

Abstract
The ability to amplify and sequence either DNA or RNA from small starting samples has only been achieved in the last five years. Unfortunately, the standard protocols for generating genomic or transcriptomic libraries are incompatible and researchers must choose whether to sequence DNA or RNA for a particular sample. Gel-seq solves this problem by enabling researchers to simultaneously prepare libraries for both DNA and RNA starting with 100 - 1000 cells using a simple hydrogel device. This paper presents a detailed approach for the fabrication of the device as well as the biological protocol to generate paired libraries. We designed Gel-seq so that it could be easily implemented by other researchers; many genetics labs already have the necessary equipment to reproduce the Gel-seq device fabrication. Our protocol employs commonly-used kits for both whole-transcript amplification (WTA) and library preparation, which are also likely to be familiar to researchers already versed in generating genomic and transcriptomic libraries. Our approach allows researchers to bring to bear the power of both DNA and RNA sequencing on a single sample without splitting and with negligible added cost.
Keywords: Bioengineering, Issue 133, Electrophoresis, Polyacrylamide hydrogel, Next generation sequencing, Genome, Transcriptome, Library preparation, DNA, RNA
Introduction
Next generation sequencing (NGS) has had a profound impact on the way genetics research is conducted. Where researchers once focused on sequencing the genome of an entire species, it is now possible to sequence the genome of a single tumor or even a single cell in one experiment.1 NGS has also made it cost effective to sequence the RNA transcripts found within a cell, a collection of data known as the transcriptome. The ability to amplify and sequence either DNA or RNA from small starting samples has only been achieved in the last five years.2,3,4 Unfortunately, standard protocols are incompatible and researchers must choose whether to sequence DNA or RNA for a given sample. When a starting sample is large enough, it can be split in half. At smaller scales, however, loss of material due to splitting samples can affect library quality, and pooling of samples can average out interesting variations between cells.5 Furthermore, researchers are increasingly interested in examining samples that cannot be split, such as single cells or small heterogeneous tumor biopsies.6
To address this problem, three protocols have recently been developed to sequence both DNA and RNA from the same starting sample: Gel-seq7, G&T-seq8, and DR-seq9. This article presents a detailed protocol for Gel-seq, which can be used to simultaneously generate DNA and RNA libraries from as few as 100 cells at negligible added cost. The novel aspect of Gel-seq is the ability to separate DNA and RNA based exclusively on size using low cost hydrogel matrices. The core innovation of the Gel-Seq protocol is the physical separation of DNA from RNA. This separation is achieved electrophoretically using a combination of polyacrylamide membranes that take advantage of the size differences between these molecules. To put these size differences in context, consider how DNA and RNA are imaged: while DNA exists on the micron-scale and can be viewed using traditional microscopes, RNA exists on the nanometer scale and must be imaged using complex techniques such as cryo-electron microscopy.10
The approach to separating DNA and RNA in this protocol is shown in Figure 1. The left panel shows DNA and RNA free floating in solution near a membrane. When an electric field is applied, as shown in the right panel, DNA and RNA experience an electrophoretic force that induces migration through the membrane. By tuning the membrane properties, we have created a semi-permeable membrane that separates DNA from RNA. The DNA molecules are pushed against the membrane, but become entangled at the edge because of their large size. Small RNA molecules, on the other hand, can reconfigure and weave their way through the membrane. This process, known as reptation, is similar to the way a snake moves through grass. Eventually these RNA molecules are stopped by a second, high-density membrane that is too difficult for even smaller polymers (>200 base pairs) to wriggle through. Once physically separated, DNA and RNA can be recovered and processed to generate information about both the genome and transcriptome. While we can separate DNA and RNA, we have found better results are obtained if the RNA is reverse transcribed to cDNA before separation. The cDNA/RNA hybrids are more stable than RNA alone and can still pass through the low-density membrane.
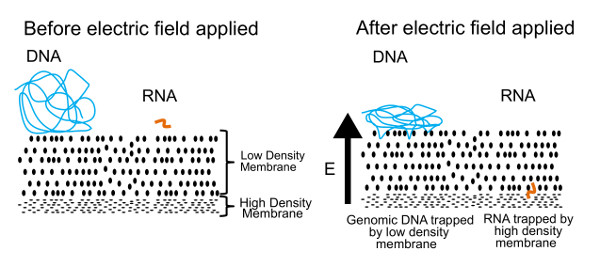
Figure 1. Gel-seq Operating Principle. The underlying principle used to physically separate DNA and RNA. In an applied electric field, small RNA molecules migrate through the low-density membrane but large DNA molecules are trapped at the surface. This figure was reproduced from Ref. 7 with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
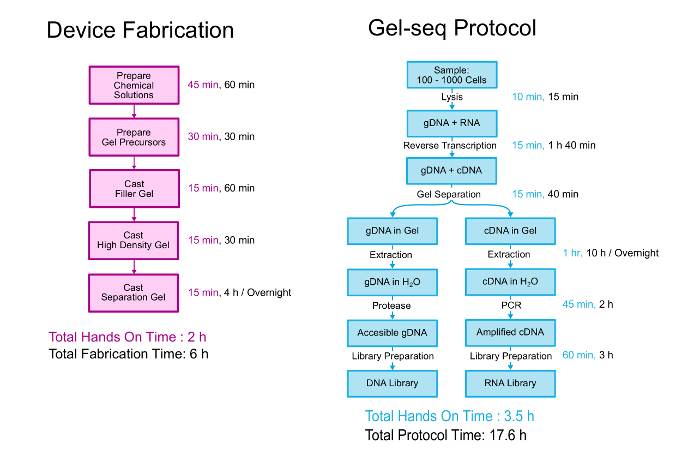
This paper describes in detail both the fabrication of the Gel-seq device and the biological protocol to generate paired DNA and RNA libraries. An overview of both is shown in Figure 2. The device is fabricated by layering three different density polyacrylamide gels on top of each other in a process similar to creating standard stacking gels.11 The biological protocol starts with 100 - 1000 cells suspended in PBS. The cells are lysed and the RNA is converted into cDNA before the device is used to separate the genomic DNA from the cDNA/RNA hybrids. After separation and recovery, genomic and transcriptomic libraries are prepared using a process that closely follows the standard whole-genome library preparation kit protocol. Further detail about the development and validation of Gel-seq can be read in the Lab on a Chip publication "Gel-seq: whole-genome and transcriptome sequencing by simultaneous low-input DNA and RNA library preparation using semi-permeable hydrogel barriers."7
Figure 2. Gel-seq Protocol. An overview of the steps to fabricate the Gel-seq device and the protocol to generated paired DNA and RNA libraries. Portions of this figure were reproduced from Ref. 7 with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
To generate DNA and RNA libraries from single cells, researchers should consider using either G&T-seq or DR-seq. G&T-Seq, like Gel-seq, relies on a physical separation of RNA from genomic DNA. This approach relies on messenger RNA's (mRNA) 3′ polyadenylated tail as a pull-down target. The mRNA is captured on a magnetic bead using a biotinylated oligo-dT primer. Once the mRNA has been captured the beads are held in place with a magnet and the supernatant containing the genomic DNA can be removed and transferred to another tube. After this physical separation is complete, separate libraries can be generated from the mRNA and DNA.8 This approach works well if the RNA of interest is polyadenylated, however it cannot be used to study non-polyadenylated transcripts, such as ribosomal RNA, tRNA, or RNA from prokaryotes.
DR-seq relies on a pre-amplification step where both DNA and cDNA derived from RNA are amplified in the same tube. The sample is then split in two and processed in parallel to prepare DNA- and RNA-seq libraries. To distinguish between genomic DNA and the cDNA derived from RNA, DR-seq takes a computational approach. Sequences where only exons are present are computationally suppressed in the genomic DNA data, as those could have originated from either DNA or RNA.9 An advantage of this approach is that the DNA and cDNA/RNA need not be physically separated as is done in Gel-seq and G&T-seq. The drawback, however, is that DR-seq requires a priori knowledge of the genome and transcriptome (i.e., exons versus introns), and might not be ideal for applications such as sequencing of nuclei, in which many transcripts are not yet fully spliced and still contain introns.12
The novel aspect of Gel-seq is the ability to separate DNA and RNA in hundreds of cells based exclusively on size. This method requires no a priori knowledge of the genome or transcriptome, is robust against incomplete splicing, and is not limited to poly-adenylated transcripts. For applications where a researcher can start with at least 100 cells, Gel-seq provides a straightforward approach using cheap and widely-available materials.
Protocol
1. Chemical Solution Preparation
NOTE: The following steps are for preparing chemical solutions required in later steps. These can be made in bulk and stored for several months.
To begin, prepare 50 mL of purified, deionized water by sterilizing in a 254 nm UV crosslinking oven for 15 min (15 mJ/cm2 total exposure) to neutralize any contaminating DNA. Heat to 37 °C for use in following steps.
Make 10 mL of a 40% total (T) and 3.3% crosslinker (C) (29:1) polyacrylamide precursor solution. Weigh 3.867 g of acrylamide monomer and 0.133 g of bis-acrylamide monomer. Combine and bring volume up to 10 mL with warm purified water and vortex until dissolved. Store protected from light at room temperature. NOTE: Premixed 40% T, 3.3% C (29:1) polyacrylamide solutions can be purchased commercially.
Make 10mL of 50% T, 5% C gel solution. Weigh 4.750 g of acrylamide monomer and 0.250 g of bis-acrylamide monomer. Combine and bring volume up to 10 mL using warm purified water and vortex until dissolved. Store protected from light at room temperature.
Make 10 mL of a 50% (w/v) sucrose solution. Add 5 g sucrose to a graduated cylinder and add warm purified water up to a total volume of 10 mL. Vortex until dissolved and store at room temperature.
Make 10 mL of 10% APS (w/v). Add 1 g of ammonium persulfate (APS) to a graduated cylinder. Add cold (~4° C) purified water up to a total volume of 10 mL and vortex until dissolved. Immediately freeze in aliquots of 200 µL.
2. Gel-seq Cassette Fabrication
Note: Gel-seq was originally developed with upright cassettes (see Table of Materials for more information); however, this protocol can be adapted to work with any standard gel electrophoresis cassette.
Prepare gel precursors in three separate plastic tubes by adding reagents as shown below in Table 1. Do not add either APS or Tetramethylethylenediamine (TEMED) until directed in the following steps. Vortex ingredients to mix thoroughly.
| Filler Gel Precursor | High Density Gel Precursor | Low Density Gel Precursor | |||
| 40%T, 3.3%C Acrylamide Bisacrylamide Solution | 1.6 mL | 50%T, 5%C Acrylamide Bisacrylamide Solution | 2.4 mL | 40%T, 3.3%C Acrylamide Bisacrylamide Solution | 0.6 mL |
| Deionized Water | 10.2 mL | Deionized Water | 1.0 mL | Deionized Water | 4.8 mL |
| Sucrose Solution (50% w/v) | 2.6 mL | Sucrose Solution (50% w/v) | 0.6 mL | ||
| 10X Tris-Borate-EDTA | 1.6 mL | 10X Tris-Borate-EDTA | 0.6 mL | ||
| Ammonium persulfate (10% w/v) | 104.0 µL | Ammonium persulfate (10% w/v) | 50.0 µL | Ammonium persulfate (10% w/v) | 39.0 µL |
| TEMED | 6.0 µL | TEMED | 1.0 µL | TEMED | 2.2 µL |
| Total Volume | 16.1 mL | Total Volume | 4.1 mL | Total Volume | 6.0 mL |
Table 1. Gel Synthesis Reagents. Polyacrylamide gel precursor reagents sufficient for fabrication of 2 cassettes.
De-gas gel monomer solutions by inserting a needle through the cap of the tube and connecting this needle to a house vacuum line. Submerge this assembly in an ultrasonic bath set to high and wait until bubbles stop emerging from the liquid before moving to next step (~60 seconds / tube). NOTE: The quality of the vacuum and power of the ultrasonic bath is not critical so long as bubbles can be seen emerging from the solution.
Add TEMED and APS to the filler gel precursor and vortex briefly. Immediately add 6 mL of the filler gel precursor to each gel cassette by pipetting the solution into the top of the cassette. Use a 1 mL micropipette to add the precursor in six increments to avoid spilling. The total time for this step should be less than 3 minutes.
Ensure the fluid is level by positioning the cassettes upright on a level table. Fill the remainder of the cassette with deionized, degassed water. Pipette the water, again using 1 mL increments, slowly into the center of the cassette to minimize mixing. Allow the polymer to cure for at least one hour, up to overnight.
After the polymer has cured, invert the gel cassettes over a sink to remove the water overlay. A compressed air gun can be used to gently dry the interface by blowing air through the top opening of the cassette from a distance of 6 inches.
Add TEMED and APS to the high-density gel precursor and vortex briefly. Immediately add 320 µL of the high-density precursor to the cassette, again using a 1 mL pipette. Ensure the fluid evenly coats the filler gel layer by rocking the cassette back and forth around 3 times. This step should take less than three minutes.
Fill the remainder of the cassette with deionized, degassed water. Pipette slowly with a 1 mL pipette into the center of the cassette to minimize mixing. Allow the polymer to cure for at least fifteen minutes, preferably an hour.
Again, invert the gel cassettes to remove the water overlay. Compressed air can be used to gently dry the interface. Add TEMED and APS to the low-density gel precursor and vortex briefly. Immediately fill the remainder of the cassette (~1.65 mL) with the low-density gel precursor and insert the gel comb.
Pipette an excess of reserve precursor at the top of the comb as it will be absorbed during polymerization. Allow the polymer to cure at least 4 hours, preferably overnight. Gels can be stored for a week or more in a buffer of Tris-Borate-Ethylenediaminetetraacetic acid (EDTA) (TBE buffer).
3. Sample Preparation and Reverse Transcription
Starting with a suspension of the cells of interest, and working in a polymerase chain reaction (PCR) laminar flow hood, use a hemocytometer or an automatic cell counter to calculate the cell concentration. Dilute the cells to a concentration of 100 to 1000 cells per µL in phosphate-buffered saline (PBS). NOTE: This protocol has been validated on a range of cells including PC3, HeLa, and mouse liver cells.
Using reagents provided in the WTA kit (see Table of Materials), mix 19 µL of lysis buffer and 1 µL of RNase inhibitor to prepare a 10X stock solution of reaction buffer. Create a lysis master mix of sufficient volume containing 0.5 µL of reaction buffer and 2.75 µL of nuclease free water for each sample.
Pipette the cell suspension up and down 5 times to re-suspend settled cells and then pipette 1 µL of sample into a 200 µL nuclease free strip tube sterilized by UV. Repeat as necessary depending on number of samples. Be sure to include a negative control by pipetting 1 µL of nuclease free water instead of cells for one reaction. Next, add 3.25 µL of lysis master mix to each sample and mix by gently pipetting up and down 5 times.
Preheat a thermal cycler (with heated lid) to 72 °C. Add 1 µL of RT primer and 1 µL of 20 µM random hexamer with WTA adapter (5′-AAGCAGTGGTAT-CAACGCAGAGTAC-NNNNNN-3′) to each sample. Reserve at least one tube as a positive control for gDNA library preparation and add 2 µL of water instead of primers. NOTE: Random hexamer with WTA adapter is optional and has minimal impact on sequencing results.
Incubate samples at 72 °C in the preheated thermal cycler for 3 minutes to lyse the cells. Remove cells from thermal cycler and place on ice for 2 minutes. Store the positive control at 4 °C until step 5.
While the cells are lysing, create a sufficient volume of reverse transcription master mix for all RNA samples containing the following reagent ratios: 2 µL of first strand buffer, 0.5 µL of template switch oligonucleotide (TSO), 0.25 µL of RNase inhibitor, and 1 µL of reverse transcriptase (100 U/µL).
Preheat thermal cycler to 42 °C. Add 3.75 µL of reverse transcription master mix to the remaining samples, bringing the total sample volume to 10 µL. Mix by pipetting up and down 5 times.
Perform reverse transcription by immediately placing samples in a preheated thermal cycler. Run the following program: 42 °C for 90 min, 70 °C for 10 min, 4 °C forever. This is a safe stopping point.
4. Gel Separation and Sample Recovery
Carefully clean a gel electrophoresis chamber using a DNA removal product. Apply several mL of the liquid cleaning agent to a disposable lint free wipe and wipe across all surfaces of the chamber, and then fill the chamber with clean 0.5x TBE. For optimal results, place the entire apparatus in a 254 nm UV crosslinking oven and sterilize for 15 minutes (15 mJ/cm2).
Insert the Gel-seq cassette into the gel electrophoresis chamber and lock it into place. Slowly remove the gel comb by pulling straight up. Move slowly to avoid tearing the gel or ripping any of the arms.
Keeping the samples from step 3 on ice, reserve at least one sample as a positive control for cDNA library generation. Store this control at 4 °C until step 6. Add 2 µL of 6X loading dye to the remaining samples, bringing the total volume to ~12 µL. Thoroughly mix the samples by pipetting up and down 5 times.
Combine 1 µL of a DNA ladder with 2 µL of 6x loading dye and 7 µL of water. Pipette this mixture into lane 1 of the Gel-seq cassette as an electrophoresis control. Pipette the samples from the previous step into separate lanes of the Gel-seq cassette. Be careful to prevent contamination between wells by inserting the pipette fully into each well and removing it using only a vertical motion.
Using a standard gel electrophoresis power supply, apply an electric field of 250 V across the Gel-seq cassette for 30 minutes to separate the gDNA from the cDNA/RNA hybrids. Once separated, remove the Gel-seq cassette from the gel electrophoresis chamber and open the two halves of the cassette by prying the edges with a scraping tool.
Using a scalpel, cut the gel in half just below the high-density layer. Discard the half that contains filler gel by picking it up with your gloved hand. Gently peel the remaining gel off of the cassette by scraping it with a paint scraper or other similar tool. Place this section of the gel into a dish containing ~30 mL of 0.5x TBE with 3 µL of gel stain.
Cover the container to minimize photobleaching and soak the gel while gently shaking the container for 5 minutes. Place the gel on plastic wrap and take a UV image using a gel documentation system (for further detail see Ref. 13). A 30 second exposure typically produces clear images. Verify that separation has occurred.
Move the gel to a UV transilluminator to facilitate visualization of the nucleic acids. Wearing appropriate UV glasses, confirm the results from the gel documentation system. The gDNA should be located at the start of the low-density gel and the cDNA at the interface of the low-density and high-density regions.
Use a scalpel, cut out the regions of the gel containing the gDNA and cDNA. Samples are best recovered by cutting a 4 mm by 10 mm rectangular section of gel; however, the exact geometry will depend on the gel electrophoresis system used. Remember to also cut the lane loaded with the negative control.
Place each excised section of gel into a strip tube by using a pair of blunt end tweezers. Be careful not to apply too much force or the gel will split into multiple pieces. Should this happen, simply pick up each piece and add it to the tube.
Grind the gel in each tube using the tip of a pipette (200 µL pipette tips work well) by moving the pipette tip in a circular fashion against the bottom of the tube. Add nuclease free water (40 µL into the gDNA samples and 80 µL into the cDNA samples) to each tube before removing the pipette tip used to grind the gel to minimize sample loss.
Place the strip tubes to a vortex mixer inside of a 37 °C incubator and shake for 8 - 12 hours. This allows the nucleic acids to diffuse out of the gel and is a natural stopping point for this multi day protocol.
Pipette the samples into an 8 µm mesh filter plate and spin the plate at 2600 x g for 5 minutes to strain out the gel fragments. Lift the mesh filter plate away from the housing plate and pipette the gel-free water samples into a new 200 µL strip tube.
Add 1 µL of protease (0.9 AU/mL) to each sample containing gDNA, mix well by pipetting up and down, and incubate at 50 °C for 15 min followed by a heat inactivation at 70 °C for 15 min. This step is critical for depleting nucleosomes and makes the gDNA accessible for subsequent reaction steps.
Using an 18 gauge needle, poke holes in the caps of all sample tubes. Place the samples in a vacufuge to reduce liquid volume. gDNA samples should be reduced to 5 µL and cDNA samples reduced to 10 µL.
Depending on the vacufuge and number of samples, the total evaporation time will vary between 30 and 60 minutes. If the sample volume falls below the target volume, simply add nuclease free water to increase the sample volume.
5. gDNA Library Preparation
Using a fluorometer or similar technology, quantify the DNA concentration in each gDNA sample from step 4 as well as the positive control from step 3. For a detailed protocol see the fluorometer reference manual.14
Dilute the samples to 0.2 ng/µL of DNA. Depending on the starting cell type and quality of results required, lower concentrations may still produce viable libraries. Some experimentation will be required; however, the authors had success with libraries as low as 0.1 ng/µL.
Complete gDNA library preparation by following the library preparation half reaction volume protocol in Step 7.
6. cDNA Library Preparation
Starting with the 10 µL cDNA samples and positive control from step 4, add 12.5 µL of 2X qPCR mix, 0.5 µL cDNA PCR primer, and 2 µL nuclease-free water.
Perform PCR in a real-time thermocycler using the following protocol: hot-start at 95 °C for 3 min, followed by 20-30 cycles of 98 °C for 10 s, 65 °C for 30 s, and 72 °C for 3 min. Total sample volume is 25 µL. Monitor the reaction curves and stop the amplification before the reactions leave the exponential phase (linear signal increase versus cycle number) in order to avoid PCR artifacts due to overamplification. For more on avoiding overamplification, see the discussion section of this paper as well as Ref. 15.
After amplification, clean the product using solid phase reversible immobilization (SPRI) beads following the protocol in step 8. Once completed, proceed to the next step.
Using a fluorometer or similar technology, quantify the DNA concentration in each cDNA sample as well as the positive control from step 4. Dilute the samples if necessary to contain approximately 0.2 ng/µL of DNA. Slightly lower concentrations still produce viable libraries. Some experimentation will be required, however the authors had success with libraries as low as 0.1 ng/µL.
Optional step: Perform a standard polyacrylamide gel electrophoresis separation on 1 - 2 µL of each the qPCR product to validate the library generation reaction worked. A sample image of successful result is shown in Figure 4. For a detailed protocol on how to conduct polyacrylamide gel electrophoresis, see Ref. 16.
Complete cDNA library preparation by following the library preparation kit half volume reaction protocol in Step 7.
7. Library Preparation with Half Volume Reactions
Library preparation follows the library preparation kit protocol using half volume reactions.17 All reagents referenced in this section of the protocol are from the library preparation kit (see Table of Materials). Begin by UV sterilizing a sufficient number of strip tubes for the number of samples to be processed.
Perform the transposase reaction by adding 5 µL transposase buffer to each strip tube to be used in the assay. Then add 2.5 µL input DNA at 0.2 ng/µL (0.5 ng total) followed by 2.5 µL transposase. Mix by pipetting up and down 5 times.
Incubate at 55 °C for 5 minutes, then hold at 10 °C. After the sample reaches 10 °C, remove it from the thermocycler and immediately add 2.5 µL transposase stop buffer to each sample. Keep the samples at room temperature for 5 minutes.
Prepare the PCR reaction to amplify the transpose treated sample by adding 7.5 µL library prep PCR mix, 2.5 µL of an index 1 primer, and 2.5 µL of an index 2 primer. These primers are proprietary and are supplied by the manufacturer of the library preparation kit. Mix well by pipetting up and down 5 times. NOTE: Ensure that unique primer combinations are used for each sample. There are 12 different index 1 primers and 8 different index 2 primers, making it possible to uniquely label up to 96 different samples. Choose a unique combination of primers for each sample.
Perform PCR using the following program on a thermocycler. The sample volume is 25 µL. 72 °C for 3 minutes 95 °C for 30 seconds 12 cycles of: 95 °C for 10 seconds 72 °C for 30 seconds 55 °C for 30 seconds 72 °C for 5 minutes Hold at 10 °C
Clean the prepared libraries using SPRI beads following the protocol in step 8. The libraries should be validated using gel electrophoresis or a similar assay. Refer to the library preparation kit manual for details on how to validate libraries.17
8. Solid Phase Reversible Immobilization Bead Library Cleaning
Solid phase reversible immobilization (SPRI) beads should be stored in 1.5 mL aliquots and should be brought to room temperature before each use. It is also recommended to prepare fresh 80% ethanol for each experiment. The following steps are based the SPRI bead protocol in the WTA kit manual.18
Add 1 µl of the WTA kit lysis buffer to each PCR product. Vortex the SPRI beads until evenly mixed, then add 50 µl of SPRI beads to each sample. Mix by pipetting the sample up and down 10 times and then incubate the samples at room temperature for 8 minutes.
Briefly spin the samples to collect the liquid from the side of the tubes. Place the samples on a magnetic separation device for ~5 minutes until the liquid appears completely clear.
While the samples are on the magnetic separation device, slowly pipette off the supernatant and discard - be careful not to disturb the ring of beads on the tube. Next add 200 µL of 80% ethanol to each sample without disturbing the beads. Wait for 30 seconds and then carefully pipette off the supernatant. Repeat this step (ethanol wash) once.
Allow the samples to dry for 30 s - 1 min. Do not over-dry the samples as large fragments will become permanently bound to the beads. NOTE: A brief dry time is recommended to ensure all traces of ethanol are removed. Should trace amounts of ethanol remain behind they can slightly inhibit downstream reactions.
Remove the samples from the magnetic separation device and add 15 µL of water to each sample to elute the DNA from the beads. Pipette up and down and ensure the beads are removed from the sides of the tubes. Incubate at room temperature for 2 minutes. Briefly spin the samples and then place them on the magnetic separation device for ~1 minute until the solution appears clear.
While the samples are on the magnetic separation device, slowly pipette up the supernatant and transfer it to clean tubes. Be careful not to disturb the ring of beads on the tube. Discard the tubes with beads.
Representative Results
The physical separation of gDNA and cDNA/RNA hybrids in the Gel-seq device can be visualized through fluorescent gel imaging; a representative result is shown in Figure 3. Panel A shows the fabricated Gel-seq device; false color has been added to distinguish the different gel regions. Panel B shows a close up of four different separations used for validation. The third lane, a negative control, represents background and shows that there is no autoflourescence of the gel at the interfaces. We loaded the first and second lanes with DNA ladders. These lanes show only a dark band at the interface between the low- and high-density membranes, revealing that small fragments can pass through the low-density gel. The fourth lane shows the behavior of a biological sample of interest: 500 PC3 cells. We loaded lane four as described in step 3 of the protocol. The image shows separation of genomic DNA and cDNA/RNA hybrids. A dark band at the top of the low-density membrane is megabase-scale genomic DNA while the cDNA/RNA hybrids are stacked at the interface of the low and high-density regions. Unlike the lanes loaded with ladder, there are also several bands present within the high-density region of the gel. These fragments, smaller than 100 bp, are off-target products generated from primer oligonucleotides during reverse transcription. Panel C shows a representative image of the entire Gel-seq device from a successful experiment. Lanes labeled RNA/cDNA were processed with the Gel-seq protocol, while lanes labeled RNA show a separation of just gDNA and RNA. Panel D shows a failed experiment with black bands at the top of every lane of the low-density membrane. This was caused by electrophoresis buffer contaminated with fragmented DNA. At this step, researchers should be looking for both clean negative controls and two distinct black bands indicating the presence of separated gDNA and RNA/cDNA hybrids.

Figure 3. Gel-Seq Separation Results. The Gel-seq device (A) and a fluorescent image showing separation of DNA and RNA/cDNA hybrids (B). False color has been added to more easily distinguish between the different regions of density within the gel. (C and D) Representative fluorescent images of the entire Gel-seq device from a successful (C) and failed (D) experiment. NTC = No Template Control. Portions of this figure were reproduced from Ref. 7 with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
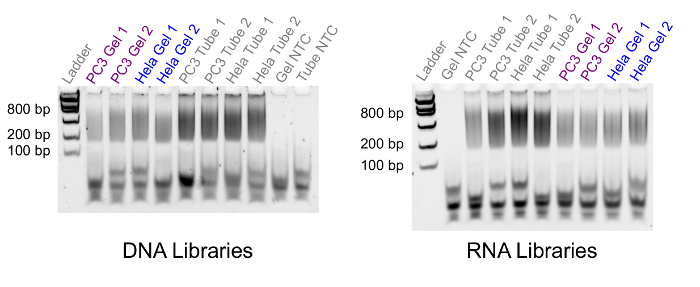
Once the DNA and RNA/cDNA hybrids have been separated and the remainder of the Gel-seq protocol completed, it is possible to generate sequencing libraries. To validate the prepared libraries, we run either a standard gel electrophoresis experiment (Figure 4) or a bioanalyzer. The results in Figure 4 show libraries generated from 500 PC3 cells and 750 HeLa cells. The figure shows the fragment distributions for matched libraries generated from Gel-seq (labeled 'Gel') compared to unmatched samples generated with standard protocols (labeled 'Tube'). The fragment sizes for Gel-seq appear between 200 and 800 basepairs as expected when preparing libraries using the standard whole-genome library preparation kit. If the library fragments do not appear in the correct size range in this step, library preparation has failed.
Figure 4. Library Fragment Size Comparison. A fluorescent gel electrophoresis image comparing library size distribution between Gel-seq (Gel) and standard controls (Tube). The left lane contains a low mass DNA ladder with fragment sizes 100, 200, 400, 800, 1200, and 2000 basepairs. The fragment sizes for all libraries fall between 200 and 800 basepairs, as expected for libraries prepared with this kit. Please click here to view a larger version of this figure.
The ultimate validation of the Gel-seq protocol is based on the analysis of sequencing results. We selected PC3 cells for our validation experiments, as these cells have homogenous expression profiles that allow samples to be split and processed using both Gel-seq and traditional methods; see Figure 5. A comparison between genomic DNA for PC3 cells is shown in Figures 5A and 5B. Figure 5A shows a comparison of genome-wide copy number variation (CNV) profiles generated from PC3 using either Gel-seq or a standard whole-genome library prep reaction (tube control). Each point is a mean normalized bin count; bins are defined from reference genome data such that each bin has equal expected count in a healthy diploid cell, i.e., a flat line, representing equal copies for each region of all autosomal (excluding X and Y) chromosomes. PC3 contains multiple copies of the same regions which show up as spikes above a background copy number of two. Gel-seq yields a qualitatively similar CNV profile as standard tube reaction. Agreement between the two plots can be assessed quantitatively by linear regression, as shown in panel B. A Pearson correlation of R = 0.90 indicates that genomic data gathered from either method is functionally equivalent.
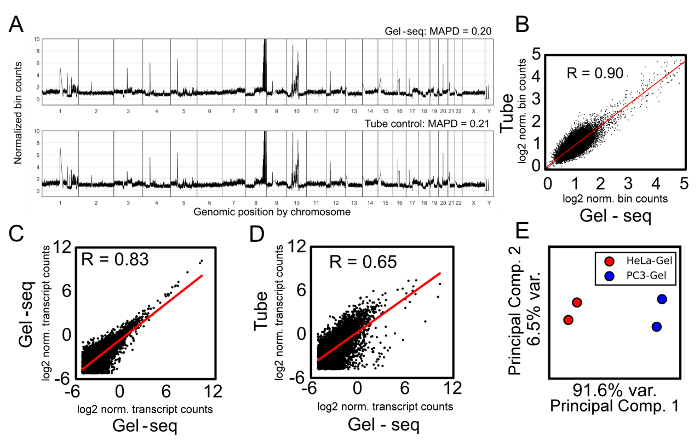
Figure 5. Library Validation. Gel-seq validation for the genomic (A and B) and transcriptomic (C, D, and E) data generated from PC3 and Hela cells. Panel A shows a comparison of genome-wide CNV profiles generated from PC3 using either Gel-seq (Gel-seq) or a standard reaction (tube). MAPD = Median Absolute Pairwise Difference. Panel B is a linear regression between the two samples in Panel A, with R = 0.90 indicating the genomic data are functionally equivalent. The axes show the log2 normalized bin counts. Panels C and D compare transcriptomic data from PC3 cells, with each point showing a count in transcripts per kilobase per million (TPM). The axes show the comparison between two samples as log2 normalized transcript counts. Panel C shows a comparison of technical replicates generated using Gel-seq, and Panel D shows a comparison between Gel-seq and traditional RNA-seq. Panel E shows that Gel-seq can resolve cell type based on RNA expression using a principal component analysis. The x axis shows that the first principal component accounts for 91.6% of the variation between the samples while the y axis shows that the second principal component accounts for only 6.5% of the variance. Portions of this figure were reproduced from Ref. 7 with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
Similarly, we compared transcriptomic data from our Gel-seq protocol to the standard in-tube Smart-Seq protocol. Figure 5 shows the correlation between both Gel-seq technical replicates (Figure 5C) and between Gel-seq and the standard method (Figure 5D). Each point is a count in transcripts per kilobase per million (TPM) for each gene detected at TPM > 5 in both datasets. The linear regressions are shown as red lines, and the Pearson correlation coefficient is shown in the upper left corner. Technical replicates from Gel-seq agree (R ∼ 0.8), but correlate less well with the standard method (R < 0.7). This suggests that Gel-seq introduces a bias in gene counts. Fortunately, this bias is systematic and, as can be seen by the principal component analysis in Figure 5E, meaningful conclusions can still be drawn between different biological samples.
Discussion
There are several critical steps associated with the Gel-seq device fabrication as well as the protocol itself. During fabrication, we recommend starting with the prescribed layer thicknesses for the various regions of the gel. We spent significant time testing different fabrication options and the protocol described here produces the best devices for the cassettes listed in the Table of Materials and Reagents. If researchers use an alternative cassette system, they may find it necessary to tweak the volumes used when creating the devices. The major challenge in fabrication is that if the high-density gel region is too large it can delaminate from the edges of the cassette and create air pockets on the interior of the cassette that will disrupt electrophoresis. By casting several cassettes with several different layer volumes, researchers should be able to quickly determine the optimal configuration for their specific hardware.
The Gel-seq protocol also has several critical steps that can be validated before the protocol is complete. One potential failure point is the separation of gDNA and RNA/cDNA hybrids. This can be validated by imaging the Gel-seq device after separation (see Figure 3B). In one set of experiments, we found that our lab supply of buffer had become contaminated with DNA and was causing substantial autoflourescence in our device (see Figure 3D). This made it difficult to determine if the separation had taken place. Fluorescent imaging helped us identify and correct this problem before using any costly reagents to generate sequencing libraries.
Another critical point is step 6.2, the qPCR amplification of cDNA after separation. Researchers should pay careful attention not to overamplify in this step as it will reduce the quality of the RNA-seq data. This consideration is not unique to Gel-seq, but is a common aspect of low-input RNA-seq library preparation. PCR amplification during sequencing library preparation is often necessary, but it can introduce sequence errors and biases. The required number of cycles for PCR depends on sample quantity and complexity. It is generally advisable to limit PCR cycle number to the bare minimum required to yield sufficient clustering when the libraries are sequenced. In theory, a protocol can be optimized to determine the exact cycle number that yields sufficient copy number without introducing excessive artifacts. In practice, however, inconsistencies in sample quality, loading, or handling early in the protocol can dramatically affect the distribution of molecular templates available for library prep PCR, which in turn affects the optimal PCR cycle number. The most general solution we have found to monitor the progress of the amplification reactions using a fluorescent dye, run the reactions on a real-time PCR thermocycler, and stop the reactions in the exponential (linear versus cycle number) phase. In our experience, real-time monitoring is especially relevant when developing, adapting, or adopting a new protocol.
The last critical step is generating genomic and transcriptomic libraries. The key to this step is to set the starting sample concentration for the DNA library prep reaction as close to 0.2 ng/µL (0.5 ng total) as possible. This is relatively straightforward for the qPCR amplified cDNA as there is usually an excess of cDNA, but it can be more challenging for the gDNA samples. We found careful attention to the vacufuge step was required while the samples were being concentrated. As expected, in experiments with 1000 cells, the vacufuge step could be stopped much sooner than experiments with 100 cells. The number of samples in the vacufuge also impacted the evaporation rate in our experiments. We found that using a flourometer to validate DNA content midway through the concentration step could be helpful when performing the protocol with unfamiliar samples. Fortunately, if researchers over concentrate a sample, nuclease free water can be added to dilute the sample. Theoretically, it is possible to use the vacufuge to dry the DNA and then resuspend it in the desired volume; however, we suggest avoiding complete evaporation.
We view the three current methods for generating simultaneous DNA and RNA libraries, Gel-seq7, G&T-seq8, and DR-seq9, as complimentary. Gel-seq is ideal for samples in the 100 - 1000 cell range and requires no pull-down targets or a priori knowledge of the genome. The other two methods are better suited for single cell applications. One of our goals in developing Gel-seq was to create a protocol that could be easily implemented by other researchers. We therefore decided to fabricate devices within the standard form factor of a polyacrylamide gel cassette. While the technique we used to define our different membranes is novel, most genetics labs already have all the necessary equipment to fabricate the Gel-seq device. Furthermore, the cost of the device is trivial - just $5.25 for a device that can process 12 samples. That said, as with any library preparation protocol using commercial reagents, the overall cost for generating libraries remains high. Our reagent cost per sample was $50 for whole-transcript amplification and $28 for library preparation for both DNA and RNA. Fortunately, the Gel-seq device itself is protocol agnostic. For example, during development we successfully tested the device using cells from culture and an older RNA library amplification protocol19, although we found it was not suitable for tissue samples from mice. Looking towards the future, as cheaper alternatives for library preparation are developed, our protocol can be adapted to work with these new techniques. We believe researchers will find it straightforward to implement Gel-seq in their own labs. We hope this will facilitate the rapid adoption of the technology.
Disclosures
KZ is co-founder and Scientific Advisor of Singlera Genomics Inc.
Acknowledgments
Funding for this work was provided by the University of San Diego, the National Science Foundation Graduate Research Fellowship Program, NIH grant R01-HG007836, and by the Korean Ministry of Science, ICT and Future Planning.
Earlier versions of a several figures were first published in "Hoople, G. D. et al. Gel-seq: whole-genome and transcriptome sequencing by simultaneous low-input DNA and RNA library preparation using semi-permeable hydrogel barriers. Lab on a Chip 17, 2619-2630, doi:10.1039/c7lc00430c (2017)." Lab on a Chip has sanctioned the reuse of figures in this publication.
References
- Mawy T. Single-cell sequencing. Nat Methods. 2014;11(1):18. doi: 10.1038/nmeth.2771. [DOI] [PubMed] [Google Scholar]
- Gole J, et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotech. 2013;31(12):1126–1132. doi: 10.1038/nbt.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa Y, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA-Seq reveals non-genetic gene expression heterogeneity. Genome Biol. 2013;14(4):31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14(9):618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoople GD, et al. Gel-seq: whole-genome and transcriptome sequencing by simultaneous low-input DNA and RNA library preparation using semi-permeable hydrogel barriers. Lab Chip. 2017;17:2619–2630. doi: 10.1039/c7lc00430c. [DOI] [PubMed] [Google Scholar]
- Macaulay IC, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12(6):519–522. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol. 2015;33(3):285–289. doi: 10.1038/nbt.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal A, Zhou ZH, Knobler CM, Gelbart WM. Visualizing large RNA molecules in solution. RNA. 2012;18(2):284–299. doi: 10.1261/rna.027557.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SDS-PAGE Gel. Cold Spring Harb Protoc. 2015;2015(7):087908. [Google Scholar]
- Lake BB, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352(6293) doi: 10.1126/science.aaf1204. at http://science.sciencemag.org/content/352/6293/1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PY, Costumbrado J, Hsu C-Y, Kim YH. Agarose Gel Electrophoresis for the Separation of DNA Fragments. J Vis Exp. 2012. p. e3923. [DOI] [PMC free article] [PubMed]
- Qubit 3.0 Fluorometer Manual. MAN0010866. Life Technologies. 2017. Available from: https://tools.thermofisher.com/content/sfs/manuals/qubit_3_fluorometer_man.pd.
- Vitak SA, et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat Methods. 2017;14(3):302–308. doi: 10.1038/nmeth.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A Guide to Polyacrylamide Gel Electrophoresis and Detection. Bulletin 6040 Rev B. Bio Rad. 2017. Available from: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040.pdf.
- Illumina. Nextera ® XT DNA Library Preparation Kit. 2017. [DOI] [PubMed]
- Takara Bio USA Inc. SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing User Manual. 2016.
- Kurimoto K, Yabuta Y, Ohinata Y, Saitou M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat Protoc. 2007;2(3):739–752. doi: 10.1038/nprot.2007.79. [DOI] [PubMed] [Google Scholar]