

Abstract
The DNA Damage Response (DDR) uses a plethora of proteins to detect, signal, and repair DNA lesions. Delineating this response is critical to understand genome maintenance mechanisms. Since recruitment and exchange of proteins at lesions are highly dynamic, their study requires the ability to generate DNA damage in a rapid and spatially-delimited manner. Here, we describe procedures to locally induce DNA damage in human cells using a commonly available laser-scanning confocal microscope equipped with a 405 nm laser line. Accumulation of genome maintenance factors at laser stripes can be assessed by immunofluorescence (IF) or in real-time using proteins tagged with fluorescent reporters. Using phosphorylated histone H2A.X (γ-H2A.X) and Replication Protein A (RPA) as markers, the method provides sufficient resolution to discriminate locally-recruited factors from those that spread on adjacent chromatin. We further provide ImageJ-based scripts to efficiently monitor the kinetics of protein relocalization at DNA damage sites. These refinements greatly simplify the study of the DDR dynamics.
Keywords: Genetics, Issue 133, DNA damage, double-stranded DNA breaks, micro-irradiation, confocal microscopy, immunofluorescence, real-time imaging, automated image analysis
Introduction
Cells are constantly exposed to endogenous and exogenous sources of DNA damage that threaten their genomic integrity. The DDR is an ensemble of signaling pathways that detect, signal, and repair DNA lesions to sustain genome stability. At DNA double-stranded breaks (DSBs), the DDR occurs mainly on two complementary platforms: γ-H2A.X-labeled chromatin and a resected single-stranded DNA (ssDNA) region typically coated with the ssDNA-binding complex RPA1,2.
UV-laser micro-irradiation of cells pre-sensitized with the thymidine analogue 5-bromo-2'deoxyuridine (BrdU) or the bisbenzimide ethoxide trihydrochloride (BBET, Hoechst 33342) DNA dye creates a mixture of DNA lesions including single-stranded breaks (SSBs) and DSBs which elicit a localized DDR on both the chromatin and ssDNA platforms3,4. Previous work showed that recruitment of genome maintenance factors to these two distinct platforms at DSB can be discriminated using high energy UV-A lasers (335-365 nm) combined with IF2,5. Microscopes equipped with such lasers are costly as they require a high energy laser and dedicated UV-A transmitting objectives making them far less prevalent in academic and pharmaceutical settings than laser-scanning confocal microscopes with 405 nm laser lines. The study of protein recruitment and exchange at micro-irradiated sites is also precluded by the laborious manual image analysis required to describe the dynamic behavior of genome maintenance factors.
Here, we show that the pre-sensitization of cells with BrdU or BBET nucleic acid stain followed by micro-irradiation using a 405 nm laser on a common confocal microscope allows the monitoring of genome maintenance factor dynamics at DNA lesions. Using γ-H2A.X or RPA complex subunits as platform markers together with z-stacking for greater depth of field and deconvolution for improved resolution allows the experimenter to discriminate factors that are locally recruited to DSBs from those that spread to large chromatin domains surrounding the initial lesion. This sub-classification according to distinct intra-nuclear compartments helps refine the potential roles of uncharacterized proteins recruited to micro-irradiated sites. Moreover, we provide convenient protocols and optimized pipelines to rapidly analyze the dynamics of genome maintenance factors using the open-source software Fiji (an ImageJ distribution)6,7,8. These refinements to current micro-irradiation methods render the study of the DDR possible in virtually any laboratory setting.
Protocol
1. Pre-sensitization of Cells
24 h prior to the micro-irradiation experiment, seed 40,000 U2OS cells per well in 1x DMEM containing 10% FBS in 8-well culture slides with 170 µm-thick coverslip-like glass/polymer bottoms to ensure maximal transmittance of 405 nm laser light and excellent imaging with a laser-scanning inverted confocal microscope. If using an 8-well microslide, use 500 µL of media or solutions per well for all subsequent treatments and wash steps of the protocol. NOTE: Due to their flat morphology, good adherence, and large nuclei, U2OS cells facilitate the detection of protein recruitment at micro-irradiated sites but other adherent cell types can be used as needed. Cells should ideally be around 80% confluency at the time of micro-irradiation to increase the number of irradiated cells per experiment. Very high confluence may result in cell cycle alterations that can perturb specific repair pathways such as homologous recombination which takes place during the S and G2 phases of the cell cycle9.
Incubate the cells overnight at 37 °C with 5% CO2, and then pre-sensitize the cells with (step 1.3) BrdU or (step 1.4) BBET (Hoechst 33342). NOTE: To observe live recruitment of a protein of interest (POI) to DNA lesions, transfect or transduce plasmid DNA carrying a fusion between a fluorescent protein and a POI 24 h after cell seeding. 24 h post-transfection/transduction, pre-sensitize and micro-irradiate the cells to monitor the recruitment of the POI at DNA damage sites in real time.
To increase the number of DSBs generated by exposition to the 405 nm laser, add 10 µM of BrdU to the media for 24 h prior to micro-irradiation. Prior to micro-irradiation, using a micropipette, rinse the cells twice with medium without phenol red to ensure maximal exposure of the cells to the 405 nm laser.
Alternatively, treat cells for 15 min with BBET at 10 µg/mL in the appropriate medium in a cell-culture incubator at 37 °C. After treatment, rinse twice with medium without phenol red prior to micro-irradiation.
2. Micro-irradiation of Cells
Select a suitable laser-scanning confocal microscope. NOTE: The experiments presented here were performed on a microscope system equipped with a 50 mW 405 nm laser line and a 63X 1.4 numerical aperture oil objective. To generate DSBs, cells were micro-irradiated at 1X scan zoom at 1,024 x 1,024 pixels/field (0.21 µm/pixel at 63X magnification) and in unidirectional mode at 8 µs/pixel. The laser power used was 25% and 80% for BBET- and BrdU-pre-sensitized cells, respectively. To help users configure their system, the laser power post-objective on the presented system was measured using a power meter according to manufacturer's instructions. The measurements correspond to 2.6 mW and 7.0 mW for BBET- and BrdU-pre-sensitized cells, respectively.
- Turn on the microscope and the environmental chamber.
- Set the chamber to 37 °C with 5% CO2 before samples are placed into the on-stage incubator to avoid unnecessary stress to the cells and to ensure homogeneous DDR kinetics across experiments.
- Select field(s) with well-distributed cells, adjust the focus, and register their position to facilitate image acquisition after the IF protocol. Acquire at least one image that will serve as a pre-damage reference point to monitor the kinetics of protein recruitment after micro-irradiation. NOTE: Gridded culture vessels may also be used to facilitate the localization of micro-irradiated cells at subsequent steps.
- For IF experiments using BBET-labeled cells, adjust the focus on cells using the 405 nm laser at the lowest laser power sufficient to visualize the cells in order to avoid unwarranted DNA damage.
- Avoid using widefield fluorescence to adjust the focus on BBET-stained cells as the UV light emitted by the lamp will induce DNA damage at illuminated sites.
- If using BrdU-labeled cells, adjust the focus using differential interference contrast (DIC) or phase-contrast imaging by looking at sub-nuclear features such as nucleoli.
- If using cells expressing fluorescently-labeled POIs, select a focal plane with representative fluorescence intensity. NOTE: Avoid cells that express very high levels of the POI as strong overexpression may result in artifactual localization. Additionally, select cells that express the POI at comparable levels (selection of stable clones is strongly recommended to minimize variations in POI expression and recruitment levels).
Configure the microscope for the micro-irradiation step using the fluorescence-recovery after photobleaching (FRAP)_module of the system. NOTE: The most important parameters to consider when setting-up the microscope system for micro-irradiation experiments are: the output power of the 405 nm laser, number of iterations (i.e., number of times the laser will go over the same coordinates), number of pixels/field (at 63X magnification and 1,024 x 1,024 resolution, 1 pixel = 0.21 µm), and dwell time per pixel. All of these factors will impact the amount of energy that the cells experience and thus the amount and type of DNA damage generated (see Discussion for further dose optimization information).
Using the FRAP module of the microscope system, micro-irradiate the cells. NOTE: On most systems, micro-irradiation of multiple cells can be performed in a single field as spots or lines/rectangles (typically 1-2 µm wide). When performing live-imaging of fluorescently-labeled proteins immediately post-damage, limit the micro-irradiation to a small number of cells per field because the time taken to micro-irradiate each cell will result in a delayed induction of the DDR. This is particularly important when monitoring proteins that are recruited very rapidly to DNA lesions.
- After micro-irradiation, put back the multi-well slides or plates in an incubator at 37 °C with 5% CO2 for the required duration prior to processing samples for IF (Section 5) or proceed immediately with live-imaging (Section 3).
- As a first step, fix the cells within a few minutes and up to a few hours post-irradiation for the IF protocol. Depending on the POI, recruitment kinetics may vary. Typically, proteins that are chromatin-associated at DSBs and SSBs will localize very rapidly to the lesion and are sometimes removed within 5 min. Some factors such as those that assemble at ssDNA will be recruited at later time points once the resection machinery has been engaged and remain there for hours (See Figure 1 and reference4).
3. Live-imaging of Fluorescently-labeled Proteins
Perform time-lapse imaging of the micro-irradiated cells. For proteins that are recruited very rapidly to lesions, a few seconds per frame will be ideal, but for proteins that localize to the damaged area later (e.g., resection factors), acquisition may be performed every 1–2 min until a plateau is achieved. NOTE: Minimize the laser power for image acquisition to avoid bleaching as much as possible and to ensure that protein recruitment at damage sites does not reach saturation which would preclude signal quantification and subsequent analyses (see data analysis Section 4). On the presented system, the imaging was done with a 63X/1.4 oil objective using an 8 µs/pixel dwell time, 1,024 x 1,024 resolution, 1X zoom, and averaging of 2. Note that these parameters may need to be optimized for other systems and/or different experiments.
Micro-irradiate the cells in additional fields until kinetics measurements are obtained for 15-30 cells. Repeat the experiments at least 3 times in biological replicates to ensure the accuracy and reproducibility of the recruitment kinetics. NOTE : When adapting this protocol to a specific confocal system, it may be useful to start by monitoring the recruitment kinetics of a known fluorescently-labeled genome maintenance factor (e.g., 53BP1-GFP, RPA32-GFP, etc.). Using live-imaging to optimize micro-irradiation conditions will allow the user to test multiple parameters rapidly.
4. Recruitment Kinetics Analysis
Install the Microirradiation Analysis plugin10 in the Fiji image analysis software11.
- Open the acquired images in Fiji. Select "The Damage Analyzer" from the pull-down plugins menu in the Microirradiation Analysis suite. Follow the prompts to define a background region of interest (ROI) (typically 3 x 3 µm), and non-irradiated and irradiated ROIs in each micro-irradiated cell.
- Use ROIs of the same size to minimize potential biases in mean signal intensities. For acquired images, also include at least one pre-irradiation frame which will be used as a baseline to monitor recruitment at micro-irradiated sites.
- Once all ROI data have been collected over the full time-lapse series, save the file created by the plugin containing the results as a csv (comma delimited file) or txt file (tab delimited file). This file contains measurements of the mean intensity for each time point and each ROI (Mean Intensity Background, Mean Intensity Non-irradiated, and Mean Intensity Irradiated). NOTE: The plugin automatically subtracts the background intensity (Ib) from the intensity of the irradiated ROIs (Is) and the non-irradiated ROIs (In). It also calculates a ratio between both values (see the formula below) to correct for photobleaching (Irradiated/Non-irradiated).
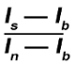 The plugin also calculates a normalization between the first time point (value set to 1) and all the other time points for each nucleus (ratio relative to first time point). Additional information can be found on the plugin website10.
The plugin also calculates a normalization between the first time point (value set to 1) and all the other time points for each nucleus (ratio relative to first time point). Additional information can be found on the plugin website10.
- Perform the analysis using the plugin for all micro-irradiated cells (at least 15–30 cells) and graph the mean relative enrichment at micro-irradiated regions over time. Calculate the standard errors of the mean for each data point and plot them.
- Confirm differences in recruitment kinetics between two experimental conditions by performing Student's t-tests for each data point.
5. IF Protocol
- Prepare IF solutions. Prepare pre/post-extraction solution (ice-cold 1x PBS containing 0.25% Triton X-100), washing solution (1x PBS containing 0.05% Tween-20), blocking solution (1x PBS containing 3% BSA and 0.05% Tween-20), and fixation solution (Paraformaldehyde 3% + sucrose 2% in PBS 1x) in conical polypropylene tubes. NOTE: For a single 8-well microslide, 10 mL of pre/post-extraction solution, 50 mL of washing solution, 10 mL of blocking solution, and 5 mL of fixation solution are required.
- Prepare the fixation solution.
- Under a chemical hood, mix 400 mL of double-distilled water (ddH2O) with 15 g of paraformaldehyde and 30 µL of 10 N NaOH in a 1 L Erlenmeyer flask.
- Close the container and heat at 65 °C on a magnetic stirrer hot plate while stirring to solubilize the paraformaldehyde completely.
- Using a 25-mL pipette, add 50 mL of 10X PBS, stir, and filter using a 0.45 µm filter.
- Add 10 g of sucrose and complete to 500 mL with ddH2O.
- Aliquot the solution in 15 mL conical tubes; store at -20 °C until use.
- Prepare the nuclei staining solution by diluting a stock solution of 1 mg/mL of 4,6-Diamidino-2-phenylindole (DAPI) 1:1,000 in 1x PBS.
Using a micropipette, carefully remove the media from the wells of the multi-well microscopy slides or plates and immediately wash with 500 µL of IF washing solution by changing the solution twice without waiting. NOTE: The IF data presented here were obtained using cells grown in 8-well microscopy slides that were washed and incubated in 500 µL of all solutions. Volumes should be adjusted depending on the cell culture containers used by experimenters. Additionally, perform the IF protocol with extreme care as cells can easily detach from the glass/polymer bottom. Use a micropipette for all solution changes to minimize cell loss.
Incubate on ice for 5 min with 500 µL of ice-cold IF pre/post-extraction solution and swirl gently by hand for 15–30 s to perform the pre-extraction step that removes most cytoplasmic and nucleoplasmic proteins and maximizes the signal from chromatin/DNA-associated factors.
Wash twice with 500 µL of IF washing solution.
Incubate for 15 min at room temperature in 500 µL of IF fixation solution. Alternatively, use a pre-made paraformaldehyde solution (3–4%) for the fixation step.
Wash twice with 500 µL of IF washing solution.
Incubate on ice for 5 min in 500 µL of ice-cold IF pre/post-extraction solution and swirl the slide gently by hand to perform the permeabilization step.
Wash twice with 500 µL of IF washing solution.
Incubate at least 30 min at room temperature with 500 µL of IF blocking solution.
Remove blocking solution using a micropipette and incubate overnight in a humidified chamber at 4 °C with 250 µL primary antibodies diluted in blocking solution. NOTE: It is good practice to perform the incubation with primary antibodies sequentially to avoid potential artifacts of co-localization. Once appropriate controls have been performed, simultaneously incubate primary antibodies to speed up the process.
Wash four times for 5 min in 500 µL of IF washing solution with gentle agitation (60 rpms on a rotator).
Incubate for 1 h at 37 °C with 250 µL of secondary antibodies diluted in blocking solution in a humidified chamber. NOTE: Protect the samples from light upon addition of fluorescently-labeled secondary antibodies.
Wash four times each for 5 min in 500 µL of IF washing solution with gentle agitation.
Incubate at room temperature for 5 min in 500 µL of 1x PBS containing 1 µg/mL DAPI to stain nuclei.
Wash twice with 500 µL of 1x PBS and leave cells in 500 µL of 1x PBS solution for imaging.
Using gridded slides or registered stage positions, find the micro-irradiated cells using a well-characterized DNA damage marker (e.g., γ-H2A.X, RPA32, etc.).
Image the multi-well culture slides or plates in all relevant channels using the appropriate settings on a laser-scanning confocal microscope. NOTE: On the presented system, the imaging was done with a 63X/1.4 oil objective using an 8 µs/pixel dwell time, 1,024 x 1,024 resolution, 1X zoom, and averaging of 2. Note that these parameters may need to be optimized for other systems and/or different experiments.
Representative Results
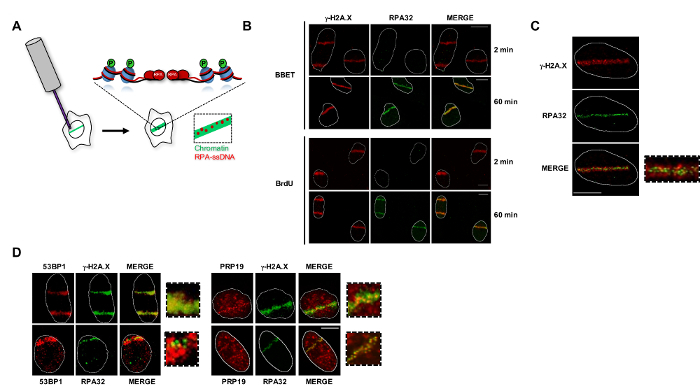
Following micro-irradiation, cells are allowed to recover for a specific period of time depending on the nature of the genome maintenance proteins1,12. DSBs will be processed by nucleases, most extensively during the S and G2 phases of the cell cycle, to create a limited region of ssDNA which is rapidly coated by RPA and other genome maintenance factors9. This ssDNA region is surrounded by chromatin which is extensively modified during the DDR to regulate the recruitment and exchange of other DDR factors13. Protein recruitment to micro-irradiated regions can be monitored by IF (Figure 1A). Typically, chromatin-associated factors (e.g., γ-H2A.X, 53BP1, etc.) are detectable at earlier (1–5 min) time points than ssDNA-bound proteins because nuclease-mediated resection of DSB ends is required to generate the RPA-ssDNA platform (≥10 min). To facilitate image analysis and publication, we have devised a Fiji script (The Outliner)10 which automatically outlines cell nuclei using a DNA dye-associated channel (e.g., DAPI), making it easier to distinguish micro-irradiation stripes and individual cells by leaving out uninformative DNA staining from the final images while preserving nuclear definition (Figure 1B).
γ-H2A.X and RPA complex subunits function as markers for chromatin- and ssDNA-associated factors, respectively. RPA-ssDNA appears as punctate foci surrounded by large fluorescent chromatin stripes decorated by the γ-H2A.X antibody (Figure 1C). Co-localization with these markers can be used to precisely define the position of genome maintenance factors at DNA lesions. For instance, 53BP1, a chromatin-associated protein which controls the DSB repair pathway choice, co-localizes well with the γ-H2A.X signal13. Conversely, PRP19, an E3 ubiquitin ligase that functions on RPA-ssDNA to promote ATR activation and DNA repair is recruited as punctate foci that closely match the RPA32 distribution14,15,16,17 (Figure 1D). Of note, some factors may also be recruited on the chromatin adjacent to the initial lesions but do not spread to large domains surrounding the DSBs like γ-H2A.X and 53BP1. In such cases, these factors would appear as punctate foci but would not perfectly co-localize with ssDNA-bound proteins.
For live cell imaging, cells expressing a POI tagged with a fluorescent marker can be micro-irradiated and imaged in real-time to obtain highly detailed recruitment kinetics (Figure 2A-C). Multiple different proteins can be observed simultaneously provided that their fluorescent labels can be spectrally separated from each other. To facilitate analysis of time series, we have created two Fiji scripts which allow the user to produce movie files containing time-series information (Movie Maker, see movies of Figure 2C as Online Supplementary Material) and to quantify protein recruitment at micro-irradiated sites (Damage Analyzer)10. Instead of the time-consuming manual image curation normally required to describe the behavior of a protein, the Damage Analyzer script guides the user through streamlined steps which monitor signal enrichment at micro-irradiated sites, while automatically correcting for cell movement, image drift, background signal, and bleaching at all time points of the experiment. The generated data can then be easily compiled into a spreadsheet application and plotted as needed (Figure 2D).
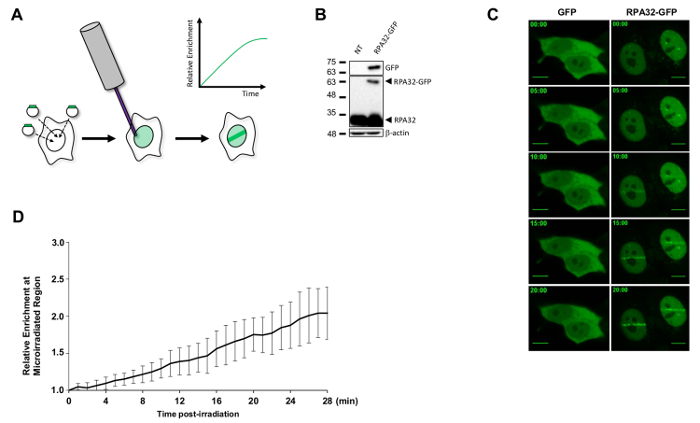
Figure 1: Localization of genome maintenance factors to the chromatin and RPA-ssDNA platforms at micro-irradiation-induced lesions. (A) Laser micro-irradiation in pre-sensitized cells leads to the production of DNA double-stranded breaks, which are resected by endo- and exo-nucleases to generate ssDNA. The ssDNA sub-compartment can be visualized as punctate foci using antibodies against Replication Protein A or other ssDNA-localized factors. In contrast, proteins or modifications that spread to large chromatin domains appear as large stripes at the micro-irradiated site similar to the pattern observed with antibodies targeting the phosphorylated histone variant H2A.X (γ-H2A.X). (B) Cells pre-sensitized with BBET or BrdU were micro-irradiated, pre-extracted, fixed, and stained for the indicated proteins. 12-bit images were collected at 1X zoom and processed using the Outliner Fiji script. (C) Z-stacking of BBET or BrdU pre-sensitized cells allows the resolution of ssDNA- and chromatin-associated factors (maximum intensity Z-projection shown). (D) Using this method, 53BP1 and PRP19 can be classified as chromatin- and ssDNA-associated genome maintenance factors, respectively. Scale bars = 10 µm; images in D are all at the same scale. Please click here to view a larger version of this figure.
Figure 2: Live-imaging of RPA complex recruitment to sites of DNA damage. (A) Stable or transient transfection of plasmids encoding candidate proteins tagged with fluorescent reporters followed by micro-irradiation of transfected cells allows the monitoring of protein recruitment to DNA lesions in real-time. (B) Cells were transfected with a pDEST47-RPA32-GFP plasmid and lysed 24 h later. Protein expression was confirmed by immunoblotting. (C) Cells transfected with pDEST-SFB EGFP or pDEST47 RPA32-GFP plasmids pre-sensitized with BrdU were micro-irradiated and imaged at 12-bit at 1X zoom once per minute post-irradiation. Timing is in minutes post-irradiation. The 00:00 time point image was taken prior to micro-irradiation. Movie files were created using the Movie Maker Fiji script and key frames are shown. Scale bars = 10 µm. (D) Recruitment of RPA32-GFP to micro-irradiation stripes was quantitatively monitored using the Damage Analyzer Fiji script. Output data were plotted using Microsoft Excel. The black line is the mean enrichment-fold of RPA32-GFP at micro-irradiated sites relative to the pre-irradiation signal and the error bars represent the standard error of the mean of 15 independently micro-irradiated cells. Please click here to view a larger version of this figure.
 Movie 1: Micro-irradiation time-lapse of SFB-GFP expressing cells. Cells transiently transfected with a pDEST-SFB GFP plasmid were pre-sensitized 24 h with BrdU, micro-irradiated, and imaged once per minute for 29 min. Please click here to download this movie.
Movie 1: Micro-irradiation time-lapse of SFB-GFP expressing cells. Cells transiently transfected with a pDEST-SFB GFP plasmid were pre-sensitized 24 h with BrdU, micro-irradiated, and imaged once per minute for 29 min. Please click here to download this movie.
 Movie 2: Micro-irradiation time-lapse of RPA32-GFP expressing cells. Cells transiently transfected with a pDEST47 RPA32-GFP plasmid were pre-sensitized 24 h with BrdU, micro-irradiated, and imaged once per minute for 29 min. Please click here to download this movie.
Movie 2: Micro-irradiation time-lapse of RPA32-GFP expressing cells. Cells transiently transfected with a pDEST47 RPA32-GFP plasmid were pre-sensitized 24 h with BrdU, micro-irradiated, and imaged once per minute for 29 min. Please click here to download this movie.
Discussion
Using the methods outlined above, 405 nm laser micro-irradiation of cells pre-sensitized with BBET or BrdU allows the localized generation of DNA lesions including DSBs within the nuclei of adherent human cells. Kinetics of DDR at these DSBs are similar to those generated with other methods in eukaryotic cells9,18,19. DSB resection at micro-irradiated sites leads to ssDNA-generation which can be followed using a RPA32-targeting antibody or an RPA32-GFP fusion. Chromatin-associated factors, on the other hand, can be located using γ-H2A.X as reference. Some chromatin-associated factors may not spread away from the initial DSB as much as γ-H2A.X and will appear as punctate foci that may be distinct from ssDNA-binding proteins foci.
Once DSBs are generated, protein recruitment and clearance from damage sites can be monitored by IF on fixed cells. Performing these experiments requires only a suitable IF antibody against the POI. If IF-compatible antibodies are unavailable, plasmids can be engineered carrying genes of interest in frame with specific epitopes (e.g.: FLAG, His-tag) or fluorescent proteins. Transient or stable transfection/transduction of these plasmids into adherent cells can then be used to determine whether a specific factor is actively recruited to damaged DNA. Overexpression of proteins can lead to mis-localization and may artificially boost their recruitment to micro-irradiated stripes. To obtain results that are more representative of endogenous proteins, CRISPR-Cas9 technology can now be used routinely to tag any gene of interest with suitable epitopes20,21.
The Python scripts provided in the micro-irradiation Analysis Fiji Plugin facilitate the visualization, presentation, publication, and analysis of DDR proteins kinetics. This analysis package is open-source and functions irrespective of the microscope platform used to perform micro-irradiation experiments and image acquisition. All that is required is that the images taken on the microscopy platform be saved as Bio-Formats compatible files22. Since background, drifting, and bleaching corrections are automatically performed, analysis of protein levels at DNA damage sites with the provided scripts is straightforward and very fast compared with the typical manual signal quantification performed with microscope image acquisition software.
As described in the protocol, users must carefully configure their respective laser-scanning confocal microscope systems in order to achieve the appropriate amount of damage and induce DSBs that follow normal DDR kinetics. Indeed, the quantity and type of DNA lesions generated by micro-irradiation will depend on the laser wavelength and dose as well as on the pre-sensitization methods. Users need to keep in mind that even though DSBs are generated by the conditions described here, 405 nm laser micro-irradiation of photosensitized cells will also generate a mixture of other DNA lesions, including cyclopyrimidine dimers (CPDs), SSBs, and oxidized bases, that will vary between microscope systems and settings23,24,25. Thus, a careful monitoring of the lesions at micro-irradiated sites needs to be performed to obtain a comprehensive picture of the damage generated and the response elicited. Importantly, to study the response to DSBs, each microscope system should be optimized by monitoring the kinetics of canonical DNA damage proteins and post-translational modifications at micro-irradiated sites. For instance, γ-H2A.X and the RPA complex subunits (RPA70, RPA32 or RPA14) can be used conveniently as they are quite easy to detect and rapidly accumulate at DSBs. As a rule of thumb, γ-H2A.X signal should be visualized as stripes at early (5 min) and late (up to 3–4 h) time points post-damage whereas RPA should start accumulating as punctate foci approximately 10-15 min post-micro-irradiation. If a pan-nuclear γ-H2A.X signal is observed, the amount of energy received by the cells is non-physiological and will result in rapid cell death.
There are some limitations that have to be considered when using a 405 nm laser for the study of specific DNA repair pathways. For example, although the BBET-405 nm combination readily produces a combination of SSBs and DSBs, it does not generate 6–4 photoproducts (6–4 PPs) and consequently, the kinetics of recruitment of nucleotide excision repair proteins are different than those observed using UV-C micro-irradiation. The kinetics and extent of recruitment of various proteins involved in the response to DSBs may also vary according to the DNA damaging method that is being used23. Because of these variations, we recommend that users adapt their systems to maximize the type of lesions that are potentially recognized by their POIs. Although such systems are less broadly available, using other types of lasers including UV-A (337–355 nm) or femtosecond near-infrared (800 nm) along with different sensitization reagents (e.g.: tri-methyl psoralen) can allow users to generate more specific DNA lesions depending on the DNA repair pathways under study24,26.
Whenever new conditions are being tested, it is convenient to use fluorescently-labeled well-established DNA repair proteins to test doses, wavelengths, and pre-sensitizer combinations, and to ensure that the lesion of interest is efficiently generated. For example, PARP1 and XRCC1 can function as markers for SSB and base-excision repair, XPA is a good indicator of nucleotide excision repair, and 53BP1 will be recruited to DSBs and function in non-homologous end-joining (references23,25,27,28). Antibodies that recognize modifications associated with specific types of DNA damage can also be used to quantify the levels of DNA adducts or breaks. It is thus possible to monitor oxidized bases, cyclopyrimidine dimers, 6–4 PPs, SSBs (using poly-ADP ribose antibody), and DSBs (γ-H2A.X and 53BP1) by IF and determine the parameters that will achieve the highest level of the desired lesions23,27,29. With careful monitoring, users should be able to study their repair pathway of interest using micro-irradiation.
Altogether, the use of wide-spread laser-scanning confocal microscopes equipped with 405 nm laser lines to generate localized DSBs, combined with platform-specific DNA damage markers to sub-classify genome maintenance factors, and streamlined data processing with the help of the micro-irradiation Analysis Fiji plugin, should render the study of DDR factors dynamics more accessible than ever before to both experienced and novice investigators of the genome maintenance field.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada [Discovery grant 5026 to A.M] and by a Next-Generation of Scientists Scholarship from the Cancer Research Society [21531 to A.M]. We thank Jean-François Lucier for providing excellent quality control of the Fiji Python scripts and advice on statistical analysis. We thank Dr. Stephanie A. Yazinski for the IF fixation solution recipe. We thank Paul-Ludovic Karsenti for help with laser power measurements.
References
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen S, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. The Journal of Cell Biology. 2006;173(2):195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli ACL, Ward JF, May N. A New Method for Introducing Double-Strand Breaks into Cellular DNA. Radiation Research. 1993;134(2):160–169. [PubMed] [Google Scholar]
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes & development. 2011;25(5):409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nature cell biology. 2003;5(3):255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ. ImageJ for microscopy. BioTechniques. 2007;43(1 Suppl):25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annual review of genetics. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Garneau D, Maréchal A. Microirradiation Analysis Fiji Plugin. 2017. Available from: https://bitbucket.org/daniel_garneau/microirradiation_analysis/
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhar L, et al. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Reports. 2015;11(9):1–15. doi: 10.1016/j.celrep.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nature Reviews Molecular Cell Biology. 2013;15(1):7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Maréchal A, et al. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation via a Ubiquitin-Mediated Circuitry. Molecular Cell. 2014;53(2):235–246. doi: 10.1016/j.molcel.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois J-C, et al. A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Research. 2017;45(15):8859–8872. doi: 10.1093/nar/gkx571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Huang J. The PSO4 Complex Associates with RPA and Modulates the Activation of ATR. The Journal of Biological Chemistry. 2014;289(10):6619–6626. doi: 10.1074/jbc.M113.543439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas M, Shanmugam I, Bsaili M, Hromas R, Shaheen M. The role of the human Psoralen 4 (hPso4) complex in replication stress and homologous recombination. The Journal of Biological Chemistry. 2014;289(20):14009–14019. doi: 10.1074/jbc.M113.520056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WH, Zhu Z, Papusha A, Malkova A, Ira G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genetics. 2010;6(5) doi: 10.1371/journal.pgen.1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WWM, Yamaguchi M, Haber JE. Real-time analysis of double-strand DNA break repair by homologous recombination. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(8):3108–3115. doi: 10.1073/pnas.1019660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner DH, et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nature Communications. 2015;6:10237. doi: 10.1038/ncomms10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Scientific Reports. 2015;5(9592) doi: 10.1038/srep09592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkert M, et al. Metadata matters: Access to image data in the real world. Journal of Cell Biology. 2010;189(5):777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C, et al. Activation of multiple DNA repair pathways by sub-nuclear damage induction methods. Journal of Cell Science. 2007;120(15):2731–2740. doi: 10.1242/jcs.004523. [DOI] [PubMed] [Google Scholar]
- Kong X, et al. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Research. 2009;37(9):e68. doi: 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton NW, Andrews JF, Gassman NR. Application of Laser Micro-irradiation for Examination of Single and Double Strand Break Repair in Mammalian Cells. Journal of Visualized Experiments. 2017. [DOI] [PMC free article] [PubMed]
- Thazhathveetil AK, Liu ST, Indig FE, Seidman MM. Psoralen conjugates for visualization of genomic interstrand cross-links localized by laser photoactivation. Bioconjugate Chemistry. 2007;18(2):431–437. doi: 10.1021/bc060309t. [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Amé JC, Schreiber V, Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Research. 2007;35(22):7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM. Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response. Nature Chemical Biology. 2015;11(10):807–814. doi: 10.1038/nchembio.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanakis RP, et al. Fluorescence detection of 8-oxoguanine in nuclear and mitochondrial DNA of cultured cells using a recombinant Fab and confocal scanning laser microscopy. Free Radical Biology and Medicine. 2000;28(6):987–998. doi: 10.1016/s0891-5849(00)00185-4. [DOI] [PubMed] [Google Scholar]