

Abstract
Binding of hepatocyte growth factor (HGF) to the c-MET receptor has mitogenic, motogenic, and morphogenic effects on cells. The versatile biological effects of HGF and c-MET interactions make them important contributors to the development of malignant tumors. We and others have demonstrated a therapeutic value in targeting the interaction of c-MET and HGF in epithelial ovarian cancer (EOC). However, both HGF and c-MET are expressed in the normal ovary as well. Therefore, it is important to understand the differences in mechanisms that control HGF signaling activation and its functional role in the normal ovary and EOC. In the normal ovary, HGF signaling may be under hormonal regulation. During ovulation, HGF-converting proteases are secreted and the subsequent activation of HGF signaling enhances the proliferation of ovarian surface epithelium in order to replenish the area damaged due to expulsion of the ovum. In contrast, EOC cells that exhibit epithelial characteristics constitutively express both c-MET and HGF-converting proteases such as urokinase-type plasminogen activator. In EOC, mechanisms to control the activation of HGF signaling are absent since HGF is provided locally from the tissue microenvironment as well as remotely throughout the body. Potential incessant HGF signaling in EOC may lead to an increase in proliferation, invasion through the stroma, and migration to other tissues of cancer cells. Therefore, targeting the interaction of c-MET and HGF would be beneficial in treating EOC.
Keywords: ovarian cancer, c-MET, hepatocyte growth factor, targeted therapy, tissue microenvironment
Introduction
Hepatocyte growth factor (HGF) is a multifunctional cytokine that stimulates cell proliferation, migration, and extracellular matrix (ECM) invasion. Versatile biological functions of HGF are mediated through its binding to the specific receptor, c-MET.1 Hepatocyte growth factor is primarily produced by mesenchymal cells and acts on cells of mainly epithelial origin, mediating mesenchymal–epithelial interactions.1 Mesenchymal cells secrete HGF in an inactive form that initially binds to heparan sulfate proteoglycans within the ECMs or cell surface.2–4 This inactive single-chain polypeptide requires proteolytic cleavage into an active heterodimer via hepatocyte growth factor activator (HGFA) to acquire c-MET binding activity.5 Hepatocyte growth factor activator is a serine protease that also requires protease-mediated cleavage for its activation.4,6 Other serine proteases including urokinase-type plasminogen activator (uPA), tissue plasminogen activator (tPA), and plasmin also convert pro-HGF to the active form, although they are less effective compared to HGFA.6 Matriptases have been shown to activate HGF in ovarian cancer cells.7,8 In addition, HGF activation is negatively regulated by HGFA inhibitors such as HGFA inhibitor types 1 and 2 (HAI-1 and HAI-2).4,9
Due to the pleiotropic effects of HGF as a mitogen, motogen, and morphogen, it has been implicated in morphogenesis and the organization of tissues during embryonic development and regeneration after tissue injury. Studies have demonstrated that HGF plays an integral role in the repair of liver, lung, skin, and kidney epithelium.10,11 Upon tissue damage, HGFA undergoes proteolytic cleavage by downstream proteases (eg, thrombin) of the blood coagulation cascade and becomes activated.6 Many studies have suggested that HGF activation rather than local production of HGF is the rate-limiting step in HGF-induced signaling at the damaged areas.4,9,12 As such, HGF signaling can be influenced by the following 3 states: (1) activation of HGF by HGFA, (2) inhibition of HGFA by HAI-1 and HAI-2, or (3) maintenance of the balance in levels of active HGFA and HGFA inhibitors.4 This mode of regulation may make HGF activation selective at the site of tissue damage.4
Versatile functions of HGF also make it an important contributor to the onset and progression of cancer as demonstrated in many different types of cancer.13,14 c-MET expression has been shown to be high in a variety of tumors.13,15 c-MET activation is primarily associated with the migratory and invasive property of cancer cells.16,17 However, it has also been demonstrated that cancer cell lines with c-MET amplification are dependent on c-MET for their growth, and subsequently, c-MET inhibition induces cell death.15,18,19 c-MET overexpression is also related to the resistance to antiangiogenic therapy.20 Therefore, binding of HGF induces activation of c-MET, which triggers cancer cells to proliferate, invade through the stroma, and migrate. In addition, c-MET activation is often related to resistance to cell death and cancer therapy.
Ovarian cancer is the seventh most common cancer in women worldwide, with the highest incidence rate in North America and Europe.21 There are 2 forms of the disease: “nonepithelial” and “epithelial.” Nonepithelial types are divided into either germ cell or sex cord tumors and account for ∼10% of all ovarian cancers.22 The epithelial type is a more common and deadly form of ovarian cancer. About 90% of all ovarian cancers are epithelial; however, the origin of this disease is still a subject of debate.23–25 A growing number of molecular-based studies have suggested that these epithelial ovarian cancer can develop from cells not directly related to the ovary, including the fallopian tube, gastrointestinal tract, cervix, and endometriosis.25,26 Other scientists still believe that a subset of these epithelial tumors are derived from a single-cell mesothelial layer, termed the “ovarian surface epithelium” (OSE).27 Epithelial ovarian cancer encompasses several histologically and molecularly distinct malignancies in the ovaries, including serous, mucinous, endometrioid, and clear cell carcinomas.28,29 Each of these distinct subtypes has been traced back to independent cell origins. In the case of serous ovarian cancer, there is growing evidence that epithelial cells within the fallopian tube are the origin of many of these tumors and grow in the ovary as a result of ovulation: The repeated rupture and repair process during a woman’s menstrual cycles creates a local inflammatory microenvironment in which signals are produced at the ovulatory wound site of the ovary that can recruit extraovarian premalignant and malignant cells to the ovary.25,26 This may also explain why incessant ovulation is a risk factor for ovarian cancer and why the inhibitory factors of ovulation such as multiple pregnancies, breast-feeding, late menarche, and the use of oral contraceptives reduce ovarian cancer risk.30 The origins of mucinous ovarian tumors are thought to be metastatic tumors from the gastrointestinal tract or cervix. For clear cell and endometrioid ovarian cancers, they are considered to originate from endometriosis. But for all of these EOC, the interplay between tumor cells and the microenvironment of the ovary is critical in its progression. We and others have demonstrated the importance of the tumor–stromal interaction mediated by c-MET and HGF and the therapeutic value of targeting their interaction in EOC.31–35 However, both molecules are expressed in the normal ovary as well. Therefore, it is important to understand the differences in the regulation of their expression and the function mediated by their interaction at the normal ovary and EOC.
In this review, we examine the expression levels of c-MET, HGF, and HGF-converting enzymes in both the normal ovary and EOC and discuss their functional importance. The goal is to highlight the importance of targeting the interactions of c-MET and HGF in EOC and provide a rationale for targeting c-MET in the clinic.
Expression and Functional Roles of HGF Signaling Molecules in the Normal Ovary
The ovary is derived from multiple embryonic structures including the coelomic epithelium, the subcoelomic mesoderm, and the primordial germ cells from the yolk sac endoderm. The remaining components of the female genital tract, including the fallopian tubes, uterus, cervix, and upper vagina, are derived from the Müllerian ducts. As a result of its complex embryologic development, the ovary is composed of various cell types that serve specific structural, hormonal, or reproductive functions. Additionally, each cell type can develop into a distinctly different neoplasm as described earlier.
Hepatocyte growth factor and c-MET are expressed in normal ovaries of various mammalian species, including human, mouse, cow, sheep, and rat.32,36–40 Hepatocyte growth factor signaling in the normal ovary is largely studied in granulosa cells and theca cells. Both cell types contain HGF signaling molecules, including c-MET, HGF, and HGFA.38,40 The local secretion of HGF in human follicles was further demonstrated by Osuga et al.37 In those cells, the expression levels of c-MET, HGF, and HGFA are hormonally regulated.38,40,41 Furthermore, studies have suggested that HGF signaling in granulosa cells functions to enhance cell proliferation and to prevent apoptosis.38,40,42 Hepatocyte growth factor signaling downregulates ovarian steroidogenesis,38,43 which may contribute to the inhibition of apoptosis and support folliculogenesis.40 Thus, growing follicles contain the molecules necessary for HGF signaling, and this may play a role in folliculogenesis.
Hepatocyte growth factor is also expressed in the stroma of the ovary, and both HGF and c-MET are present in OSE, suggesting the potential importance of the autocrine/paracrine effect of HGF in OSE.39,44 Interestingly, the expression level of HGF is greater in OSE compared to the underlying stroma.39 Although the functional role of HGF signaling in human OSE is not well understood, it has been speculated that HGF signaling may be related to the involvement of OSE in ovulation.45 An ovary undergoes cyclic disruption and repair with complex remodeling during ovulation. Ovulation is induced by a surge of follicle-stimulating hormone and luteinizing hormone, which results in follicle rupture mediated by proteolytic enzymes of a plasmin-generating system and collagenases.46 Studies suggest that OSE, which is derived from the coelomic epithelium, is essential in the ovulatory process and that these epithelial cells secrete uPA and other proteolytic enzymes in response to the rise in the concentration of locally delivered gonadotropins via the follicular vascular wreath.45,46 We and others have also demonstrated that immortalized OSE cells express uPA and matrix metalloproteinases.47,48
During ovulation, extensive architectural remodeling occurs in OSE, which includes simultaneous apoptotic degradation of the monolayer of OSE at the apex of the ovulating follicle and cell proliferation of OSE to cover the area affected by follicular rupture.49–52 It has been suggested that HGF may play a role in mediating mitogenic action on OSE by activating the c-MET receptor during ovulation.38,39,41,53 However, studies have found conflicting results regarding the action of HGF on cell proliferation. In one study, it was found that extraneous HGF did not promote the growth of ovine OSE,54 whereas others found that HGF could stimulate proliferation of human OSE cells.44 The factor that derived different results was later attributed to the presence of fibronectin and other extracellular components that occur in vivo.44,55 Hepatocyte growth factor induces the mitosis of rat OSE when they are grown on ECM proteins, including fibronectin, but not in a serum-free condition.55 Furthermore, physiological HGF is more potent in stimulating biological activities probably due to the presence of other extracellular components such as heparan sulfate secreted by stromal cells.33 Heparan sulfate enhances HGF association and its mitogenic activity.56 Thus, the ovulation process may resemble wound repair in vivo and HGF signaling may be involved in the regeneration of OSE after the ovarian rupture.
Regulation of HGF Signaling in Normal OSE
Hepatocyte growth factor production is greatly enhanced in the damaged tissues and to some degree in other tissues.5,6 However, conversion of the single chain form of HGF to the active heterodimer only occurs in injured tissues.5,57 It is well established that HGFA expression is induced in response to tissue damage.5,6,58 In addition, during gastrointestinal tract development, the morphogenic action of HGF is regulated not only by a local increase in HGF production but also by an increase in HGFA levels.12 Sisson et al59 have demonstrated that the activation of HGF by uPA is necessary for muscle regeneration and myoblast proliferation. Likewise, uPA and tPA are required during the reepithelialization process in keratinocytes after tissue injuries.53 Thus, studies suggest that activation of HGF by HGF-converting enzymes primarily regulates HGF action and acts as a mechanism for localizing HGF activities to damaged tissues during wound repair.
The ovulatory process is considered hormone-induced tissue injury. Hepatocyte growth factor activities during ovulation appear to be regulated similarly to other injured tissues as described above. Although studies have not been reported in human ovary, ovine OSE that is in close contact with the apical wall of preovulatory follicles secretes uPA into the underlying stroma in response to an abrupt increase in local hormone levels during ovulation.46 Proteolytic enzymes secreted by OSE weaken and degrade underneath connective tissues.45 Secreted proteases may also induce the release of HGF from degrading connective tissues, increasing locally available HGF.33,60 In addition, proteolytic enzymes may activate HGF and trigger HGF signaling selectively in the damaged area. It is not well understood whether the expression levels of HGF, c-MET, and HGF-converting enzymes are also hormonally regulated in OSE and the underneath stroma. However, studies suggest that HGF is activated during ovulation and contributes to the regeneration of OSE to replenish damaged epithelium by the extrusion of ovum,38,39,41,53 very similar to the regeneration of epithelial cells during wound repair observed in other tissues.
Expression of c-MET and HGF-Converting Enzymes in EOC
High c-MET expression has been demonstrated in many cancers of epithelial origin including ovarian cancer. c-MET expression has been shown to be enhanced in 11% to 60% of EOC patient samples.31,32,61,62 Studies have also suggested that high c-MET expression is associated with lymph node metastasis, high histologic grade, and low survival rate in patients with ovarian cancer.31,35 However, mechanisms governing aberrant upregulation of c-MET are not well understood. Somatic mutations on the c-MET are rarely found in most human cancers.15,63 Overexpression of c-MET in EOC does not appear to be related to gene amplification.32 A recent study indicated that the high expression of c-MET in cancer cells might be related to TP53 mutation, which occurs in most if not all high-grade serous ovarian cancers.64 Mutant p53 enhances c-MET trafficking mediated by Rab coupling protein-dependent receptor recycling.65 Thus, the mechanisms contributing to aberrant expression of c-MET in EOC are not fully understood, but high levels of c-MET significantly correlate with a poor prognosis in patients.35
Hepatocyte growth factor-converting enzymes are upregulated in EOC as well. Although HGFA has not been reported to be aberrantly expressed in EOC cells, matriptase, a serine protease of epithelial cells, is highly expressed in most malignant ovarian cancers.7,8 Another serine protease, hepsin, was reported to be overexpressed in over 80% of ovarian carcinomas.66 Urokinase-type plasminogen activator levels are enhanced in epithelial tumors, including EOCs,67 and are associated with tumor progression.68 In addition, studies have shown coexpression of c-MET and HGF-converting proteases in epithelial cells during tumorigenesis and morphogenesis. Matsubara et al12 demonstrated that HGFA messenger RNA (mRNA) is present only in epithelia that coexpress c-MET mRNA, and Kwon and colleagues reported that EOC cells expressing c-MET also contain uPA.48 Furthermore, the caseinolytic activity of the cells that express both uPA and c-MET is enhanced when they are cultured within 3-dimensional ECMs derived from fibroblasts,48 suggesting that the proteases secreted by EOC cells are functional and secretion can be enhanced when cells are in contact with ECMs. Therefore, c-MET and HGF-converting proteases are coexpressed in EOC cells instead of increasing the protease expression upon tissue damage as is expected in the normal ovary (Figure 1).
Figure 1.

Comparison of c-MET and hepatocyte growth factor (HGF)-converting protease expression in the normal ovary and epithelial ovarian cancer (EOC). Both c-MET and HGF-converting proteases are expressed at low levels in the normal ovary, and the expression of HGF-converting proteases is induced and secreted upon ovulation while both molecules are constitutively high in EOC.
Expression of HGF in EOC
The enhancement of c-MET expression in EOC has been well documented39; however, cancer progression may also alter HGF expression. Nontumorigenic OSE expresses undetectable levels of c-MET31,44 but exhibits strong expression of HGF.39 In comparison, EOC cells contain high levels of c-MET but little or no HGF.31,39,69 Thus, c-MET expression is enhanced while HGF expression is diminished during ovarian cancer progression. There are no suggested mechanisms to explain these peculiar changes in expression levels of c-MET and HGF as ovarian progenitor cells become malignant. However, these changes may be associated with epithelial characteristics of EOC. Human OSE exhibits both epithelial and mesenchymal phenotypes,70 whereas they often lose mesenchymal characteristics and increase E-cadherin with cancer progression.48,70–73 Another explanation may be that serous ovarian tumors originated from dysplastic lesions in the distal fallopian tube and these progenitor cells express higher c-MET compared to OSE and have more differentiated epithelial cell characteristics.62
In addition, EOC cells do not express both c-MET and HGF simultaneously; EOC cell lines that demonstrate epithelial cell phenotypes48 express c-MET and respond to extraneous HGF.33 In contrast, the cells with mesenchymal characteristics produce HGF but do not either express c-MET or respond to added HGF.33 Moreover, EOC cell lines that contain constitutively active c-MET receptor require extracellular HGF for the activation of downstream signaling pathways, including AKT and extracellular signal-regulated kinases (ERK).33 Epithelial ovarian cancer cell lines express phospho-c-MET (Tyr1349), a multifunctional docking site for the recruitment of multiple transducers and adapters, only in response to the added recombinant HGF or fibroblast HGF.33 This is in agreement with the observation that c-MET activation in cancer cells occurs mostly via an HGF-dependent manner.15,74 Therefore, c-MET activation is dependent on HGF provided from the tumor microenvironment in EOC, and paracrine regulation of HGF is important in EOC cells that gain features of epithelial cells and lose their mesenchymal properties.
Considering the lack of HGF expression in EOC cells and dependence of c-MET activation on environmental HGF, local production of HGF in the tumor microenvironment is expected. However, HGF expression does not appear to be prevalent in the tumor microenvironment; its levels are higher in the stroma of the normal ovary when compared to the tumor stroma.33,39 The mechanism associated with decreased levels of HGF in the tumor stroma is unknown, while HGF can be provided to EOC cells both locally and distantly. Hepatocyte growth factor is expressed throughout the body,15 and high levels of circulating HGF are found in patients with ovarian cancer.75 Ovarian ascites contain significant levels of HGF to stimulate cell migration,76 and therefore, HGF might be provided by tissues distant from the EOC cell. Another possibility is that selective tumor-associated fibroblasts at the invasive front may express high levels of HGF, thereby contributing to the invasive and migratory behavior of cancer cells.33 In addition, HGF secreted from selective fibroblasts can be accumulated within the focalized area of ECMs, leading to increase in the local concentration of HGF and the activation of HGF signaling upon interaction with cancer cells that contain both c-MET and HGF-converting proteases.33 Therefore, HGF can be provided to EOC cells either locally or distantly throughout the body, even though HGF expression is not high in the entire stromal compartment of EOC (Figure 2).
Figure 2.
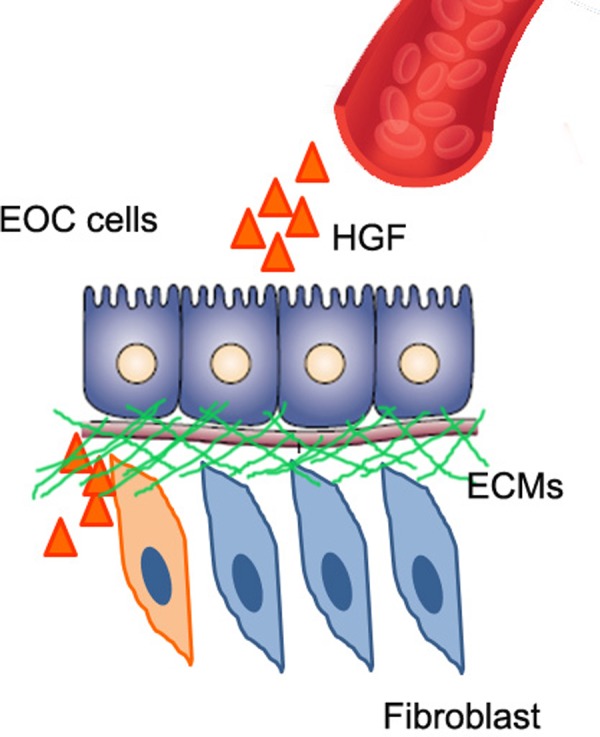
Possible mechanisms for providing hepatocyte growth factor (HGF) to the tumor microenvironment. Hepatocyte growth factor can be delivered from remote tissues through the bloodstream. In addition, HGF can be provided locally. Selective tumor fibroblasts adjacent to cancer cells may secrete high levels of HGF. Hepatocyte growth factor secreted by a subpopulation of tumor fibroblasts can be accumulated within extracellular matrices (ECMs), and the release of HGF by interaction with epithelial ovarian cancer (EOC) cells may increase the concentration of HGF available.
Regulation of HGF Signaling and Therapeutic Importance of Targeting c-MET-HGF Interaction in EOC
In OSE, HGF signaling is activated upon secretion and activation of HGF-converting proteases during ovulation.46 In contrast, typical EOC constitutively express both c-MET and HGF-converting enzymes (eg, uPA) as described earlier (Figure 1). Thus, EOC cells do not have mechanisms to control HGF activation since HGF can be constantly provided from other tissues through the bloodstream and the release of HGF upon pericellular proteolysis of the local ECM by EOC cells.77,78 Whether HGF comes from the local area or distant tissues, serine proteases secreted by EOC cells activate HGF and activated HGF binds to the c-MET receptor of EOC cells. Therefore, HGF and c-MET interaction constitutively occurs and signaling mediated by their interaction is activated in EOC cells.
Activation of c-MET via the binding of HGF triggers signaling cascades that activate mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways,79 resulting in the enhancement of cell migratory and invasive properties of EOC cells.69 In addition, c-MET overexpression induces resistance to apoptosis in EOC. High expression of c-MET was linked with high expression of antiapoptotic proteins, X-chromosome-linked inhibitors of apoptosis, and Bcl-xL, partly due to AKT activation by c-MET-HGF interaction in Saudi EOC patient samples.61 Further, c-MET inhibition resulted in loss of the mitochondrial membrane potential and caspase activation, concomitantly enhancing the activation of the mitochondrial apoptotic pathway.61 c-MET-expressing EOC cells also acquired a notable resistance to anoikis and apoptosis induced by chemotherapeutic drugs, cisplatin and paclitaxel, when grown in nonadherent cell cultures.80 This drug resistance is mediated through the PI3K/AKT and ERK signaling pathways.80
Studies also suggest that proliferation of EOC cells at least partly depends on the interaction of c-MET and HGF. The use of multikinase inhibitors targeting c-MET, for example, PF-2341066,81 foretinib,82 and DCC-2701,33 exhibited effective antitumor activities against ovarian cancer in animal models. Furthermore, the effectiveness of a c-MET inhibitor, DCC-2701, appears to be dependent on the availability of HGF from the tumor microenvironment.33 These studies suggest that c-MET and HGF interaction, which occurs constitutively in EOC cells, activates AKT and MAPK signaling cascade that contributes to the invasive and migratory behavior of EOC cells. Therefore, targeting the interaction of HGF and c-MET is important in inhibiting development of aggressive properties in EOC where HGF signaling is constitutively activated.
Concluding Remarks
Hepatocyte growth factor binding to the c-MET receptor induces mitogenic, motogenic, and morphogenic activities of the cells. These versatile biological effects of the HGF and c-MET interaction play an important role in OSE regeneration during ovulation in the normal ovary. However, this multifunctional effect of HGF also contributes to the acquirement of malignant behavior in EOC cells. In contrast to the regulation of c-MET activation at the level of HGF-converting enzymes by hormonal changes in the normal ovary, c-MET activation is indigenous due to the capability of secretion of HGF-converting proteases by EOC cells. Therefore, inhibition of c-MET and HGF interaction is necessary to prevent the development of proliferative, invasive, and metastatic behavior of EOC cells where HGF activation is not regulated.
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.K.G. is the Chancellors Distinguished Chair in Biomedical Sciences endowed Professor.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported in part by a grant from the National Cancer Institute (R01 CA140323) and the Kansas Bioscience Authority Eminent Scholar Program to A.K.G. and the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2014R1A1A3050916) to Y.K.
References
- 1. Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–804. [DOI] [PubMed] [Google Scholar]
- 2. Appleman LJ. MET signaling pathway: a rational target for cancer therapy. J Clin Oncol. 2011;29(36):4837–4838. [DOI] [PubMed] [Google Scholar]
- 3. Naldini L, Vigna E, Narsimhan RP, et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 1991;6(4):501–504. [PubMed] [Google Scholar]
- 4. Parr C, Jiang WG. Hepatocyte growth factor activators, inhibitors and antagonists and their implication in cancer intervention. Histol Histopathol. 2001;16(1):251–268. [DOI] [PubMed] [Google Scholar]
- 5. Miyazawa K, Shimomura T, Kitamura N. Activation of hepatocyte growth factor in the injured tissues is mediated by hepatocyte growth factor activator. J Biol Chem. 1996;271(7):3615–3618. [DOI] [PubMed] [Google Scholar]
- 6. Miyazawa K. Hepatocyte growth factor activator (HGFA): a serine protease that links tissue injury to activation of hepatocyte growth factor. FEBS J. 2010;277(10):2208–2214. [DOI] [PubMed] [Google Scholar]
- 7. Jin JS, Hsieh DS, Loh SH, Chen A, Yao CW, Yen CY. Increasing expression of serine protease matriptase in ovarian tumors: tissue microarray analysis of immunostaining score with clinicopathological parameters. Modern Pathol. 2006;19(3):447–452. [DOI] [PubMed] [Google Scholar]
- 8. Oberst MD, Johnson MD, Dickson RB, et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: correlation with clinical outcome and tumor clinicopathological parameters. Clin Cancer Res. 2002;8(4):1101–1107. [PubMed] [Google Scholar]
- 9. Kataoka H, Miyata S, Uchinokura S, Itoh H. Roles of hepatocyte growth factor (HGF) activator and HGF activator inhibitor in the pericellular activation of HGF/scatter factor. Cancer Metastasis Rev. 2003;22(2-3):223–236. [DOI] [PubMed] [Google Scholar]
- 10. Abdulla S. Hepatocyte growth factor, tissue repair and cancer. Mol Med Today. 1997;3(6):233. [DOI] [PubMed] [Google Scholar]
- 11. Bevan D, Gherardi E, Fan TP, Edwards D, Warn R. Diverse and potent activities of HGF/SF in skin wound repair. J Pathol. 2004;203(3):831–838. [DOI] [PubMed] [Google Scholar]
- 12. Matsubara Y, Ichinose M, Yahagi N, et al. Hepatocyte growth factor activator: a possible regulator of morphogenesis during fetal development of the rat gastrointestinal tract. Biochem Biophys Res Commun. 1998;253(2):477–484. [DOI] [PubMed] [Google Scholar]
- 13. Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109(7):863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li M, Xin X, Wu T, Hua T, Wang H. HGF and c-Met in pathogenesis of endometrial carcinoma. Front Biosci. 2015;20:635–643. [DOI] [PubMed] [Google Scholar]
- 15. Blumenschein GR, Jr, Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol. 2012;30(26):3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suarez-Causado A, Caballero-Diaz D, Bertran E, et al. HGF/c-Met signaling promotes liver progenitor cell migration and invasion by an epithelial-mesenchymal transition-independent, phosphatidyl inositol-3 kinase-dependent pathway in an in vitro model. Biochimica et biophysica acta. 2015;1853(10 pt A):2453–2463. [DOI] [PubMed] [Google Scholar]
- 17. Usatyuk PV, Fu P, Mohan V, et al. Role of c-Met/phosphatidylinositol 3-kinase (PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation, and motility of lung endothelial cells. J Biol Chem. 2014;289(19):13476–13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006;103(7):2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li M, Xin X, Wu T, Hua T, Wang H, Wang H. Stromal cells of endometrial carcinoma promotes proliferation of epithelial cells through the HGF/c-Met/Akt signaling pathway. Tumour Biol. 2015;36(8):6239–6248. [DOI] [PubMed] [Google Scholar]
- 20. Jahangiri A, De Lay M, Miller LM, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19(7):1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. [DOI] [PubMed] [Google Scholar]
- 22. Colombo N, Peiretti M, Garbi A, et al. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7):vii20–vii26. [DOI] [PubMed] [Google Scholar]
- 23. Ahmed AA, Becker CM, Bast RC., Jr The origin of ovarian cancer. BJOG. 2012;119(2):134–136. [DOI] [PubMed] [Google Scholar]
- 24. Kurman RJ. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol. 2013;24(suppl 10):x16–x21. [DOI] [PubMed] [Google Scholar]
- 25. Yang-Hartwich Y, Gurrea-Soteras M, Sumi N, et al. Ovulation and extra-ovarian origin of ovarian cancer. Sci Rep. 2014;4:6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kurman RJ, Shih Ie M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol. 2011;42(7):918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cho KR, Shih Ie M. Ovarian cancer. Ann Rev Pathol. 2009;4:287–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prat J. New insights into ovarian cancer pathology. Ann Oncol. 2012;23(suppl 10):x111–x117. [DOI] [PubMed] [Google Scholar]
- 30. La Vecchia C. Ovarian cancer: epidemiology and risk factors. Eur J Cancer Prev. 2017;26(1):55–62. [DOI] [PubMed] [Google Scholar]
- 31. Ayhan A, Ertunc D, Tok EC, Ayhan A. Expression of the c-Met in advanced epithelial ovarian cancer and its prognostic significance. Int J Gynecol Cancer. 2005;15(4):618–623. [DOI] [PubMed] [Google Scholar]
- 32. Di Renzo MF, Olivero M, Katsaros D, et al. Overexpression of the Met/HGF receptor in ovarian cancer. Int J Cancer. 1994;58(5):658–662. [DOI] [PubMed] [Google Scholar]
- 33. Kwon Y, Smith BD, Zhou Y, Kaufman MD, Godwin AK. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene. 2015;34(2):144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mhawech-Fauceglia P, Afkhami M, Pejovic T. MET/HGF signaling pathway in ovarian carcinoma: clinical implications and future direction. Pathol Res Int. 2012;2012:960327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sawada K, Radjabi AR, Shinomiya N, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67(4):1670–1679. [DOI] [PubMed] [Google Scholar]
- 36. Canipari R, O’Connell ML, Meyer G, Strickland S. Mouse ovarian granulosa cells produce urokinase-type plasminogen activator, whereas the corresponding rat cells produce tissue-type plasminogen activator. J Cell Biol. 1987;105(2):977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Osuga Y, Tsutsumi O, Momoeda M, et al. Evidence for the presence of hepatocyte growth factor expression in human ovarian follicles. Mol Hum Reprod. 1999;5(8):703–707. [DOI] [PubMed] [Google Scholar]
- 38. Parrott JA, Skinner MK. Developmental and hormonal regulation of hepatocyte growth factor expression and action in the bovine ovarian follicle. Biol Reprod. 1998;59(3):553–560. [DOI] [PubMed] [Google Scholar]
- 39. Parrott JA, Skinner MK. Expression and action of hepatocyte growth factor in human and bovine normal ovarian surface epithelium and ovarian cancer. Biol Reprod. 2000;62(3):491–500. [DOI] [PubMed] [Google Scholar]
- 40. Uzumcu M, Pan Z, Chu Y, Kuhn PE, Zachow R. Immunolocalization of the hepatocyte growth factor (HGF) system in the rat ovary and the anti-apoptotic effect of HGF in rat ovarian granulosa cells in vitro. Reproduction. 2006;132(2):291–299. [DOI] [PubMed] [Google Scholar]
- 41. Liu Y, Lin L, Zarnegar R. Modulation of hepatocyte growth factor gene expression by estrogen in mouse ovary. Mol Cell Endocrinol. 1994;104(2):173–181. [DOI] [PubMed] [Google Scholar]
- 42. Taniguchi F, Harada T, Deura I, Iwabe T, Tsukihara S, Terakawa N. Hepatocyte growth factor promotes cell proliferation and inhibits progesterone secretion via PKA and MAPK pathways in a human granulosa cell line. Mol Reprod Dev. 2004;68(3):335–344. [DOI] [PubMed] [Google Scholar]
- 43. Zachow RJ, Ramski BE, Lee H. Modulation of estrogen production and 17beta-hydroxysteroid dehydrogenase-type 1, cytochrome P450 aromatase, c-met, and protein kinase Balpha messenger ribonucleic acid content in rat ovarian granulosa cells by hepatocyte growth factor and follicle-stimulating hormone. Biol Reprod. 2000;62(6):1851–1857. [DOI] [PubMed] [Google Scholar]
- 44. Wong AS, Pelech SL, Woo MM, et al. Coexpression of hepatocyte growth factor-Met: an early step in ovarian carcinogenesis? Oncogene. 2001;20(11):1318–1328. [DOI] [PubMed] [Google Scholar]
- 45. Murdoch WJ, McDonnel AC. Roles of the ovarian surface epithelium in ovulation and carcinogenesis. Reproduction. 2002;123(6):743–750. [DOI] [PubMed] [Google Scholar]
- 46. Murdoch WJ, Murphy CJ, Van Kirk EA, Shen Y. Mechanisms and pathobiology of ovulation. Soc Reprod Fertil Suppl. 2010;67:189–201. [DOI] [PubMed] [Google Scholar]
- 47. Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22(2):255–288. [DOI] [PubMed] [Google Scholar]
- 48. Kwon Y, Cukierman E, Godwin AK. Differential expressions of adhesive molecules and proteases define mechanisms of ovarian tumor cell matrix penetration/invasion. PloS One. 2011;6(4):e18872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bjersing L, Cajander S. Ovulation and the role of the ovarian surface epithelium. Experientia. 1975;31(5):605–608. [DOI] [PubMed] [Google Scholar]
- 50. Clow OL, Hurst PR, Fleming JS. Changes in the mouse ovarian surface epithelium with age and ovulation number. Mol Cell Endocrinol. 2002;191(1):105–111. [DOI] [PubMed] [Google Scholar]
- 51. Singavarapu R, Buchinsky N, Cheon DJ, Orsulic S. Whole ovary immunohistochemistry for monitoring cell proliferation and ovulatory wound repair in the mouse. Reprod Biol Endocrinol. 2010;8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan OL, Fleming JS. Proliferating cell nuclear antigen immunoreactivity in the ovarian surface epithelium of mice of varying ages and total lifetime ovulation number following ovulation. Biol Reprod. 2004;71(5):1501–1507. [DOI] [PubMed] [Google Scholar]
- 53. Parrott JA, Vigne JL, Chu BZ, Skinner MK. Mesenchymal-epithelial interactions in the ovarian follicle involve keratinocyte and hepatocyte growth factor production by thecal cells and their action on granulosa cells. Endocrinology. 1994;135(2):569–575. [DOI] [PubMed] [Google Scholar]
- 54. Gubbay O, Guo W, Rae MT, et al. Anti-inflammatory and proliferative responses in human and ovine ovarian surface epithelial cells. Reproduction. 2004;128(5):607–614. [DOI] [PubMed] [Google Scholar]
- 55. Hess S, Gulati R, Peluso JJ. Hepatocyte growth factor induces rat ovarian surface epithelial cell mitosis or apoptosis depending on the presence or absence of an extracellular matrix. Endocrinology. 1999;140(6):2908–2916. [DOI] [PubMed] [Google Scholar]
- 56. Zioncheck TF, Richardson L, Liu J, et al. Sulfated oligosaccharides promote hepatocyte growth factor association and govern its mitogenic activity. J Biol Chem. 1995;270(28):16871–16878. [DOI] [PubMed] [Google Scholar]
- 57. Miyazawa K, Shimomura T, Naka D, Kitamura N. Proteolytic activation of hepatocyte growth factor in response to tissue injury. J Biol Chem. 1994;269(12):8966–8970. [PubMed] [Google Scholar]
- 58. Zachow R, Uzumcu M. The hepatocyte growth factor system as a regulator of female and male gonadal function. J Endocrinol. 2007;195(3):359–371. [DOI] [PubMed] [Google Scholar]
- 59. Sisson TH, Nguyen MH, Yu B, Novak ML, Simon RH, Koh TJ. Urokinase-type plasminogen activator increases hepatocyte growth factor activity required for skeletal muscle regeneration. Blood. 2009;114(24):5052–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matsuoka H, Sisson TH, Nishiuma T, Simon RH. Plasminogen-mediated activation and release of hepatocyte growth factor from extracellular matrix. Am J Respir Cell Mol Biol. 2006;35(6):705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bu R, Uddin S, Bavi P, et al. HGF/c-Met pathway has a prominent role in mediating antiapoptotic signals through AKT in epithelial ovarian carcinoma. Lab Invest. 2011;91(1):124–137. [DOI] [PubMed] [Google Scholar]
- 62. Huntsman D, Resau JH, Klineberg E, Auersperg N. Comparison of c-met expression in ovarian epithelial tumors and normal epithelia of the female reproductive tract by quantitative laser scan microscopy. Am J Pathol. 1999;155(2):343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3(1 suppl):S21–S35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Muller PA, Trinidad AG, Timpson P, et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene. 2013;32(10):1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tanimoto H, Yan Y, Clarke J, et al. Hepsin, a cell surface serine protease identified in hepatoma cells, is overexpressed in ovarian cancer. Cancer Res. 1997;57(14):2884–2887. [PubMed] [Google Scholar]
- 67. Mazar AP, Ahn RW, O’Halloran TV. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr Pharm Design. 2011;17(19):1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kuhn W, Pache L, Schmalfeldt B, et al. Urokinase (uPA) and PAI-1 predict survival in advanced ovarian cancer patients (FIGO III) after radical surgery and platinum-based chemotherapy. Gynecol Oncol. 1994;55(3 pt 1):401–409. [DOI] [PubMed] [Google Scholar]
- 69. Ueoka Y, Kato K, Kuriaki Y, et al. Hepatocyte growth factor modulates motility and invasiveness of ovarian carcinomas via Ras-mediated pathway. Br J Cancer. 2000;82(4):891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hudson LG, Zeineldin R, Stack MS. Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clin Exp Metastasis. 2008;25(6):643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ahmed N, Thompson EW, Quinn MA. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: an exception to the norm. J Cell Physiol. 2007;213(3):581–588. [DOI] [PubMed] [Google Scholar]
- 72. Auersperg N, Pan J, Grove BD, et al. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci U S A. 1999;96(11):6249–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sundfeldt K, Piontkewitz Y, Ivarsson K, et al. E-cadherin expression in human epithelial ovarian cancer and normal ovary. Int J Cancer. 1997;74(3):275–280. [DOI] [PubMed] [Google Scholar]
- 74. Michieli P, Basilico C, Pennacchietti S, et al. Mutant Met-mediated transformation is ligand-dependent and can be inhibited by HGF antagonists. Oncogene. 1999;18(37):5221–5231. [DOI] [PubMed] [Google Scholar]
- 75. Aune G, Lian AM, Tingulstad S, et al. Increased circulating hepatocyte growth factor (HGF): a marker of epithelial ovarian cancer and an indicator of poor prognosis. Gynecol Oncol. 2011;121(2):402–406. [DOI] [PubMed] [Google Scholar]
- 76. Sowter HM, Corps AN, Smith SK. Hepatocyte growth factor (HGF) in ovarian epithelial tumour fluids stimulates the migration of ovarian carcinoma cells. Int J Cancer. 1999;83(4):476–480. [DOI] [PubMed] [Google Scholar]
- 77. Aguirre Ghiso JA, Alonso DF, Farias EF, Gomez DE, de Kier Joffe EB. Deregulation of the signaling pathways controlling urokinase production. Its relationship with the invasive phenotype. Eur J Biochem. 1999;263(2):295–304. [DOI] [PubMed] [Google Scholar]
- 78. Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91(4):439–442. [DOI] [PubMed] [Google Scholar]
- 79. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1 suppl):S7–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tang MK, Zhou HY, Yam JW, Wong AS. c-Met overexpression contributes to the acquired apoptotic resistance of nonadherent ovarian cancer cells through a cross talk mediated by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2. Neoplasia. 2010;12(2):128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zillhardt M, Christensen JG, Lengyel E. An orally available small-molecule inhibitor of c-Met, PF-2341066, reduces tumor burden and metastasis in a preclinical model of ovarian cancer metastasis. Neoplasia. 2010;12(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zillhardt M, Park SM, Romero IL, et al. Foretinib (GSK1363089), an orally available multikinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. Clin Cancer Res. 2011;17(12):4042–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]