPA had similar ability to decrease uterine weight in mice bearing lesions from BWE as in BNV (Supplemental Figure 3; P < .05).
Figure 2.

Regression of endometriotic lesions established in nude mice with biopsies from normal volunteers (BNV) or biopsies from women with endometriosis (BWE). A, Biopsies from normal volunteers (BNV) were prepared for injection to establish lesions; 10 days after disease was induced, treatments began. Estradiol was delivered by implants, and bentamapimod (JNKi) was administered orally. Medroxyprogesterone acetate (MPA) was delivered by twice-weekly injection as described in the Methods section. Results are mean ± standard error from 2 independent experiments; n = 3 to 4 mice/group/experiment. B, Regression of endometriotic lesions established in nude mice with BWE. Results are mean ± standard error from 2 to 4 independent experiments; n = 3 to 4 mice/group/experiment. Asterisks represent analysis of variance (ANOVA) followed by Tukey test for comparisons with 17β-estradiol (E2) + progesterone (PR) (P < .05). Comparisons with E2 alone were not significant (P > .1).
The augmentation of lesion regression by the combination of MPA and bentamapimod was further evaluated in an endometrial organ culture in which MMP-3 has been validated as a marker for endometrial remodeling within lesions. Human endometrial organ cultures established from BNV produced detectable levels of MMP-3 when E2 was present (Figure 3A), replicating previous observations in this culture system.34 Similar to previous results with PR, addition of MPA at 50 or 250 nmol/L caused decreased production of MMP-3. Bentamapimod also caused decreased production of MMP-3, in the presence or absence of MPA. Human endometrial organ cultures from BWE (Figure 3B) produced large amounts of MMP-3 in response to estradiol that were not reduced by the addition of PR or MPA. Markedly different from this PR resistance, bentamapimod (15 μmol/L) caused a dramatic reduction in MMP-3 levels. Moreover, combination of a subeffective concentration of bentamapimod (5 μmol/L) restored the concentration-dependent suppression of MMP-3 by MPA.
Figure 3.
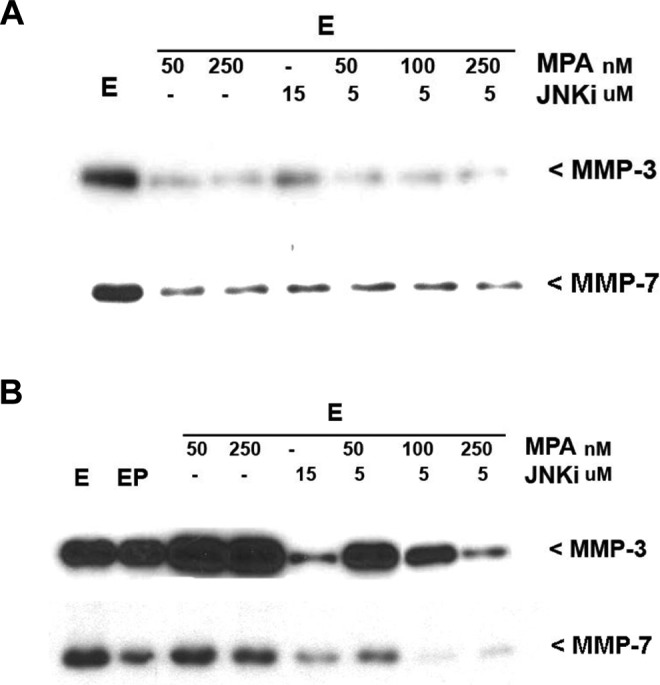
Western blots of MMP-3 and MMP-7 in conditioned media of explant cultures of human endometrium from normal volunteers (A) or patients with endometriosis (B). Biopsies for the uterine tissue were cut into small pieces and pretreated with estradiol for 24 hours. Organ cultures were challenged with various factors for further 72 hours in tissue culture inserts. Media were stored at −70°C until analyzed by Western blot. A, Bentamapimod (JNKi) reduces expression of MMP-3, and MMP-7, and markers of endometriosis in endometrial explant cultures obtained from volunteers. B, Bentamapimod (JNKi) restores progesterone regulation of MMP-3 and MMP-7 expression in endometrial explant cultures obtained from women with endometriosis. MMP indicated matrix metalloproteinase.
Human endometriotic-like lesions were recovered from the nude mice that had received biopsies from patients and had been exposed to vehicle or 10 mg/kg bentamapimod in vivo. Lesions from other bentamapimod-treated groups and combination therapy with MPA were too small for retrieval from all animals in the other treatment groups. The endometriotic-like lesions were pooled from 4 different experiments and evaluated for expression of mRNA corresponding to panels of inflammatory cytokine genes, tissue remodeling and angiogenesis, and steroid-related genes (Figure 4A-C). Primers were selected based on evidence of gene expression modification in endometriotic lesions from previous reports and for the ability to amplify specifically human sequences.35 Human-specific expression of 18 of the 22 selected genes could be detected, and their relative expression levels have been normalized to HPRT1 (±standard deviation). The expression of the respective genes in vehicle-treated mice is expressed as 1.0 and is depicted in the figure by a line. Levels of inflammatory cytokines (RANTES, GM-CSF, IL-8, TNF-α, and ICAM-1) were reduced by approximately 50% in bentamapimod-treated mice (Figure 4A). Similarly, levels of tissue remodeling enzymes and angiogenic growth factors were measured (Figure 4B), and MMP-9 and hepatocyte growth factor levels were reduced by 50% in JNK-I–treated mice compared to vehicle-treated mice. In these pooled samples, levels of estrogen and PR mRNA were not altered, whereas StAR and SF-1 were reduced by 50% (Figure 4C).
Figure 4.

Bentamapimod (AS-01) causes reduction in the expression of genes previously associated with endometriosis in endometriotic-like lesions established with biopsies from patients with disease. TaqMan quantitative polymerase chain reaction was performed on lesions from each treatment group within an experiment. The lesions from 4 independent experiments were analyzed, and the mean responses are represented. Gene expression analysis was conducted on a TaqMan low-density array. The responses are grouped according to proinflammatory cytokines (A), matrix proteins (B), and steroidogenic pathway (C). The results are normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT1) expression, and error bars represent standard deviation determind by analysis of variance (ANOVA) followed by Tukey test. Significance (P < .05) is represented by asterisks relative to control groups receiving estrogen alone (dotted line at 1.0).
To extend our understanding of the utility of JNK inhibition for the treatment of endometriosis, we evaluated bentamapimod in a rat model of endometriosis in which the immune system remains competent. In this model, endometriotic foci in rats receiving the low dose of bentamapimod (10 mg/kg orally BID) were similar to vehicle, but at 30 and 60 mg/kg, significant regression of established endometriotic lesions was observed (48% regression at 60 mg/kg) compared to the control group, as well to the pretreatment control for each group (Figure 5). Treatment with Antide caused 85% regression. This level of regression with bentamapimod is similar to regression obtained with soluble TBP in previous experiments in the rat model.36 To confirm that bentamapimod effects were mediated by inhibition of JNK activity, sections of rat endometriotic lesions were stained for end points related to JNK and immune function (Figure 6). Compared to control or Antide-treated rats, both total c-Jun (Figure 6A-C) and phospho-c-Jun staining (Figure 6D-F) were decreased with bentamapimod treatment. The reduction in phospho-c-Jun was more dramatic (Figure 6E) in animals treated with bentamapimod than the change in total c-Jun (Figure 6B). Also apparent from these sections is the compaction of endometriotic stromal tissue in the presence of Antide, while no similar compaction occurs with bentamapimod treatment, indicating a lack of inhibition by bentamapimod on endogenous ER and PR support of endometrium. As expected, signs of apoptosis as measured by TUNEL staining were evident in sections from bentamapimod-treated uteri and Antide-treated uteri, relative to control (Figure 6G-I). However, TUNEL staining was more abundant in Antide-treated rats, relative to control or bentamapimod-treated rats. Lastly, CD45 staining, reflecting a broad marker for T and B lymphocytes, was suppressed by AS602801, whereas the presence of CD45 was unaffected by Antide treatment (Figure 6L-N). These results suggest that bentamapimod has potential to reduce the number of CD45+ lymphocytes recruited to lesions (perhaps through inhibition of phosphorylated c-Jun) or alternatively to reduce the presence of T or B cells present within lesions, through processes that could include apoptosis. These results confirm an effect of JNK inhibition on the inflammatory and/or immune responses to endometriosis and demonstrate that this effect occurs through a different mechanism than endocrine regulators such as Antide.
Figure 5.

Bentamapimod at both 30 mg/kg twice daily (BID) and 60 mg/kg BID causes regression of surgically induced autologous endometriotic-like foci in rats. Uterine segments from each rat were used to establish lesions. Three weeks later, animals with confirmed endometriotic foci were assigned to treatment groups. A sham-treated group received autologous uterine segment removal and adipose transplant near the uterus; they did not develop lesions (not shown). Bentamapimod (AS-01; orally) was administered BID for 9 days, and Antide (subcutaneous injection) was delivered as a single injection. Data are mean ± standard error of the mean (SEM), **P < .05, ***P < .01, analysis of variance (ANOVA) followed by Tukey test.
Figure 6.

Histological analysis of rat endometriotic foci treated with vehicle (A, D, G, L), bentamapimod (AS602801) 60 mg/kg (B, E, H, M), and Antide 2 mg/kg (C, F, I, N). These sections have been stained for anti-total-c-Jun antibody (A, B, C), anti-phospho-c-Jun antibody (Ser73; D, E, F), TUNEL assay for apoptotic cell detection (G, H, I), and anti-CD45 antibody for leukocyte infiltration evaluation (L, M, N).
To further describe the effects of bentamapimod on immune modulation, and particularly at sites of inflammation, levels of 3 proinflammatory cytokines were measured in endometriotic foci and contralateral uterine horns and expressed as concentrations/100 μg of protein in the treated rats. Concentrations of all 3 cytokines were elevated in the endometriotic foci (Figure 7A), relative to concentrations of cytokines in the contralateral horn (Figure 7B). Treatment of rats with bentamapimod (30 mg/kg) reduced levels of IL-12p70, IFN-g, and MCP-1 (both 30 and 60 mg/kg doses) in the endometriotic foci. By comparison, there was no effect of bentamapimod on production of these cytokines in the contralateral uterine horn. These results suggest that bentamapimod inhibits an inflammatory response specifically at the site of the lesion without affecting uninvolved sites of the uteri in the same animal. This level of specificity for the treatment of endometriosis is distinctly novel compared to endocrine modulation of disease.
Figure 7.

Concentrations of proinflammatory cytokines in endometriotic-like foci (A) and in unaffected sites on contralateral uterine horn (B). Cytokines were measured in lysates (per 100 μg protein) prepared from endometriotic foci or unaffected sites (contralateral horn) for each animal; n = 3 samples/rat/group; *P < .05 and **P < .01, analysis of variance (ANOVA) followed by Tukey test. Bentamapimod = AS602801.
Natural killer cell cytotoxicity was further assessed in the animals. Cells from endometriotic animals showed reduced NK cell lytic activity compared to sham operated (42% vs 48%, respectively, at 10:1 effector to target ratio; Figure 8). On the other hand, bentamapimod treatment resulted in increased NK cell lytic activity compared to the endometriotic animals (59% vs 42%, respectively, at 10:1 effector to target ratio and 60% vs 39%, respectively, at 40:1 effector to target ratio; Figure 8).
Figure 8.
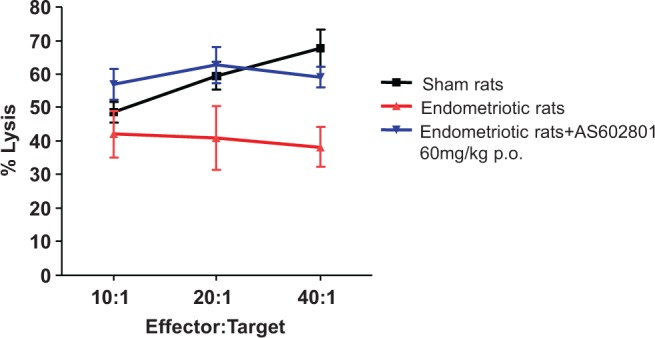
Cytotoxic activity of natural killer (NK) cells from splenocytes. Splenocytes isolated from endometriotic rat after their treatment were subjected to lytic activity of the target cells as described in the Methods section. Data are mean ± standard deviation (SD), n = 3, P < .05; Dunnett multiple comparison test, endometriotic versus sham and endometriotic versus endometriotic + JNKi treated. Bentamapimod = AS602801.
Discussion
Our studies demonstrate that kinase inhibitors selective for inhibition of JNK are potential new therapeutics for the treatment of endometriosis functioning independent of hormone regulation. These results suggest that endometriosis can be treated by release of the inhibiting effects of inflammation through cellular mechanisms proposed recently.30,37 It has been recognized that there is an immune contribution to establishment and progression of endometriosis.38 A recent review by Bianco et al39 summarizes the autoimmune basis for endometriosis and the multiple gene polymorphisms in proinflammatory pathways in patients with endometriosis. The involvement of TNF-α pathways in establishment and progression of endometriosis has been well demonstrated. Suppression of TNF-α with onercept, etanercept, or infliximab prevented establishment or induced regression of endometriotic lesions in rat and baboon models of disease.17-19,36 The JNK inhibitors decreased generation of TNF-α following LPS stimulation, decreased inflammation score in collagen-induced arthritis,22 and reduced proliferation of immune cells causing autoimmune inflammation in multiple sclerosis.23 So, there is reasonable expectation that JNK inhibitors may interrupt immune cell or endometrial cell response to proinflammatory cytokine-mediated processes and effectively reduce the number of endometriotic lesions and associated pain.
Candidate endometriosis therapeutics with anti-inflammatory pharmacology profiled in preclinical studies have either failed in human clinical trials because of lack of efficacy in human disease (infliximab)40 or have not progressed beyond early clinical trials for safety concerns (onercept).41 Relative to the broad pharmacological response of soluble receptor or antibody inhibitors of TNF-α, bentamapimod demonstrates more discrete impact on intracellular inflammatory pathways and cytokines, at the site of inflammation.37 Moreover, the inhibition of JNK regulates MMP expression, cellular adhesion, and motility31 and has potential for more convenient treatments than the biologicals because they are orally active.
The purpose of the experiments reported here was to provide sufficient preclinical evidence for a role of bentamapimod for the treatment of endometriosis and to justify further analysis of bentamapimod in primate models of endometriosis as a bridge to human clinical trials. Medroxyprogesterone acetate was used in several experiments in this report because it is an approved medication for treating endometriosis. The JNK inhibitors interrupt inflammatory processes, and one could expect that the use of these inhibitors in women would require simultaneous contraception while on therapy. Our primary goal of experiments using MPA was to demonstrate that MPA (also approved progestin for contraception) did not interfere with the action of bentamapimod in preclinical models. A secondary goal was to determine if bentamapimod and MPA complemented each other when delivered as a combination therapy. Our results suggest that the efficacy of bentamapimod treatment is complemented by PR receptor ligand, MPA, and bentamapimod restores PR receptor function in endometriotic lesions established from biopsies obtained from women with disease. Furthermore, our results demonstrate that bentamapimod does not interrupt ER action on uterine size.
Our results in nude mice suggest that the response to bentamapimod alone or in combination with MPA differed among mice bearing lesions established from BWE compared to mice with lesions from biopsies of normal women. Progesterone resistance is commonly observed in women with endometriosis.42 Previous studies in nude mice demonstrated PR resistance in animals with BWE.34 In these animals, bentamapimod was able to overcome PR resistance and reduce the disease burden either alone or in the presence of MPA. This was further demonstrated in the organ culture of BWE; PR or MPA had no effect on MMP-3 production, a hallmark for PR resistance, whereas bentamapimod was quite effective in reversing the effect. In endometriotic tissue, PR is incapable of inducing epithelial HSD17B2 expression due to the very low levels of PR in endometriotic tissue.43 In the present study, gene expression analysis of the endometriotic lesions following bentamapimod treatment showed no change in the expression of PR and ER, although expression of StAR, SF1, and aromatase was reduced. Further expression of mRNA for several inflammatory cytokine was suppressed by JNK inhibition compared to control in the lesion of nude mice. Inhibition of JNK activity with SP600125 in their stromal cell cultures decreased production of inflammatory cytokine IL-8.21
From our previous experience with TBP (onercept), 64% inhibition of endometriotic foci in the rat model36 translated into significant reductions in lesions in the baboon model, comparable to GnRH-mediated endocrine suppression.17 Therefore, 50% decrease in lesion size by bentamapimod in the rat model is a reasonable predictor of a clinically meaningful response in baboons and ultimately in humans. As another measure of efficacy in the rat model, we also measured changes in inflammatory cytokines in the endometriotic foci and contralateral uterine horns. In the foci, bentamapimod reduced concentrations of IL-12, IFN-g, and MCP-1; IL-12 and MCP-1 have been reported to be elevated in peritoneal fluid of women with endometriosis.44,45 The absence of this suppression by bentamapimod in the contralateral horn, distal from the inflamed site, is remarkable for an endometriosis therapy. These results imply that JNK inhibitors target immune or endometrial cell function at the site of inflammation rather than immune cells or endometrium at locations distant from the lesion.
Consistent with reductions of inflammatory cytokines in the endometriotic foci, bentamapimod increased the levels of TUNEL staining within lesions and reduced the number of CD45-stained cells in lesions. These results imply that elimination of a subpopulation of immune cells is an essential component of lesion regression by bentamapimod. However, it is still unresolved which specific immune cell types are modulated by bentamapimod. Women with endometriosis had reduced numbers of CD4+ and CD8+ cells and greater number of apoptotic cells in peritoneal fluid than women without disease,46 and we also observed reductions of CD45 immune cells at the endometriotic lesion. Rats treated with Antide showed stromal compaction, and decreases in epithelial cell height, consistent with ER deprivation. This observation in rats is consistent with the known effects of GnRH agonists or antagonists in the clinical management of endometriosis. These ER-deprived features were absent in rats receiving bentamapimod and confirm our observations on lack of ER interference in the nude mouse model.
In contrast to the earlier study in rat endometriosis,36 where Antide and TBP-1 showed no effect on NK cell activity, bentamapimod was able to reverse the effect of decreased NK cell cytotoxicity in endometriotic animals similar to earlier reports in mice47 and human.48 Reduced NK cell activity was observed in women with endometriosis.49,50 These results support the unique feature of bentamapimod: separation of immune cell pharmacology from endocrine function. In a parallel manuscript submission (Hussein et al, unpublished data), bentamapimod demonstrated significant reduction in all lesions and red lesions in the presence or absence of MPA, to a similar extent as Cetrotide in an induced endometriosis model in baboons.
The present work demonstrated 3 unique therapeutic benefits of JNK inhibition on endometriosis treatments compared to hormonal therapies. First, bentamapimod restored the ability of progestin (MPA) to cause regression of human ectopic endometrial cells, which would include eventual apoptosis of endometrial cells upon progestin withdrawal. Addition of bentamapimod restored the effects of MPA to suppress MMP-3 production in vitro. Second, bentamapimod therapy interrupted inflammatory cytokine production at the site of the lesion, without affecting production of cytokines at sites distal from the lesion. Third, bentamapimod therapy was independent from significant impact on the endocrine axis. The evaluation of bentamapimod in a baboon model of endometriosis confirms suppression of disease to an extent similar to that of a GnRH antagonist without impact on ovarian function (Hussein et al, 2015, unpublished data). These 2 companion reports encourage evaluation of therapies that interrupt inflammatory pathways such as the c-Jun N-terminal kinase. Further efforts are needed to demonstrate that JNK inhibitors ameliorate PR resistance in this disease.
Supplementary Material
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SSP, MA, DD, J-PG, and SGN were employees of EMD/Merck Serono during the performance of these studies. EGT was employed at RBM (Istituto Di Ricerche Biomedicine “Antoine Marxer”), an affiliate of Merck Serono. SSP, MA, EGT, and SGN are no longer employees at EMD Serono or RBM and have no continuing interest in bentamapimod (PGL5001). J-PG is no longer employee of Merck Serono but affiliated with PreGlem, who holds rights to PGL5001, and is the contact for questions regarding bentamapimod.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Merck Serono Research Institute and in part by the Specialized Cooperative Centers Program in Reproduction Research (U54 HD37321; KGO).
Supplemental Material: The online supplemental figures are available at http://rs.sagepub.com/supplemental
References
- 1. Kyama CM, Debrock S, Mwenda JM, D’Hooghe TM. Potential involvement of the immune system in the development of endometriosis. Reprod Biol Endocrinol. 2003;1:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lebovic DI, Mueller MD, Taylor RN. Immunobiology of endometriosis. Fertil Steril. 2001;75(1):1–10. [DOI] [PubMed] [Google Scholar]
- 3. Senturk LM, Arici A. Immunology of endometriosis. J Reprod Immunol. 1999;43(1):67–83. [DOI] [PubMed] [Google Scholar]
- 4. Bulun SE, Cheng YH, Yin P, et al. Progesterone resistance in endometriosis: link to failure to metabolize estradiol. Mol Cell Endocrinol. 2006;248(1-2):94–103. [DOI] [PubMed] [Google Scholar]
- 5. Hastings JM, Fazleabas AT. A baboon model for endometriosis: implications for fertility. Reprod Biol Endocrinol. 2006;4(suppl 1):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gaetje R, Kotzian S, Herrmann G, Baumann R, Starzinski-Powitz A. Nonmalignant epithelial cells, potentially invasive in human endometriosis, lack the tumor suppressor molecule E-cadherin. Am J Pathol. 1997;150(2):461–467. [PMC free article] [PubMed] [Google Scholar]
- 7. Matsuzaki S, Darcha C. Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis. Hum Reprod. 2012;27(3):712–721. [DOI] [PubMed] [Google Scholar]
- 8. Al-Sabbagh M, Lam EW, Brosens JJ. Mechanisms of endometrial progesterone resistance. Mol Cell Endocrinol. 2012;358(2):208–215. [DOI] [PubMed] [Google Scholar]
- 9. Dunselman GA, Vermeulen N, Becker C, et al. ESHRE guideline: management of women with endometriosis. Hum Reprod. 2014;29(3):400–412. [DOI] [PubMed] [Google Scholar]
- 10. Streuli I, de Ziegler D, Santulli P, et al. An update on the pharmacological management of endometriosis. Expert Opin Pharmacother. 2013;14(3):291–305. [DOI] [PubMed] [Google Scholar]
- 11. Efstathiou JA, Sampson DA, Levine Z, et al. Nonsteroidal antiinflammatory drugs differentially suppress endometriosis in a murine model. Fertil Steril. 2005;83(1):171–181. [DOI] [PubMed] [Google Scholar]
- 12. Bulun SE. Endometriosis. N Engl J Med. 2009;360(3):268–279. [DOI] [PubMed] [Google Scholar]
- 13. Herington JL, Bruner-Tran KL, Lucas JA, Osteen KG. Immune interactions in endometriosis. Expert Rev Clin Immunol. 2011;7(5):611–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barcz E, Milewski L, Dziunycz P, Kaminski P, Ploski R, Malejczyk J. Peritoneal cytokines and adhesion formation in endometriosis: an inverse association with vascular endothelial growth factor concentration. Fertil Steril. 2012;97(6):1380–1386. e1381. [DOI] [PubMed] [Google Scholar]
- 15. Braun DP, Ding J, Dmowski WP. Peritoneal fluid-mediated enhancement of eutopic and ectopic endometrial cell proliferation is dependent on tumor necrosis factor-alpha in women with endometriosis. Fertil Steril. 2002;78(4):727–732. [DOI] [PubMed] [Google Scholar]
- 16. Keenan JA, Chen TT, Chadwell NL, Torry DS, Caudle MR. IL-1 beta, TNF-alpha, and IL-2 in peritoneal fluid and macrophage-conditioned media of women with endometriosis. Am J Reprod Immunol. 1995;34(6):381–385. [DOI] [PubMed] [Google Scholar]
- 17. D’Hooghe TM, Nugent NP, Cuneo S, et al. Recombinant human TNFRSF1A (r-hTBP1) inhibits the development of endometriosis in baboons: a prospective, randomized, placebo- and drug-controlled study. Biol Reprod. 2006;74(1):131–136. [DOI] [PubMed] [Google Scholar]
- 18. Barrier BF, Bates GW, Leland MM, Leach DA, Robinson RD, Propst AM. Efficacy of anti-tumor necrosis factor therapy in the treatment of spontaneous endometriosis in baboons. Fertil Steril. 2004;81(suppl 1):775–779. [DOI] [PubMed] [Google Scholar]
- 19. Falconer H, Mwenda JM, Chai DC, et al. Treatment with anti-TNF monoclonal antibody (c5N) reduces the extent of induced endometriosis in the baboon. Hum Reprod. 2006;21(7):1856–1862. [DOI] [PubMed] [Google Scholar]
- 20. Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6(6):644–653. [DOI] [PubMed] [Google Scholar]
- 21. Yoshino O, Osuga Y, Hirota Y, et al. Possible pathophysiological roles of mitogen-activated protein kinases (MAPKs) in endometriosis. Am J Reprod Immunol. 2004;52(5):306–311. [DOI] [PubMed] [Google Scholar]
- 22. Gaillard P, Jeanclaude-Etter I, Ardissone V, et al. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J Med Chem. 2005;48(14):4596–4607. [DOI] [PubMed] [Google Scholar]
- 23. Ferrandi C, Richard F, Tavano P, et al. Characterization of immune cell subsets during the active phase of multiple sclerosis reveals disease and c-Jun N-terminal kinase pathway biomarkers. Mult Scler. 2011;17(1):43–56. [DOI] [PubMed] [Google Scholar]
- 24. Kao LC, Germeyer A, Tulac S, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144(7):2870–2881. [DOI] [PubMed] [Google Scholar]
- 25. Matsuzaki S, Canis M, Pouly JL, Botchorishvili R, Dechelotte PJ, Mage G. Differential expression of genes in eutopic and ectopic endometrium from patients with ovarian endometriosis. Fertil Steril. 2006;86(3):548–553. [DOI] [PubMed] [Google Scholar]
- 26. Matsuzaki S, Canis M, Vaurs-Barriere C, Boespflug-Tanguy O, Dastugue B, Mage G. DNA microarray analysis of gene expression in eutopic endometrium from patients with deep endometriosis using laser capture microdissection. Fertil Steril. 2005;84(suppl 2):1180–1190. [DOI] [PubMed] [Google Scholar]
- 27. Wu Y, Kajdacsy-Balla A, Strawn E, et al. Transcriptional characterizations of differences between eutopic and ectopic endometrium. Endocrinology. 2006;147(1):232–246. [DOI] [PubMed] [Google Scholar]
- 28. Shazand K, Baban S, Prive C, et al. FOXO1 and c-jun transcription factors mRNA are modulated in endometriosis. Mol Hum Reprod. 2004;10(12):871–877. [DOI] [PubMed] [Google Scholar]
- 29. Uz YH, Murk W, Bozkurt I, Kizilay G, Arici A, Kayisli UA. Increased c-Jun N-terminal kinase activation in human endometriotic endothelial cells. Histochem Cell Biol. 2011;135(1):83–91. [DOI] [PubMed] [Google Scholar]
- 30. Grund EM, Kagan D, Tran CA, et al. Tumor necrosis factor-alpha regulates inflammatory and mesenchymal responses via mitogen-activated protein kinase kinase, p38, and nuclear factor kappaB in human endometriotic epithelial cells. Mol Pharmacol. 2008;73(5):1394–1404. [DOI] [PubMed] [Google Scholar]
- 31. Miller MA, Meyer AS, Beste MT, et al. ADAM-10 and -17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proc Natl Acad Sci U S A. 2013;110(22):E2074–E2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carboni S, Boschert U, Gaillard P, Gotteland JP, Gillon JY, Vitte PA. AS601245, a c-Jun NH2-terminal kinase (JNK) inhibitor, reduces axon/dendrite damage and cognitive deficits after global cerebral ischaemia in gerbils. Br J Pharmacol. 2008;153(1):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tokushige N, Markham R, Russell P, Fraser IS. Different types of small nerve fibers in eutopic endometrium and myometrium in women with endometriosis. Fertil Steril. 2007;88(4):795–803. [DOI] [PubMed] [Google Scholar]
- 34. Bruner KL, Eisenberg E, Gorstein F, Osteen KG. Progesterone and transforming growth factor-beta coordinately regulate suppression of endometrial matrix metalloproteinases in a model of experimental endometriosis. Steroids. 1999;64(9):648–653. [DOI] [PubMed] [Google Scholar]
- 35. Bruner-Tran KL, Zhang Z, Eisenberg E, Winneker RC, Osteen KG. Down-regulation of endometrial matrix metalloproteinase-3 and -7 expression in vitro and therapeutic regression of experimental endometriosis in vivo by a novel nonsteroidal progesterone receptor agonist, tanaproget. J Clin Endocrinol Metab. 2006;91(4):1554–1560. [DOI] [PubMed] [Google Scholar]
- 36. D’Antonio M, Martelli F, Peano S, Papoian R, Borrelli F. Ability of recombinant human TNF binding protein-1 (r-hTBP-1) to inhibit the development of experimentally-induced endometriosis in rats. J Reprod Immunol. 2000;48(2):81–98. [DOI] [PubMed] [Google Scholar]
- 37. Beste MT, Pfaffle-Doyle N, Prentice EA, et al. Molecular network analysis of endometriosis reveals a role for c-Jun-regulated macrophage activation. Sci Transl Med. 2014;6(222):222 ra216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kobayashi H, Higashiura Y, Shigetomi H, Kajihara H. Pathogenesis of endometriosis: the role of initial infection and subsequent sterile inflammation (Review). Mol Med Rep. 2014;9(1):9–15. [DOI] [PubMed] [Google Scholar]
- 39. Bianco B, Andre GM, Vilarino FL, et al. The possible role of genetic variants in autoimmune-related genes in the development of endometriosis. Hum Immunol. 2012;73(3):306–315. [DOI] [PubMed] [Google Scholar]
- 40. Koninckx PR, Craessaerts M, Timmerman D, Cornillie F, Kennedy S. Anti-TNF-alpha treatment for deep endometriosis-associated pain: a randomized placebo-controlled trial. Hum Reprod. 2008;23(9):2017–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Papp K. Clinical development of onercept, a tumor necrosis factor binding protein, in psoriasis. Curr Med Res Opin. 2010;26(10):2287–2300. [DOI] [PubMed] [Google Scholar]
- 42. Bulun SE, Cheng YH, Pavone ME, et al. Estrogen receptor-beta, estrogen receptor-alpha, and progesterone resistance in endometriosis. Semin Reprod Med. 2010;28(1):36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bulun SE, Cheng YH, Pavone ME, et al. 17Beta-hydroxysteroid dehydrogenase-2 deficiency and progesterone resistance in endometriosis. Semin Reprod Med. 2010;28(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gallinelli A, Chiossi G, Giannella L, Marsella T, Genazzani AD, Volpe A. Different concentrations of interleukins in the peritoneal fluid of women with endometriosis: relationships with lymphocyte subsets. Gynecol Endocrinol. 2004;18(3):144–151. [DOI] [PubMed] [Google Scholar]
- 45. Harada T, Iwabe T, Terakawa N. Role of cytokines in endometriosis. Fertil Steril. 2001;76(1):1–10. [DOI] [PubMed] [Google Scholar]
- 46. Mier-Cabrera J, Jimenez-Zamudio L, Garcia-Latorre E, Cruz-Orozco O, Hernandez-Guerrero C. Quantitative and qualitative peritoneal immune profiles, T-cell apoptosis and oxidative stress-associated characteristics in women with minimal and mild endometriosis. BJOG. 2011;118(1):6–16. [DOI] [PubMed] [Google Scholar]
- 47. Altan ZM, Denis D, Kagan D, Grund EM, Palmer SS, Nataraja SG. A long-acting tumor necrosis factor alpha-binding protein demonstrates activity in both in vitro and in vivo models of endometriosis. J Pharmacol Exp Ther. 2010;334(2):460–466. [DOI] [PubMed] [Google Scholar]
- 48. Umesaki N, Tanaka T, Miyama M, Mizuno K, Kawamura N, Ogita S. Increased natural killer cell activities in patients treated with gonadotropin releasing hormone agonist. Gynecol Obstet Invest. 1999;48(1):66–68. [DOI] [PubMed] [Google Scholar]
- 49. Tanaka E, Sendo F, Kawagoe S, Hiroi M. Decreased natural killer cell activity in women with endometriosis. Gynecol Obstet Invest. 1992;34(1):27–30. [DOI] [PubMed] [Google Scholar]
- 50. Quaranta MG, Porpora MG, Mattioli B, et al. Impaired NK-cell-mediated cytotoxic activity and cytokine production in patients with endometriosis: a possible role for PCBs and DDE. Life Sci. 2006;79(5):491–498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.