

Abstract
Appropriate gene expression in response to extracellular cues, that is, tissue- and lineage-specific gene transcription, critically depends on highly defined states of chromatin organization. The dynamic architecture of the nucleus is controlled by multiple mechanisms and shapes the transcriptional output programs. It is, therefore, important to determine locus-specific chromatin accessibility in a reliable fashion that is preferably independent from antibodies, which can be a potentially confounding source of experimental variability. Chromatin accessibility can be measured by various methods, including the Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) assay, that allow the determination of general chromatin accessibility in a relatively low number of cells. Here we describe a FAIRE protocol that allows simple, reliable, and fast identification of genomic regions with a low protein occupancy. In this method, the DNA is covalently bound to the chromatin proteins using formaldehyde as a crosslinking agent and sheared to small pieces. The free DNA is afterwards enriched using phenol:chloroform extraction. The ratio of free DNA is determined by quantitative polymerase chain reaction (qPCR) or DNA sequencing (DNA-seq) compared to a control sample representing total DNA. The regions with a looser chromatin structure are enriched in the free DNA sample, thus allowing the identification of genomic regions with lower chromatin compaction.
Keywords: Genetics, Issue 134, Chromatin accessibility, FAIRE, regulatory elements, nucleosomes, phenol:chloroform extraction, protein-DNA crosslink
Introduction
The genomic DNA of eukaryotic cells is tightly packed into nucleosomes, which needs to be removed or remodeled in order to allow the interaction of other proteins with the DNA. The transcriptional activation of a gene typically leads to a more open configuration of its nucleosomal structure, particularly in the +1 nucleosome1, to facilitate transcription by RNA polymerase. Also, regulatory regions such as promoters and enhancers undergo dynamic changes in their chromatin accessibility to allow the interaction with chromatin modifiers or transcription factors2. Several approaches have been developed to identify these nucleosome-free regions. These include the sensitivity to nucleases such as DNase I3 or micrococcal nuclease4, which can only access the DNA when it is not tightly wrapped around nucleosomes. Other methods for the identification of open chromatin or free DNA include the transposase-mediated integration of retrotransposable elements (Assay for Transposase-Accessible Chromatin, ATAC)5, or chromatin immunoprecipitation (ChIP) using antibodies against histones. The FAIRE method is not based on the capacity of an enzyme to reach the DNA or the binding of an antibody to a particular protein, but instead relies on the purification of nucleosome-free regions by a phenol:chloroform extraction6,7. In this method, the DNA is covalently bound to chromatin proteins by the crosslinking agent formaldehyde and is sheared afterwards to small pieces by sonication. Tightly packed chromatin regions will have abundant DNA/protein crosslinks, while DNA regions with no or few nucleosomes will have little or no crosslinked DNA/protein complexes. A phenol:chloroform extraction allows purification of the non-crosslinked free DNA in the aqueous phase, while the DNA crosslinked to proteins is captured in the organic phenol phase or aqueous-organic interphase together with the proteins. The quantification of this free DNA compared to a reference of total DNA sample allows the identification of the chromatin free regions. The FAIRE method can be used for the characterization of individual genomic regions as described here, but is also suitable for the identification of genome-wide chromatin accessibility when coupled to deep sequencing as described in different models8,9,10,11,12,13. Results obtained by FAIRE-seq and other methods such as ATAC-seq are largely overlapping14. The FAIRE method is a very robust, cheap, and easy to perform method to identify nucleosome-free regions. Unlike other methods, it does not require pilot experiments to determine the appropriate level of digestion as required for nucleases (DNase-seq, MNase-seq) or transposases (ATAC-seq) and discussed in detail in a recent review15. The only parameters to be adjusted are the sonication conditions and in some cases the fixation time. This paper describes a straightforward protocol that is schematically displayed in Figure 1 and that does not require any special equipment other than a sonicator. The protocol can be performed in a reasonably short time and makes FAIRE an easy and reliable method to test chromatin accessibility in a number of different cell types ranging from lung cells11 to primary cells obtained from mouse livers16.
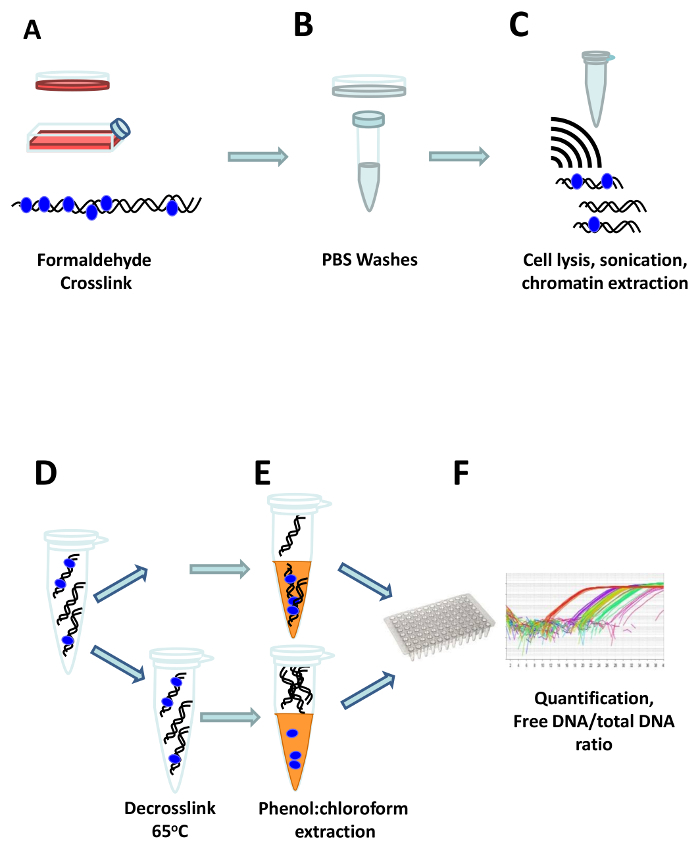
Protocol
1. Day 1: Cell Culture
Culture the cells to be analyzed to the desired amount. Cell culture conditions and media depend on the cell types under investigation. NOTE: In principle, any eukaryotic cell is amenable to an analysis of chromatin accessibility by FAIRE. For this experiment, 4 x 106 HEK-293T cells are seeded in a single 10 cm dish in DMEM medium supplemented with 10% (v/v) FBS and 1% (w/v) penicillin/streptomycin, and incubated at 5% CO2 and 37 °C for 24 h until the cells reach 70 - 80% confluence (~ 8 x 106 cells per dish).
2. Day 2: Formaldehyde Crosslinking and Cell Harvesting
CAUTION: Formaldehyde is highly toxic and must be always used under a fume hood. Wear protective clothes (gloves and lab coat). Discard the waste appropriately.
Add formaldehyde directly to the cell culture medium in a fume hood to reach a final concentration of 1% (v/v) (270 µL of 37% (v/v) formaldehyde to 10 mL of cell culture medium). Incubate for 10 min at room temperature (20 - 25 °C) and slew the plates manually every 2 min. NOTE: The crosslinking time can be adjusted according to the cell type. For the majority of cell lines, 10 min of crosslinking is adequate. For tissues, longer incubations might be required to allow the fixation of all cells.
Add glycine to a final concentration of 0.125 M to quench the formaldehyde (add 500 µL of a 2.5 M glycine stock to 10 mL of culture medium). Incubate for 5 min at room temperature and slew every 2 min.
Wash the cells 3x with ice cold PBS (8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4 · 2H2O, 0.24 g/L KH2PO4, adjust pH to 7.4 with HCl or NaOH). Aspirate the medium and wash directly in the cell culture dish or flask by adding 10 mL of PBS. Repeat this step twice being very careful in the case of loosely adherent cells such as HEK-293T. Add PBS carefully to the side of the dish in order to avoid detachment of the cells.
After the third wash, resuspend the cell in 1 mL of ice cold PBS and transfer to a 1.5 mL reaction tube on ice. Use a cell scraper to detach the cells when necessary. NOTE: When starting with a limited material, use low DNA-binding tubes to avoid sample loss during the procedure.
Centrifuge for 5 min at 300 x g at 4 °C in a cooled tabletop centrifuge. Discard the supernatant by carefully aspirating it.
3. Cell Lysis and Chromatin Fragmentation
Resuspend the cell pellet in 1 mL FAIRE lysis buffer (1% (w/v), 10 mM EDTA, 50 mM Tris HCl pH 8.1, 10 µg/mL leupeptin, 10 µg/mL aprotinin, 2 mM PMSF), and resuspend the cells by carefully pipetting up and down several times. Incubate for 20 min on ice. Store the samples at -80 °C at this step if needed.
Sonicate the crosslinked DNA to shear it to an average size of 200 - 300 bp. The specific settings of the sonicator must be determined for each source of samples and the used sonication device. In any case ensure that the DNA is efficiently sheared. An optimization step for DNA shearing is described under step 5.14. Cool the samples on ice during sonication to avoid heating of the sample.
Centrifuge the sample for 15 min and 13,000 x g at 4 °C.
Transfer the supernatant to a new reaction tube. Split the samples in 100 µL aliquots. Store the samples at -80 °C, if needed. NOTE: The protocol can be interrupted at this point.
4. De-crosslinking of Control DNA
Take a 100 µL aliquot from step 3.4 to reverse the crosslink and to be used as a reference for total DNA. Add 10 µL of RNase-A (10 mg/mL) and incubate for 1 h at 37 °C.
Add 10 µL of Proteinase K (20 mg/mL). Use a programmable thermo-block to incubate for 4 h at 37 °C and then for 6 h at 65 °C to cleave the proteins and to reverse the crosslinks. Finally, hold the samples at 4 °C until the protocol is resumed and then proceed to step 5. NOTE: Other FAIRE protocols use chromatin samples from cells which have not been crosslinked as reference, thus abolishing the need for de-crosslinking11. The fact that these samples are not crosslinked could lead to differences in the sonication efficiency or in DNA stability, therefore the use of total DNA reference samples that have undergone the same procedure as the test samples is more accurate.
5. Day 3: Phenol:Chloroform Purification of DNA
Take the non-de-crosslinked aliquot from step 3.4. Add 10 µL of RNase-A (10 mg/mL) and incubate for 1 h at 37 °C.
Take the non-de-crosslinked aliquot form step 5.1 and the de-crosslinked sample from step 4.2 and proceed in parallel. Add H2O to reach a final volume of 300 µL.
Add 300 µL of phenol:chloroform:isoamyl alcohol (25:24:1) in a fume hood and vortex vigorously. CAUTION: Phenol is corrosive and toxic and must be used always under a fume hood. Wear protective clothes (gloves and lab coat), make sure the reaction tubes are tightly closed, and dispose the waste appropriately.
Centrifuge for 10 min at 13,000 x g at 4 °C.
Carefully transfer 280 µL of the upper aqueous phase to a new reaction tube. Make sure not to take any debris from the interphase which also contains proteins. Discard the remaining lower phase as organic solvent waste.
Repeat steps 5.3 to 5.5 and carefully transfer 270 µL of the upper aqueous phase to a new reaction tube.
Add 270 µL of chloroform in a fume hood and vortex vigorously. NOTE: This step is used to eliminate any remaining phenol from the sample.
Centrifuge for 10 min at 13,000 x g at 4 °C.
Carefully transfer 250 µL of the upper aqueous phase to a new reaction tube, taking care not to carry any debris from the interphase. Discard the remaining sample as organic solvent waste.
Add 25 µL of 5 M NaCl and 250 µL of isopropanol. Add 5 µL of glycogen (20 mg/mL) as the DNA carrier. Mix well by inverting the tubes and incubate for 20 min at room temperature. Centrifuge for 10 min at 13,000 x g at 4 °C to precipitate the DNA.
Wash the DNA pellet with 400 µL of 70% (v/v) ethanol. Centrifuge for 10 min at 13,000 x g at 4 °C.
Aspirate the supernatant carefully and let the pellet dry for 10 min at room temperature. Resuspend in 100 µL TE buffer (10 mM Tris pH 8, 1 mM EDTA).
Incubate the non-de-crosslinked free DNA sample for 4 h at 65 °C to remove inter-DNA crosslinks. Store the samples at -20 °C.
Quantify the amount of DNA using a spectrophotometer and run an aliquot to check the fragment size on a 2% (w/v) agarose gel. Adjust the shearing time and intensities, depending on the results of the DNA size analysis by agarose gel electrophoresis, to obtain an average size of 200-300 bp.
6. Determination of Free Versus Total DNA by Quantitative Real Time PCR (qRT-PCR)
The ratio of nucleosome-free DNA (in the following referred to as free DNA) versus total DNA is determined by qRT-PCR17,18. Samples can also be quantitatively analyzed by microarray hybridization or deep sequencing for genome-wide determinations.
Design qPCR primers for the regions to be tested and for positive and negative controls. NOTE: Suitable positive controls are primers situated in the promoter of actively transcribed housekeeping genes such as ACTB, GAPDH, TPI1, or TUBA1A (encoding β-Actin, GAPDH, TPI or α-Tubulin). Appropriate negative controls are found in heterochromatin regions or gene deserts. Other suitable negative controls can be designed in genomic regions adjacent to the dynamically regulated locus under investigation to control for local copy number differences (Table 1).
Dilute the DNA samples to a final concentration of 10 ng/µL with TE buffer, and take the dilution factor into account for the final calculations (see step 6.5).
Set up a qPCR reaction in triplicates in a 96-well plate, with the following reaction mix per well: template DNA: 20 ng (2 µL), forward primer 200 nM (0.4 µL of a 5 µM stock), reverse primer 200 nM (0.4 µL of a 5 µM stock), a commercial ready to use reaction mix containing green dye (5 µL from a 2x stock), and H2O to 10 µL.
Run in a real time qPCR device, using the absolute quantification mode, and determine the Ct of the samples with the different primer pairs. Calculate the ratio of recovery of free DNA to total DNA for each primer using the following formula, taking into account the dilution factors from step 6.3:
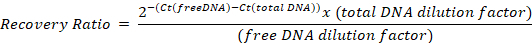 Representative results and an example calculation are given in Table 2.
Representative results and an example calculation are given in Table 2.To compensate for differences among samples, normalize to the positive control recovery ratio:
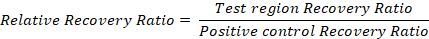
Representative Results
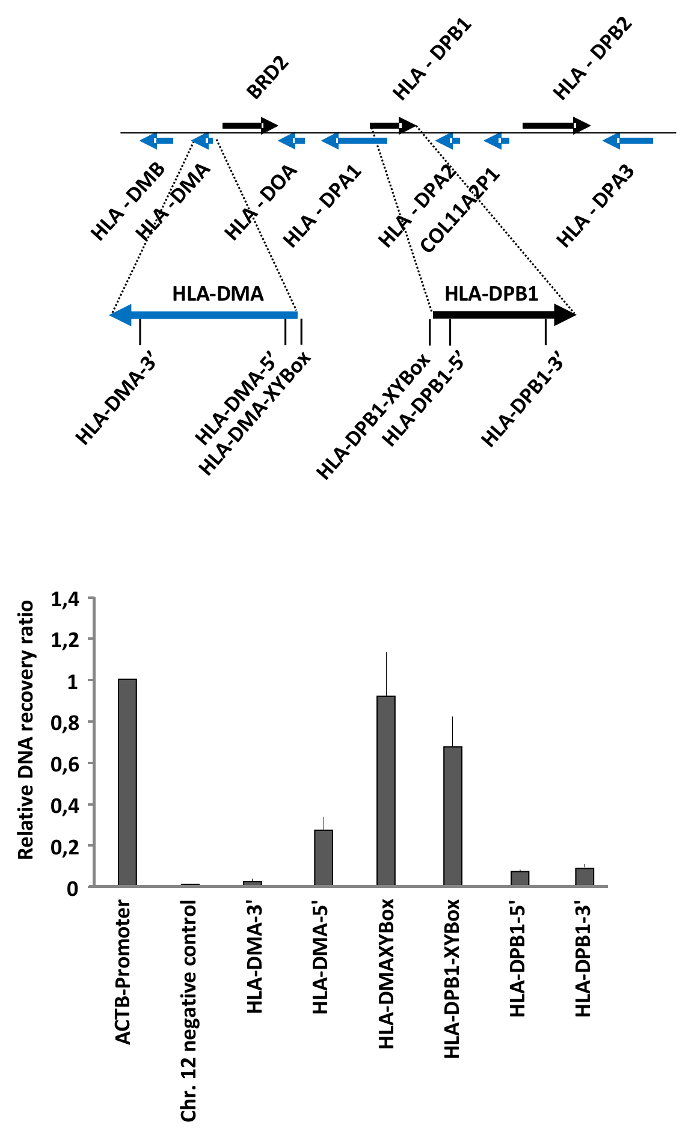
We have used the FAIRE method to measure differences in the chromatin accessibility of MHC class II genes in HEK293T cells as published19. MHC class II encoding genes are expressed in antigen presenting cells (APCs), either constitutively or in response to cytokine signaling20. The expression of MHCII genes is driven by an activator complex which binds to specific DNA elements, termed XY boxes, located in the promoter and enhancers of these genes21,22. This is reflected at the level of chromatin, as revealed by our FAIRE analysis of selected regions in the MHC class II genes HLA-DPB1 and HLA-DMA. These experiments showed that chromatin is open at the regions encompassing the XY boxes, while it is more compact in the gene bodies (Figure 2).
As an example, for the calculations, in this particular experiment, we diluted the total DNA sample 10 times (dilution factor 10), and the free DNA sample was not diluted (dilution factor 1) for the qRT-PCR. The Cts obtained for the different regions tested and the calculations to obtain the final recovery ratios and relative recovery ratios are listed in Table 2. These relative recovery ratios were obtained using the ACTB promoter as the positive control. Note that these calculations are for one of the replicates used to generate the results shown in Figure 2.
In a different context, chromatin accessibility was tested in a model for inducible gene expression. In this case, rapid changes of chromatin compaction in response to the pro-inflammatory stimulus tumor necrosis factor (TNF) were measured. HeLa cells were left untreated or stimulated with TNF for 1 h, followed by FAIRE to measure chromatin compaction at the promoter region controlling expression of the gene encoding the TNF-responsive cytokine interleukin 8 (IL8). These results showed a strongly increased chromatin accessibility at the IL8 promoter of TNF-stimulated cells (Figure 3). The accessibility at the IL-8 promoter after TNF stimulation is even higher than that found in the ACTB promoter showing a dramatic chromatin remodeling in this regulatory region. As the recovery ratio obtained in this case is higher than that obtained in the region used as the positive control (ACTB promoter) the relative recovery ratio is higher than one.
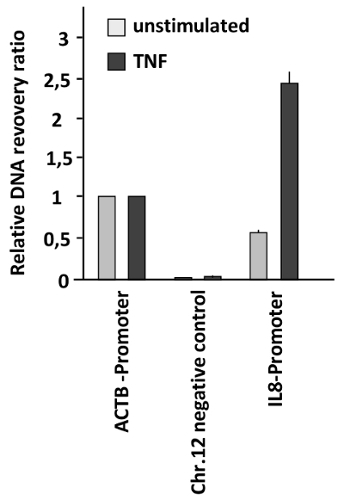
Figure 1: The FAIRE workflow. Cells are crosslinked directly in the cell culture flask or dish by adding formaldehyde (A). After quenching of the formaldehyde, cells are washed three times with ice cold PBS (B), and transferred to a reaction tube where they are resuspended in FAIRE lysis buffer and sonicated to shear the DNA (C). One aliquot of the extracted chromatin is de-crosslinked by heating at 65 °C, to get the total DNA reference (D), and afterwards the free DNA is extracted using phenol:chloroform both from the crosslinked and the de-crosslinked aliquots (E). Finally, the DNA is quantified, and the ratio of free vs. total DNA is calculated (F). Please click here to view a larger version of this figure.
Figure 2: Determination of chromatin accessibility in the MHC class II gene cluster in HEK293T cells. HEK293T cells were analyzed by FAIRE. The ratio between free versus total DNA (i.e., the relative recovery ratio) was determined for the promoter of the ACTB gene as a positive control, a heterochromatin region on Chr. 12 as negative control, and the regulatory regions and gene bodies of the MHC class II genes HLA-DMA and HLA-DPB1. The upper part shows a schematic representation of the locus and the positions of the primers. Error bars represent standard deviations from two biological replicates measured in triplicates. The figure was modified from a published report19. Please click here to view a larger version of this figure.
Figure 3: Changes of DNA accessibility at the IL8 promoter in response to TNF treatment. HeLa cells were stimulated with TNF (20 ng/mL) for 1 h and analyzed by FAIRE. The ratio between free versus total DNA was determined for the promoter of ACTB (positive control), a heterochromatin region on Chr. 12 (negative control), and the IL8 promoter. Error bars represent standard errors of the mean (SEM) from three biological replicates measured in duplicates. Please click here to view a larger version of this figure.
Figure 4: Optimization of the sonication conditions. Chromatin extracted from 293T cells was sonified using a focused sonicator with appropriate tubes (Table of Materials). The chromatin was sonicated for the indicated time with repetitions of 30 s with the following settings: peak power 150, duty factor 15, cycles per burst 500, followed by 30 s: peak power 2.5, duty factor 15, cycles per burst 500. The resulting chromatin was de-crosslinked, purified by phenol:chloroform extraction and loaded in a 2% agarose gel. An ethidium-bromide stained gel is shown, and the positions of the molecular weight markers are indicated in bp. In this experiment, the optimal chromatin size (200 - 300 bp) was obtained after 20 min of sonication. Please click here to view a larger version of this figure.
| Primer | Sequence |
| ACTB-Promoter-F | AAAGGCAACTTTCGGAACGG |
| ACTB-Promoter-R | TTCCTCAATCTCGCTCTCGC |
| Chr. 12 negative control-F | ATGGTTGCCACTGGGGATCT |
| Chr. 12 negative control- R | TGCCAAAGCCTAGGGGAAGA |
| HLA-DMA-XY-Box-F | CATCAGTCACTGGGGAGACG |
| HLA-DMA-XY-Box-R | GCTTCCCAGCCCAGTTACAT |
| HLA-DMA-5’-F | GGAGAGAACAATCTCCGCTTCA |
| HLA-DMA-5’-R | AGCTGCTATGTGTGGTTGGT |
| HLA-DMA-3’-F | TGGGGACCTAGTTAGGGAGC |
| HLA-DMA-3’-R | AGATCCATGGGAGGAGGCTT |
| HLA-DPB1-XY-Box-F | GTCCAATCCCAGGGTCACAG |
| HLA-DPB1-XY-Box-R | TGAAAAGAGCTGCAGTCAGGA |
| HLA-DPB1-5’-F | GCGTGTTCATGTCTGCATCC |
| HLA-DPB1-5’-R | TGATCCTCAGAGCCTGGACA |
| HLA-DPB1-3’-F | CCAGCCTAGGGTGAATGTTT |
| HLA-DPB1-3’-R | GCCTGGGTAGAAATCCGTCA |
| IL8 Promoter-F | GTGATGACTCAGGTTTGCCCT |
| IL8 Promoter-R | CTTATGGAGTGCTCCGGTGG |
Table 1: Primers for qPCR.
 Table 2: Example calculations of FAIRE Relative Recovery Ratios.
Please click here to download this file.
Table 2: Example calculations of FAIRE Relative Recovery Ratios.
Please click here to download this file.
Discussion
FAIRE is a robust and inexpensive method allowing the identification of nucleosome-free regions. It is based on the principle that DNA wrapped around nucleosomes can easily be crosslinked to the histones. A phenol:chloroform extraction is used to separate the non-crosslinked and protein-free DNA fragments from the protein-bound DNA, which is found in the lower phenol phase and to some extent in the interphase (Figure 1). Therefore, the nucleosome-free regions are overrepresented in the fraction of free DNA in the aqueous phase. A number of publications describe detailed protocols of the FAIRE method23,24,25,26, which can be consulted to complement the procedure described here.
Similar to other methods used to determine the chromatin structure, the FAIRE method also critically depends on optimal shearing of the DNA. If the fragments are too long, any nucleosome-free region will be masked by the adjacent chromatin-bound DNA. If the fragments are too small, the detection by qPCR or deep sequencing becomes very difficult. For this reason, the sonication of the DNA is a crucial parameter that must be controlled and optimized in order to obtain fragments around 200 - 300 bp length, which represent 1 - 2 nucleosomes (Figure 4).

Another parameter that can affect the final outcome is the crosslinking of the sample. Although the fixation procedure described here is valid for most cell lines, the researcher must carefully evaluate this step for the particular sample under study, especially when using tissues, where longer fixation times might be necessary to allow the formaldehyde to reach the cells in all the tissue sample.
Unlike other methods, such as DNase I or micrococcal nuclease hypersensitivity, ChIP, or ATAC, FAIRE does not rely on an enzymatic activity or specific antibodies, and therefore it does not need titration or optimization of the reaction conditions. This makes FAIRE an ideal method to measure chromatin accessibility for labs with little experience in the chromatin field. In addition, pilot experiments are only required in order to optimize the DNA shearing step and the crosslinking time, when necessary.
The method was initially developed in yeast6, but it has also been extended to mammalian cells. The DNA purified with the FAIRE method can be used for deep sequencing, allowing the genome-wide characterization of the chromatin status. This has already proven useful in a number of studies8,9,10,11,12,13,19,27,28.
Results from FAIRE-seq experiments show a large overlap with results obtained by other methods such as DNase I-seq14, although some differences arise. This is due to methodological differences as for example relaxed or remodeled nucleosomes could still hinder the cleavage by DNase I, while these DNA regions would be less prone to produce DNA/protein crosslinks and thus would be determined as nucleosome-free regions using FAIRE9. Also, the presence of non-histone proteins such as RNA polymerases or reader proteins for epigenetic marks could result in DNA/protein crosslinks and thus would be interpreted as protein-packed regions in FAIRE experiments. These limitations are inherent to every method used for the determination of the chromatin state, thus raising the need to use a combination of different methods to obtain a comprehensive insight.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank Tilman Borggrefe (University of Giessen) for kindly sharing the sonication device. The work from M.L.S. is supported by grants from the Deutsche Forschungsgemeinschaft (SCHM 1417/8-3), SFB/TRR81, SFB1021, SFB1213, the Excellence Cluster Cardio-Pulmonary System (ECCPS, EXC 147/2), the Deutsche Krebshilfe (111447) and the IMPRS-HLR program of the Max-Planck Society.
References
- Struhl K, Segal E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013;20(3):267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol. Cell. 2013;49(5):825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. The 5' ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature. 1980;286(5776):854–860. doi: 10.1038/286854a0. [DOI] [PubMed] [Google Scholar]
- Yuan GC, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309(5734):626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10(12):1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PL, Cleary ML, Brown PO, Lieb JD. Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc. Natl. Acad. Sci. U. S. A. 2003;100(11):6364–6369. doi: 10.1073/pnas.1131966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PL, Price DH. Formaldehyde-assisted isolation of regulatory elements. Wiley Interdiscip Rev Syst Biol Med. 2009. [DOI] [PMC free article] [PubMed]
- Krinner S, et al. CpG domains downstream of TSSs promote high levels of gene expression. Nucleic Acids Res. 2014. pp. 1–14. [DOI] [PMC free article] [PubMed]
- Gomez NC, et al. Widespread Chromatin Accessibility at Repetitive Elements Links Stem Cells with Human Cancer. Cell Rep. 2016;17(6):1607–1620. doi: 10.1016/j.celrep.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton KJ, et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010;42(3):255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17(6):877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011;43(1):27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143(2):212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie K, et al. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet. 2015;11(2):e1004994. doi: 10.1371/journal.pgen.1004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat. Rev. Genet. 2014;15(11):709–721. doi: 10.1038/nrg3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, et al. Chromatin variation associated with liver metabolism is mediated by transposable elements. Epigenetics Chromatin. 2016;9(1):28. doi: 10.1186/s13072-016-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner DZ. Real-Time PCR: Advanced Technologies and Applications. Crop Sci. 2014;54(1):455. [Google Scholar]
- Nolan T, Huggett J, Sanchez E. Good Practice Guide for the Application of Quantitative PCR (qPCR) Natl. Meas. Syst. 2013. p. 50.
- Rodríguez-Gil A, et al. The CCR4-NOT complex contributes to repression of Major Histocompatibility Complex class II transcription. Sci. Rep. 2017;7(1):3547. doi: 10.1038/s41598-017-03708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265(5168):106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- Choi NM, Majumder P, Boss JM. Regulation of major histocompatibility complex class II genes. Curr. Opin. Immunol. 2011;23(1):81–87. doi: 10.1016/j.coi.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP-Y, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109 Suppl:S21–S33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Nammo T, Rodríguez-Seguí SA, Ferrer J. Mapping open chromatin with formaldehyde-assisted isolation of regulatory elements. Methods Mol. Biol. 2011;791:287–296. doi: 10.1007/978-1-61779-316-5_21. [DOI] [PubMed] [Google Scholar]
- Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012;7(2):256–267. doi: 10.1038/nprot.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JM, Giresi PG, Davis IJ, Lieb JD. A detailed protocol for formaldehyde-assisted isolation of regulatory elements (FAIRE) Curr. Protoc. Mol. Biol. 2013. Chapter 21, Unit21.26. [DOI] [PubMed]
- Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48(3):233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Angarica VE, Andrade-Navarro MA, Buckley NJ, Del Sol A. Prediction of Chromatin Accessibility in Gene-Regulatory Regions from Transcriptomics Data. Sci. Rep. 2017;7(1):4660. doi: 10.1038/s41598-017-04929-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, et al. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011;21(3):447–455. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]