

Abstract
We have developed novel methods for the isolation and characterization of tumor-derived circulating ribonucleic acid (cRNA) for blood-based liquid biopsy. Robust detection of cRNA recovered from blood represents a solution to a critical unmet need in clinical diagnostics. The test begins with the collection of whole blood into blood collection tubes containing preservatives that stabilize cRNA. Cell-free, exosomal, and platelet-associated RNA is isolated from plasma in this test system. The cRNA is reverse transcribed to complementary DNA (cDNA) and amplified using digital polymerase chain reaction (dPCR). Samples are evaluated for both the target biomarker as well as a control gene. Test validation included limit of detection, accuracy, and robustness studies with analytic samples. The method developed as a result of these studies reproducibly detect multiple fusion variants for ROS1 (C-Ros proto-oncogene 1; 8 variants) and RET (rearranged during transfection proto-oncogene; 8 variants). The sample processing workflow has been optimized so that test results can consistently be generated within 72 hours of sample receipt.
Keywords: Medicine, Issue 134, Liquid Biopsy, Circulating RNA, NSCLC, ROS1, RET, Digital PCR
Introduction
Up to 25% of non-small cell lung cancer (NSCLC) patients may not have sufficient tissue available for testing at the time of diagnosis. Even in cases where tissue is available, it may not be of sufficient quantity or quality to perform recommended molecular tests1,2. In cases where there is enough tissue from a biopsy for molecular profiling, patients may have to wait several weeks or longer for results, or begin treatment without molecular results3,4. However, it is critical that informative molecular diagnosis be available given the advent of multiple targeted treatment options for patients diagnosed with NSCLC. Testing of circulating cell-free DNA (cfDNA) from liquid biopsy is a solution to the challenges of traditional tissue testing4,5,6. Current testing options for actionable mutations in NSCLC using cfDNA and a similar dPCR-based workflow for rapid result generation, include the epidermal growth factor receptor (EGFR) sensitizing mutations ΔE746-A750 and L858R, EGFR resistance mutation T790M, KRAS Proto-Oncogene (KRAS) variants, and B-Raf Proto-Oncogene (BRAF) variant V600E. Although not as broadly adopted by the field, circulating tumor-derived messenger RNA (mRNA) isolated from liquid biopsy can also provide important clinical information7,8,9. We have previously developed and reported on methods of multiplexed detection of the Echinoderm Microtubule Associated Protein Like 4-Anaplastic Lymphoma Receptor Tyrosine Kinase (EML4-ALK) fusion variants from blood plasma10. In this study, we extended these methods to include higher-order multiplexed RNA targets for ROS1 and RET, covering eight fusion variants within each assay. The objective was to develop a rapid, sensitive, specific, and reproducible technique for the detection of these fusion variants from the plasma of patients previously diagnosed with NSCLC.
The test process is initiated in a physician's office using RNA stabilizing blood collection tubes11. These tubes contain a cell preservative as well as RNase inhibitors. Samples are shipped priority overnight to the centralized College of American Pathologists (CAP)-accredited/Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory (Clinical Laboratory) for processing by competent personnel. Once received by the Clinical Laboratory, each step of processing is conducted under approved Standard Operating Procedures (SOP). Whole blood is centrifuged to recover plasma, which is then used to isolate circulating RNA that is either free in the blood or within encapsulating moieties, such as exosomes and platelets7,8,9. To isolate RNA from these compartments, we selected the system for RNA recovery based on comparisons of several extraction methods. The isolated RNA is concentrated and reverse transcribed to cDNA. Several reverse transcriptase enzymes and gene-specific primers were evaluated during the optimization of the cDNA synthesis method to maximize ROS1 and RET target transcript conversion10. This is critical for low abundance circulating transcripts, such as tumor-derived fusion variants. Finally, we optimized dPCR primer and probe concentrations to allow for multiplexed detection of RET or ROS1 fusion variants and the control gene, glucuronidase-β (GUSB). We then combined the best conditions from each of the optimization studies into a final locked protocol before performing the analytic validation studies described in this report. This protocol and these results provide the basis for a rapid and sensitive workflow for the routine detection of rare fusion variants in circulation.
Protocol
The manufacturers' instructions are followed for the reagents listed below, unless otherwise described. The PCR assays are commercially available products designed to detect ROS1 and RET fusions.
1. Working with RNA in Preparation for Reverse Transcription (RT)-dPCR: Laboratory Best Practices
- Create an RNase-free environment when working with RNA.
- Use commercially available sprays designed to inactivate contaminating RNases.
- Use certified RNase-free reagents, tips, and tubes. Use barrier tips for pipettors to prevent introduction of RNases or cross-contamination of samples.
Always wear a laboratory coat to prevent particulates from falling from clothing into your sample. Designate a lab coat specific to use with RNA processing.
Wear gloves to prevent sample contamination from RNases in skin. Change gloves frequently. Note: Assume laboratory surfaces are contaminated with RNase since they are exposed to the environment. Gloves that contact skin, hair, doorknobs, freezer handles, pens/markers, etc. are assumed to no longer be RNase-free.
Decontaminate pipettors, benchtops, centrifuges, and other work surfaces with a RNase inactivation spray prior to use.
If possible, maintain a set of equipment for use with RNA only.
Minimize disruption of air flow in lab areas when working with RNA samples to prevent particulates from falling into samples or contaminating the work area.
Store purified RNA at -80 ˚C.
Avoid multiple freeze-thaws of RNA samples, as this can cause degradation.
2. Generation of Analytic RNA Material for the Positive Controls
- Design synthetic DNA using published mRNA sequences for fusion variants of interest10.
- For a given fusion variant, select an mRNA fusion sequence that includes the fusion site plus sufficient length flanking on each side to cover the PCR amplicon.
- Select nucleotide sequences between 50 - 250 nt to mimic the size of circulating RNA captured using platelet-enriched plasma.
- Add a T7 promoter sequence (5'-CAGAGATGCATAATACGACTCACTATAGGGAGA-3') to the 5' end of the target sequence.
Order synthetic sequences as double stranded deoxyribonucleic acid (DNA) fragments.
Reconstitute synthetic DNA in Tris-EDTA (TE) buffer to a final concentration of 10 ng/µL.
Convert 60 ng synthetic DNA to RNA using in vitro transcription.
- Purify RNA transcripts using phenol/guanidine-based reagent12.
- Include DNase I, RNase-free to remove residual template DNA.
Measure concentration of purified in vitro RNA using a commercially available fluorometer with RNA-specific dyes and standards. Ensure the RNA is within the range acceptable for the chosen standards. Dilution may be required.
- Confirm successful transcription by gel electrophoresis using a 2% agarose gel mixed with RNA gel stain and a high range RNA ladder including 50 - 250 nt size range.
- Load 500 ng of each in vitro RNA onto a gel.
- Run the gel at 5 V/cm.
- Visualize single bands using illumination, and document the results.
- Confirm expected transcript size for each of the fusion variants (based on design in step 2.1.2).
Confirm detection of each in vitro RNA by RT-dPCR using matched variant-specific PCR assay (see steps 5 - 8 of this protocol).
Optional: Prepare an equimolar mixture of in vitro RNA that contains each of the fusion variants and the control gene GUSB.
If Step 2.9 is performed: Confirm detection of each of the fusion variants included in the control mixture by dPCR using variant-specific PCR assays (see steps 5 - 8 of this protocol).
Determine desired input concentration for analytic positive controls by testing concentrations ranging from 0.25 to 2.5 fg10. Choose concentration based on desired copy number output.
Following confirmation, prepare 10 µL single-use aliquots of analytic RNA for use in the positive control (step 4.4) and store at -80 ˚C.
3. Donor Specimens
Collect 10 mL human whole blood specimens in 10 mL blood collection tubes (BCT) containing a cell-free RNA preservative. Note: All human donors shall consent to research use and no donor-specific identifying information shall be collected or used during testing.
Process whole blood samples within the timeframe specified by the BCT manufacturer.
Pooled normal human plasma can be purchased from a commercial source for use within the analytic positive control. Prepare single-use, 1 mL aliquots of the pooled normal human plasma and store at -80 ˚C for use with the positive control (step 4.4).
4. Recovery of Circulating RNA from Plasma
Note: It is important to work quickly during this procedure.
Centrifuge whole blood tubes at 200 x g for 20 min.
Collect up to 4 mL of plasma from centrifuged blood collection tube using a serological pipette. Be careful not to disturb or aspirate the buffy coat layer.
Isolate circulating RNA using a commercially available kit that can capture exosomes, platelets and cell-free RNA from plasma. Isolate RNA from positive control sample alongside each batch.
- Prepare the Positive Control for each batch of clinical samples as follows:
- Thaw 1 mL pooled normal human plasma aliquot (Step 3.3).
- Thaw 10 µL analytic RNA aliquot (Step 2.12).
- Prepare positive control by adding 10 µL analytic RNA into the normal human plasma sample once ethanol has been added to the plasma lysate.
- Elute samples with 100 µL nuclease-free water. Proceed immediately with RNA clean up and concentration.
- Samples may be stored on wet ice, and covered, for up to one hour.
- Concentrate RNA using column based method and elute in 9 µL of RNase-free water.
- Proceed immediately to Step 5, or keep samples on wet ice for up to one hour.
5. Reverse Transcription of RNA to cDNA
- Convert concentrated circulating RNA sample to cDNA using a commercially available reverse transcription reaction kit, including random primers (see Table 1 for components). Note: Gene specific primers are optional and can be designed for test variants. Primers are designed based on the target RNA sequence. Use fusion variant sequences from Step 2.1.
- Include no reverse transcriptase control sample and no RNA control sample (see Table 1).
Isolate cDNA from reverse transcription reaction using a commercially available DNA concentrator spin column. Note: This step facilitates removal of enzymes, primers, and free deoxynucleotide triphosphates (dNTPs).
Use cDNA immediately in PCR reactions or store at -80 ˚C.
6. Digital PCR
Note: This PCR is specific to droplet digital PCR (See Table of Materials).
- PCR mix precautions.
- Wear a disposable lab coat and nitrile gloves.
- Use PCR mix reagents in a dedicated reagent preparation area. Do not handle cDNA in the reagent-only preparation area.
- Cover probes while working to protect them from light. Excessive light can photo bleach the fluorescent dye attached to the probe.
- Transport mixes, covered and protected from light, into a separate pre-amplification area before cDNA is to be added.
- Add cDNA to be tested to PCR mix in a PCR clean hood located in pre-amplification area.
Prepare PCR mixes for a final reaction volume of 20 µL according to Table 2.
Distribute PCR mixes + cDNA to PCR plates. Note: Use of a plate layout as a guide is recommended.
Cover the plate using a removable plate sealer.
Centrifuge plates briefly to collect samples at the bottom of the wells.
Mix on plate shaker on a low setting for 10 s.
Centrifuge plates briefly to collect sample at the bottom of the well.
- Remove plate sealer. Perform droplet generation for PCR-cDNA mix with either a manual or automated droplet generation system.
- For manual droplet generation, transfer 20 µL PCR mix to sample wells on the droplet generation cartridge. Add 70 µL of droplet generation oil. Cover with rubber gasket and transfer cartridge to manual droplet generator to initiate droplet generation. Following droplet generation, transfer droplets to a fresh PCR plate using tips recommended by the manufacturer. Aspirate and dispense the droplets slowly, over 5 to 6 s each, without touching the opening of the tip to the droplet cartridge or plate.
- For automated droplet generation, seal the plate with a foil seal and transfer to the droplet generator. Ensure all tips, cartridges, and plates are in place before starting droplet generation.
Following droplet generation and transfer of droplets to a fresh PCR plate, seal with a foil plate sealer, and thermal cycle plates using the settings in Table 3.
After the thermal cycler run is complete, read the plate using a droplet reader. Create a plate layout for reader software that identifies the location of controls, samples, etc., and load into software to start read.
7. Data Analysis and Review, and Generation of Results
Analyze plate read results using commercially available software.
Navigate to the Analyze menu to view two-dimensional (2D) amplitude plots.
- Evaluate the overall quality of the data by examining the droplet data.
- Evaluate data for total accepted event numbers using the Events menu. If there are fewer than 10,000 events per well, carefully evaluate the data for additional problems.
- Check data for aberrant fluorescence amplitudes. Significant amplitude differences and concentration differences between replicate samples indicate poor handling or mixing of samples.
- Make notes of droplet clusters with spray patterns on a 45-degree axis, which is indicative of poor quality droplets or problematic samples.
- Examine Positive Control, No Reverse Transcriptase (No RT), and No RNA Control (NRC) data first. Select all control samples and examine cluster quality by 2D plot. For proper thresholding, a clear separation between droplet clusters should be apparent.
- For each assay variant, set the threshold based on control wells.
- Set thresholds on 2D plots using the crosshairs tool to separate the double negative droplet population from the control gene population (labeled with 5'-hexachloro- fluorescein-CE phosphoramidite probe), y-axis, and variant gene population, if present (labeled with fluorescein amidite OR 6-carboxyfluorescein probe), x-axis.
- Sum copies from each replicate well for a single sample.
- Express test results as the number of variant copies detected. Note: To determine the analytic cutoff value for calling a positive or negative sample, run a normal healthy donor sample set (at least 10 individual samples) through the finalized process and establish the cutoff above any detectable background signal for the mutation of interest. Additionally, establish the number of control gene copies required to call a positive or negative result. This control gene cut-off functions as an internal quality control (QC) to evaluate the quantity and quality of each RNA sample that is processed.
8. Verification of RT-dPCR Reaction Conditions Using Cell-lines (Optional)
- To verify detection of fusion variants, use commercially available cell-lines expressing the ROS1 or RET fusion mRNA of interest. Proceed as follows:
- Homogenize flash-frozen cells in a guanidinium-based lysis solution directly from the frozen state. Even brief thawing prior to homogenization can cause RNA degradation and loss.
- Isolate RNA using silica-membrane spin columns designed for RNA.
- Measure the concentration of RNA samples using a fluorometer with RNA-specific reagents and standards.
- Dilute isolated RNA into a background of wild-type RNA from plasma or another commercial source.
Carry out steps from Reverse Transcription of RNA to cDNA, Digital PCR, and Data Analysis and Review, and Generation of Results listed in this protocol to confirm detection of the desired variant.
Representative Results
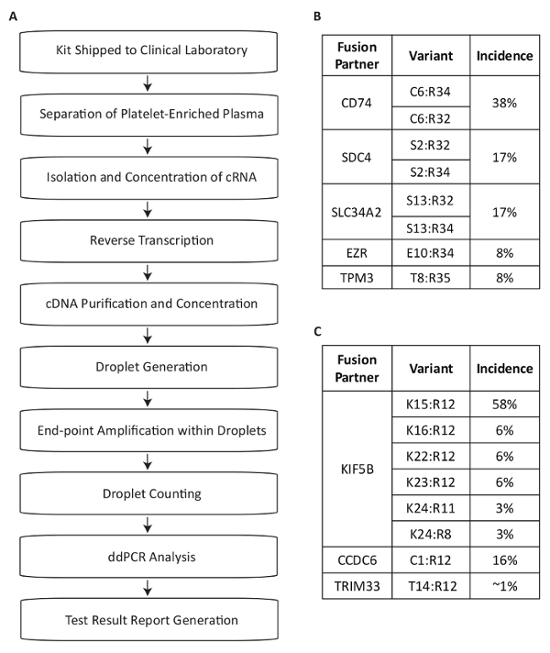
This protocol describes a test system developed for the detection of RNA fusion variants for use in the measurement of driver mutations within the plasma of NSCLC patients (Figure 1A). Fusion mRNA products from the expression of the most common RET and ROS1 rearrangements in the NSCLC population were identified13,14,15,16,17. Multiplexed PCR assays were then designed to detect the eight most common transcript variants for each target in NSCLC within a single reaction. The most common translocations at the ROS1 locus generate associations with the 5' portions of the CD74, SDC4, SLC34A2, EZR or TPM3 genes (Figure 1B). The most common translocations at the RET locus lead to juxtaposition with KIF5B, for which the assay covers six exon junctions. Additional RET partners that are covered include those with CCDC6 and TRIM33 (Figure 1C). In total, the assays cover approximately 88% of ROS1 and 99% of RET alterations known to occur in the NSCLC patient population17.
The specificity of the assay components was first evaluated using eight individual in vitro RNAs that contain the mRNA sequence for the fusion transcripts covered by the ROS1 or RET multiplexed assays. Each RNA species was tested against each individual variant assay that comprises the multiplexed version. There was no cross-reactivity of these assays, thus demonstrating 100% analytic specificity within the designed multiplexed assays (data not shown). To determine the lower limit of detection of the test protocol, total RNA derived from cell lines expressing a fusion variant included in the assay were mixed into a background of normal RNA at 5%, 1%, 0.2%, and 0.04% concentrations. The multiplexed RET and ROS1 variant PCR assays detected as little as 0.2% fusion variant (Figure 2A-B). Additionally, a preparation of 5% off-target cell line derived RNA (expressing an EML4-ALK fusion transcript) was not detected with the multiplexed ROS1 and RET assays, further demonstrating specificity (Figure 2A-B).
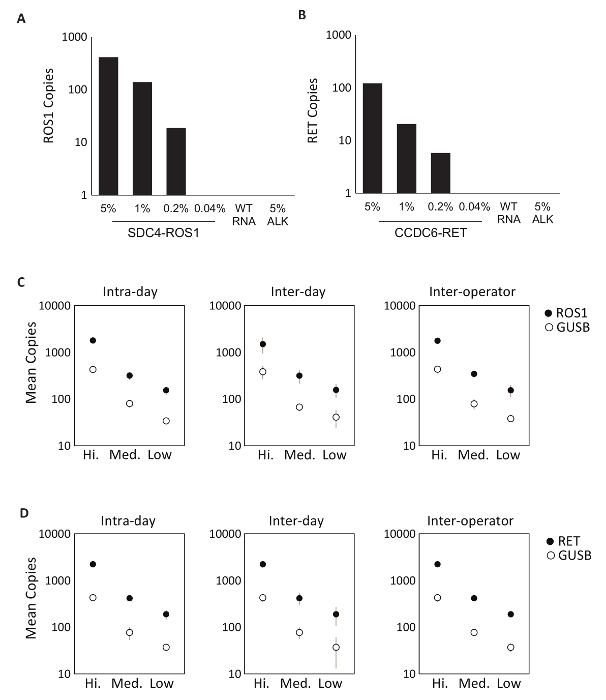
Precision testing of the RT-dPCR process was performed for both ROS1 and RET. Analytic control material comprised of equimolar in vitro RNAs was processed at three concentrations (High, Medium, and Low) through reverse transcription and dPCR on three different occasions within the same day (intra-day), on three consecutive days (inter-day), and with two operators (inter-operator). Results from precision testing demonstrated accurate detection of both the fusion transcript of interest, as well as a control gene, GUSB, which is included as an internal QC metric (Figure 2C-D).
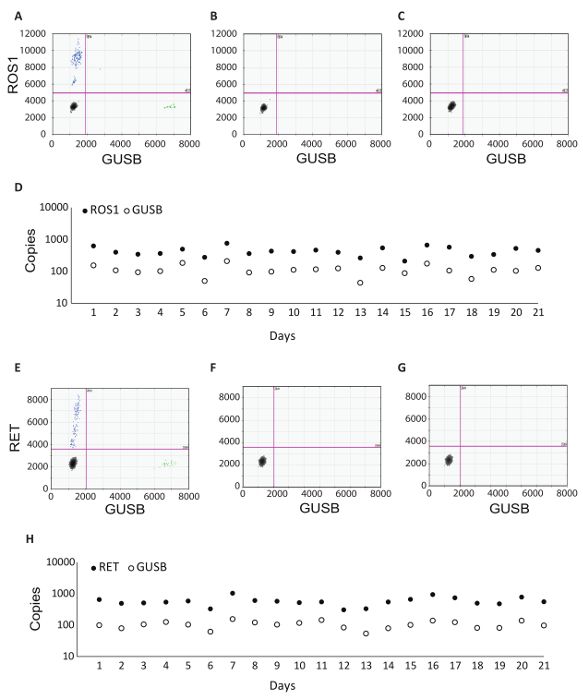
In addition to the GUSB internal control, each batch of clinical samples was run with a set of batch controls. A positive control was developed from a mixture of analytic in vitro RNA that represented each of the fusion variants tested in RT-dPCR, as well as analytic in vitro RNA for GUSB. This RNA was spiked into normal human plasma lysate during RNA extraction and was processed alongside the clinical samples throughout the protocol. The no reverse transcriptase (No RT) control was a negative control to confirm the absence of contaminating material in the RNA extraction workflow and demonstrate the specificity of the primers for RNA. The No RT control was generated using the same material as the positive control, but it does not include enzyme within the cDNA synthesis reaction. The no RNA control (NRC) is a negative control to confirm the absence of contaminating transcripts in the reverse transcription reaction components. This control was introduced into the workflow at the cDNA synthesis step, and water was added in the reaction instead of an RNA template. The No RT and NRC controls must be negative in both channels, if accurate results are to be delivered. Table 1 lists the reverse transcription reaction components for each control. Examples of the 2D plots for each of these controls are shown for the ROS1 (Figure 3 A-C) and RET (Figure 3 E-G) multiplex assays. Fusion variants were detected using a fluorescein amidite (FAM) probe and are represented along the y-axis, while the control gene, GUSB, was detected using a 5'-hexachloro-fluorescein-CE phosphoramidite (HEX) probe and is on the x-axis. These batch controls were assessed over the course of 21 days to determine assay robustness. Fusion positive droplets and GUSB control gene droplets were observed for ROS1 and RET in all 21 runs executed over the course of the study (Figure 3D,H). All negative controls (No RT and NRC) yielded negative results across the entire 21 days (data not shown).
The ability to troubleshoot is a critical component of any test protocol to be run in the clinical laboratory setting. Here, we provide real world examples of sub-optimal results using the RT-dPCR protocol. The first is an example 2D plot demonstrating the importance of the no reverse transcriptase control (Figure 4A). In this example, mutant positive droplets were present even though there was no cDNA conversion due to lack of enzyme. This outcome was likely due to dPCR primers amplifying off-target genomic DNA. In this case, design of an intron-spanning assay will prevent amplification of genomic DNA. Alternatively, an RNase-free DNase enzyme can be used to eliminate the contaminating DNA, but this is not recommended for detection of rare targets, as some RNA degradation may occur during incubation with the enzyme. The next example 2D plot was a NRC with positive droplets in both channels (Figure 4B). This indicated contamination at some point in the RT-dPCR setup. In this case, the recommendation is to discard any potentially contaminated reagents used in testing, thoroughly decontaminate all equipment, and re-test with fresh reaction components. The third example 2D plot presented as a spray of droplets along a 45° line (Figure 4C). This is often caused by shearing and coalescing of droplets. Careful droplet handling prior to thermal cycling is essential, as droplets are prone to damage. We recommend the use of automated droplet generation, when available. If transferring manually generated droplets, be certain to choose the recommended wide-bore tips and employ careful pipetting technique. Droplet transfer requires slow aspiration and dispensing, with each taking place over 5-6 seconds, and it is essential that the pipette tip opening does not touch the droplet cartridge or well. When dispensing, keep the pipette tip at the liquid level and raise it slowly as droplets are dispensed (view video for demonstration). The final 2D plot example demonstrates a lack of separation between the positive and negative droplet populations (Figure 4D). This can have several causes. Strong PCR inhibitors, such as detergents used in lysis buffers and an excess of highly degraded DNA, can cause loss of separation. In this case, consider adding a clean-up step between cDNA synthesis and dPCR (such as described within Step 5 of this protocol). Finally, lack of separation can also be due to sub-optimal amplification conditions, and optimization of the PCR step should also be considered.

Data within Figure 5 represent 984 real world patient sample turn-around times and demonstrates the rapid nature of this test workflow. Results were reported to the treating physician as early as within 48 hours (79% of cases) of sample receipt and in 95% of cases, within 72 hours. In conclusion, the use of stabilized circulating RNA blood collection tubes, optimized RNA extraction procedures from blood, and RT-dPCR run according to an optimized protocol with the appropriate internal and batch controls, can provide a rapid test system for the accurate detection of fusion RNA variants relevant in NSCLC.
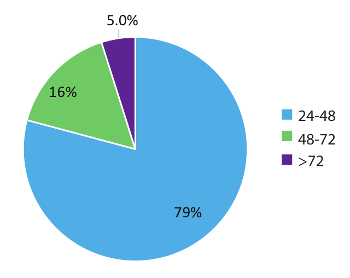
Figure 1: Overview of Blood Sample Processing Steps for Fusion Variant Detection using Assays Specific to the Most Prevalent RET and ROS1 Variants in NSCLC. (A) Sample testing is initiated when whole blood is drawn and a BCT is shipped within the specimen collection kit to the Clinical Laboratory. Circulating RNA is recovered from multiple sources within the platelet-enriched plasma, reverse transcribed with gene specific priming, and purified for use in dPCR. Samples are processed using a commercially available system which consists of droplet generation (emulsion), amplification, and droplet counting. Data is analyzed using commercially available software. The test results are then documented and reported back to the test-requesting physician. The process is designed to work within a timeframe of 72 hours from sample receipt to result release. Eight variants for (B) ROS1 and (C) RET are covered within the multiplexed assays. Adapted from Biodesix website with permission. Please click here to view a larger version of this figure.
Figure 2: Analytic Validation. Cell-lines expressing (A) SDC4-ROS1 fusion and (B) CCDC6-RET fusion were diluted in a background of total human wild-type RNA (WT RNA). With each fusion variant, the limit of detection was established at 0.2% variant frequency using pre-defined criteria for each variant assay. All samples above this threshold also contained at least 21 copies of control gene. 5% EML4-ALK (ALK) standard in a background of wild-type RNA was tested to demonstrate assay specificity, which was confirmed by a negative result. Analytic multiplexed RNA standards were measured at high, medium, and low concentrations for (C) ROS1 and (D) RET. Precision was evaluated over three runs on the same day (Intra-day), three runs on three consecutive days (Inter-day), and with two independent operators (Inter-operator). The means of copy number and standard deviations are shown. Adapted from Biodesix website with permission. Please click here to view a larger version of this figure.
Figure 3: Batch Processing Control Examples and Robustness Data. 2D plot of the ROS1 multiplexed assay dPCR results for (A) positive control, (B) no reverse transcriptase control, and (C) no RNA template control. (D) Controls were run on 21 consecutive days (excluding weekends and holidays). Mean copy number +/- standard deviations for ROS1 positive control were 439 +/- 141. No reverse transcriptase and no template controls were also run on each day, and these were all negative (data not shown). 2D plot of the RET multiplexed assay dPCR results for (E) positive control, (F) no reverse transcriptase control and (G) no RNA template control. (H) Controls were run on 21 consecutive days (excluding weekends and holidays). Mean copies +/- standard deviations for RET positive control were 586 +/- 182. Not shown are no reverse transcriptase and no template controls that were also run on each day and were all negative. Adapted from Biodesix website with permission. Please click here to view a larger version of this figure.
Figure 4: Troubleshooting RT-dPCR. 2D plots representing sub-optimal dPCR results obtained when there is (A) contamination within the no reverse transcriptase control, (B) contamination within the no RNA control, (C) shearing and coalescing of droplets, and (D) poorly optimized PCR conditions or PCR inhibition. Please click here to view a larger version of this figure.
Figure 5: Turnaround time (TAT). TAT (in hours) was compiled for tests requesting an RNA variant (n = 984). Data excludes weekends, holidays, and samples held for >24 h due to incomplete clinical information on the laboratory Test Request Forms. Please click here to view a larger version of this figure.
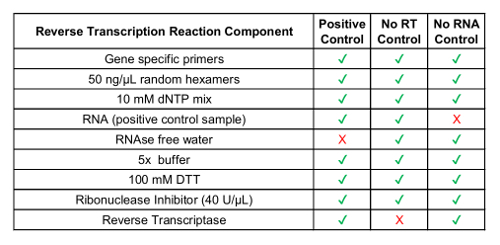 Table 1: Preparation of Reverse Transcription Reagents for Process Controls.
Table 1: Preparation of Reverse Transcription Reagents for Process Controls.
| Component | Volume |
| 2x dPCR supermix for probes (no 2’-deoxyuridine 5’-triphosphate) | 10 µL |
| 20x variant target primers/probes set (450 nmol/L primers, 250 nmol/L FAM probe) | 1 µL |
| 20x control target primers/probe set (450 nmol/L primers, 250 nmol/L HEX probe) | 1 µL |
| nuclease-free water | 1 µL |
| cDNA | 7 µL |
Table 2: Preparation of the master mix for dPCR.
| Cycling Step | Temperature | Time | # Cycles | Ramp Rate |
| Enzyme activation | 95 °C | 10 min | 1 | ~2 oC/s |
| Denaturation | 94 °C | 30 s | 40 | |
| Annealing/extension | 55 °C | 1 min | ||
| Enzyme deactivation | 98 °C | 10 min | 1 | |
| Hold (optional) | 4 °C | infinite | 1 | ~1 oC/s |
Table 3: Thermal Cycling Conditions.
Discussion
RET and ROS1 rearrangements together make up ~3% of the driver mutations within the NSCLC population18. Although rare, the detection of these genetic alterations is vital. NSCLC patients with these alterations may benefit from targeted therapeutics that specifically inhibit the aberrant kinase activity that results from the onco-protein13. Some such therapies are already approved by the FDA for use in ROS1 positive NSCLC, while others have been demonstrated to be efficacious against RET in clinical trials19.
Digital PCR technology provides sensitivity that is ideal for liquid biopsy applications20. There has been significant adoption of this technology for use with circulating cell-free DNA for the measurement of tumor mutations in patients with NSCLC4,6,21,22,23. In addition to cfDNA, we developed a protocol designed for robust measurement of the most prevalent fusion variants in patients with NSCLC from circulating tumor RNA (Figure 1A)10.
Our established protocol allows for analytic limits of detection down to 0.2% (Figure 2). While the RT-dPCR is exceptionally specific and sensitive, the assays are limited to the panel of known fusion variants that are chosen and multiplexed for detection in the PCR assay. Thus, fusions to be included in multiplexed assays must be carefully selected to ensure proper coverage within the population of patients with NSCLC. We have successfully designed assays for RET and ROS1 that simultaneously detect eight fusion variants resultant from rearrangements of the RET or ROS1 loci and cover 99% and 88% of the RET and ROS1 positive population, respectively (Figure 1B-C)17.
The final test workflow as described in this study includes batch controls to ensure consistency of results. These controls include both a positive analytic standard as well as two negative controls, which together ensure there is no contamination or PCR inhibition occurring within the batch (Figure 3). To ensure robustness of the assay, a study was performed using the batch controls over a 21-day period (Figure 3D,H). These data demonstrate the consistency of the RNA process as established within this protocol.
Good laboratory practices and appropriate RNA handling are key components of ensuring robust and accurate results. Laboratory space and equipment dedicated to use with RNA, cleaning of equipment after each use, using RNase-free reagents and consumables, and applying an RNase inactivation spray to the work space all help to reduce contaminating RNases. Conscientious handling of RNA samples by technicians, including a dedicated lab coat, frequent glove changes, working quickly through the RNA extraction procedure, and keeping samples on ice are of utmost importance to preserving sample integrity. Once RNA has been reverse transcribed to cDNA, the sample is in a more stable form that is less prone to degradation. In addition to practices that support RNA integrity, PCR components and samples should be maintained in segregated areas to prevent cross-contamination that could lead to false positive results. The stock PCR reagents and preparation of PCR master mixes should be kept separate from PCR templates and great care should be taken to segregate amplified template (post-PCR) from all pre-amplified materials including reagents, RNA, and cDNA samples. Finally, proper generation and handling of emulsified PCR mixes prior to amplification is central to maintaining droplet integrity and optimal dPCR conditions. Precautions like these are critical during execution of this protocol to obtain consistent and accurate results. All data should be examined by trained personnel before the release of results to be sure that all QC metrics have been met. In the case of suboptimal results (Figure 4), the batch must be reviewed by technical staff and the Laboratory Director and may require re-processing.
RT-dPCR results can be produced as early as 24 hours from sample receipt and 95% of sample results within the test set used in this study (n = 984) were reported to the ordering physician in less than 72 hours from the time of receipt (Figure 5). This turn-around time provides physicians with much needed molecular information in a time frame that allows initiation of the appropriate therapy. These results are typically available earlier than those obtained using a conventional tissue biopsy. Additional biomarkers for NSCLC and other cancers could be developed using similar circulating RNA-based approaches, and would benefit from the same rapid time-to-results. For example, measurement of the Programmed Death Ligand 1 (PD-L1) mRNA transcript using RT-dPCR could inform physicians about immunotherapy options. There is also a growing interest in the utility of liquid biopsy and dPCR in monitoring for therapeutic efficacy. Earlier indications of resurgence of the tumor using genomic testing for specific variants could allow physicians to adjust treatment regimens before patients are symptomatic by standard of care measures such as imaging24. Protocols such as the one reported in this study are ideal for monitoring due to their non-invasiveness, sensitivity, rapid turn-around time, and cost effectiveness. The assay described herein provides results within 72 hours from sample receipt, with minimal false-positive detection rates, which facilitates rapid treatment decisions and circumvents some limitations experienced with tissue-based testing4.
Our protocol and data demonstrate a robust test system for identifying low abundance RNA variants as well as the potential for blood-based mutation testing in clinical practice. For those patients who do not have an actionable driver mutation identified by rapid targeted liquid biopsy approaches like this one, the addition of more extensive genome and proteome testing from both tissue and blood may provide even broader clinical information to support treatment planning.
Disclosures
H.M., L.J., K.A., and G.A.P. are employees of and hold shares in Biodesix, Inc. H.M., L.J., and G.A.P. are co-inventors on a patent application filed by Biodesix, covering a diagnostic test system for the detection of circulating genetic variants in non-small cell lung cancer.
Acknowledgments
We thank our collaborators, Stephen Jones, Nia Charrington, Dr. Dianna Maar, and Dr. Samantha Cooper from the Digital Biology Center (Bio-Rad Inc. CA) for their assay design support; Nezar Rghei and Dr. Moemen Abdalla (Norgen Biotek, Canada) for critical advice when optimizing the RNA extraction protocol; and Shannon Campbell, Scott Thurston, Jeff Fensterer, Shannon Martello and Joellyn Enos for assistance with test requirements and commercial monitoring.
References
- Ignatiadis M, Lee M, Jeffrey SS. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin Cancer Res. 2015;21(21):4786–4800. doi: 10.1158/1078-0432.CCR-14-1190. [DOI] [PubMed] [Google Scholar]
- Alix-Panabieres C, Pantel K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016. [DOI] [PubMed]
- Paxton A. Is Molecular AP testing in sync with guidelines. CAP Today. 2014.
- Sacher AG, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016. [DOI] [PMC free article] [PubMed]
- Sozzi G, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 2003;21(21):3902–3908. doi: 10.1200/JCO.2003.02.006. [DOI] [PubMed] [Google Scholar]
- Oxnard GR, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20(6):1698–1705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best MG, et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 2015;28(5):666–676. doi: 10.1016/j.ccell.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, et al. Different exosome cargo from plasma/bronchoalveolar lavage in non-small-cell lung cancer. Genes Chromosomes Cancer. 2014;53(9):713–724. doi: 10.1002/gcc.22181. [DOI] [PubMed] [Google Scholar]
- Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–1215. doi: 10.1172/JCI81135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellert H, et al. Development and Clinical Utility of a Blood-Based Test Service for the Rapid Identification of Actionable Mutations in Non-Small Cell Lung Carcinoma. J Mol Diagn. 2017;19(3):404–416. doi: 10.1016/j.jmoldx.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Qin J, Williams TL, Fernando MR. A novel blood collection device stabilizes cell-free RNA in blood during sample shipping and storage. BMC Res Notes. 2013;6:380. doi: 10.1186/1756-0500-6-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15(3):532–537. [PubMed] [Google Scholar]
- Kohno T, et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 2015;4(2):156–164. doi: 10.3978/j.issn.2218-6751.2014.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, et al. ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- Rimkunas VM, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 2012;18(16):4449–4457. doi: 10.1158/1078-0432.CCR-11-3351. [DOI] [PubMed] [Google Scholar]
- Tsuta K, et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer. 2014;110(6):1571–1578. doi: 10.1038/bjc.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(1):777–783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgia R. Diagnostic challenges in non-small-cell lung cancer: an integrated medicine approach. Future Oncol. 2015;11(3):489–500. doi: 10.2217/fon.14.275. [DOI] [PubMed] [Google Scholar]
- Cagle PT, Raparia K, Portier BP. Emerging Biomarkers in Personalized Therapy of Lung Cancer. Adv Exp Med Biol. 2016;890:25–36. doi: 10.1007/978-3-319-24932-2_2. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxnard GR, et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34(28):3375–3382. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reckamp KL, et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J Thorac Oncol. 2016;11(10):1690–1700. doi: 10.1016/j.jtho.2016.05.035. [DOI] [PubMed] [Google Scholar]
- Yanagita M, et al. A prospective evaluation of circulating tumor cells and cell-free DNA in EGFR mutant non-small cell lung cancer patients treated with erlotinib on a phase II trial. Clin Cancer Res. 2016. [DOI] [PubMed]
- Abbosh C, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–451. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques. 1993;15(3):532–534. [PubMed] [Google Scholar]