

Abstract
We present a method for preparing thioester molecules as the masked form of the thiol linkers and their utilization for accessing a semiconducting and porous metal-dithiolene network in the highly ordered single crystalline state. Unlike the highly reactive free-standing thiols, which tend to decompose and complicate the crystallization of metal-thiolate open frameworks, the thioester reacts in situ to provide the thiol species, serving to mitigate the reaction between the mercaptan units and the metal centers, and to improve crystallization consequently. Specifically, the thioester was synthesized in a one-pot procedure: an aromatic bromide (hexabromotriphenylene) reacted with excess sodium thiomethoxide under vigorous conditions to first form the thioether intermediate product. The thioether was then demethylated by the excess thiomethoxide to provide the thiolate anion that was acylated to form the thioester product. The thioester was conveniently purified by standard column chromatography, and then used directly in the framework synthesis, wherein NaOH and ethylenediamine serve to revert in situ the thioester to the thiol linker for assembling the single-crystalline Pb(II)-dithiolene network. Compared with other methods for thiol synthesis (e.g., by cleaving alkyl thioether using sodium metal and liquid ammonia), the thioester synthesis here uses simple conditions and economical reagents. Moreover, the thioester product is stable and can be conveniently handled and stored. More importantly, in contrast to the generic difficulty in accessing crystalline metal-thiolate open frameworks, we demonstrate that using the thioester for in situ formation of the thiol linker greatly improves the crystallinity of the solid-state product. We intend to encourage broader research efforts on the technologically important metal-sulfur frameworks by disclosing the synthetic protocol for the thioester as well as the crystalline framework solid.
Keywords: Chemistry, Issue 134, Thiol Synthesis, Porous Materials, Semiconductors, Metal-Organic Frameworks, Metal Dithiolene Compounds, Gyroid
Introduction
There is currently great interest in employing strong, polarizable metal-sulfur (e.g., metal-thiolate) links for constructing open framework materials with enhanced electrocatalytic and conductive properties1,2,3,4,5,6,7,8,9,10. In addition to promoting electronic interaction and transport in the extended state, the soft and covalent metal-sulfur links also impart better stability for applications in aqueous environments. Among the sulfur-equipped building blocks, symmetrical, multidentate polycyclic aromatic building blocks like 2,3,6,7,10,11-triphenylene hexathiol (HTT)9,11,12,13,14 not only supply highly polarizable π-electrons, but also offer distinct advantages with respect to framework design and synthesis. First, the rigid and symmetrical triphenylene core, in conjunction with the chelating dithiolene groups of HTT, serves to lock in metal ions in regular bonding motifs, simplifying the structural prediction of the prospective network7,15. Together with the rigid and open geometry of the sulfur linker, framework structures with substantial porosity features can often be achieved in the solid state.
One major challenge in assembling thiol-equipped metal orangic framework (MOF) materials is rooted in the synthesis of the organic linker molecules. In a classical protocol, the thiol group had to be derived from the phenol group using the Newman-Kwart rearrangement of the O-aryl thiocarbamate precursor16,17,18. This approach, however, involves elaborate preparative steps for the phenol precursor molecule as well as potential complications of a high-temperature solid phase transformation. Another way of making thiols utilizes reductive dealkylation of thioethers under the harsh conditions of, for example, sodium metal in liquid ammonia19,20,21,22, and is not compatible with the carboxyl and many other donor functions for network construction.
By comparison, the protocol presented here has multiple advantages: safety, convenience, cost-effectiveness, and compatibility with other functional groups (e.g., carbonitrile and pyridinyl). By vigorously heating the generally inexpensive aromatic halide (e.g., hexabromotriphenylene) and thiomethoxide anion, the thiolate anion was generated (via the methyl thioether intermediate product) and then acylated to give the stable and easy-to-handle thioester product-all in one pot.
We will also describe a procedure for utilizing the thioester molecules as the masked form of the thiol linkers for accessing a single-crystalline semiconducting and porous metal-dithiolene network. Unlike the highly reactive free-standing thiols, which tend to decompose and complicate the crystallization of metal-thiolate open frameworks, the thioester can be readily cleaved (e.g., by NaOH or ethylenediamine) in situ to provide the thiol species, serving to mitigate the reaction between the mercaptan units and the metal centers, and to consequently improve the crystallization.
This protocol of preparing thiol/thioester has not been widely used by other groups for the emerging field of metal-sulfur frameworks, even though nucleophilic dealkylations of alkyl aryl thioethers by thiolate anions have already been well documented by organic chemists23,24,25,26. By showcasing this efficient synthetic method for thioesters and their use for facilitating the crystallization of metal-sulfur networks, we wish to promote further efforts to bridge the intellectual and practical divide between synthetic organic chemistry and solid state chemistry, so as to help the speedy and healthy development of porous frameworks.
Protocol
Caution: Please consult all relevant material safety data sheets before use. Methyl disulfide and sodium thiomethoxide are strongly malodorous and should be handled in a fume hood. Sodium metal is highly reactive and requires special safety precautions against potential fire and explosion hazards. In addition to the use of a fume hood, personal protective equipment (safety glasses, gloves, lab coat, full length pants, and closed-toe shoes) should be properly employed. Portions of the following procedures involve standard, air-free handling techniques.
1. Preparation of Sodium Thiomethoxide (CH3SNa)
Connect a 200-mL Schlenk flask to a vacuum gas manifold. Evacuate the flask and then backfill it with N2 three times, such that the flask is filled with a slightly positive N2 pressure. NOTE: A similar method of preparing CH3SNa was mentioned briefly in the literature, but no detailed procedures were provided27,28.
Take out a block of metallic sodium from the kerosene oil reservoir. Use a paper towel to wipe off the residual oil on the surface, and use a knife to scrape off the oxide layer on the surface. Quickly cut 6.7 g of the metallic sodium (0.29 mol) into small pieces (e.g., about soybean size) and immediately transfer the small pieces to the 200-mL Schlenk flask under a counter flow of N2. Seal the flask with a septum immediately. NOTE: To reduce the exposure to air, one can cut out the sodium with some variation from the amount of 6.7 g, and adjust the amounts of THF and dimethyl disulfide according to the actual amount of sodium used.
Transfer 80 mL of an anhydrous, airless THF into the flask via cannula under N2 protection from the Schlenk line. Withdraw 14.0 mL of dimethyl disulfide (0.158 moles; pre-purged with N2) into a syringe and inject it into the flask dropwise under N2 protection. Caution: Dimethyl disulfide is malodorous and should be handled in a fume hood.
Replace the septum with a ground glass stopper. Stir the reaction mixture at room temperature for 24 h and then at 60 °C for 3 h. NOTE: The reaction mixture became viscous while stirring at room temperature; when heated up to 60 °C, the mixture can be stirred more easily and thus speed up the reaction towards completion.
With the temperature maintained at about 60 °C, use a stream of N2 (0.2 L/min) to blow off the majority of the THF solvent and the excess dimethyl disulfide, until a dry solid appears. Use a cold trap (e.g., acetone/dry ice) to collect the THF and dimethyl disulfide in the outflow.
Evacuate the remaining solid mixture with an oil pump for about 2 h to remove the residual THF and dimethyl disulfide and then backfill it with N2 to obtain a light-yellow solid (19.8 g, 97%). Store the solid product CH3SNa in a nitrogen atmosphere in the dark. NOTE: To protect the vacuum oil pump, a cold trap should be used during the pumping.
2. Preparation of 2,3,6,7,10,11-hexakis(pentanoylthio)triphenylene (HVaTT) as a protected thiol linker
Load 0.664 g of CH3SNa (9.0 mmol) into a 50-mL Schlenk flask under a counter flow of N2 (i.e., connected as above to a vacuum gas manifold under N2 protection). NOTE: CH3SNa is sensitive to air and easily absorbs water. To minimize exposure to air, one can quickly weigh out a specific amount and add it to the flask, and then adjust the quantity of the other reactant, 2,3,6,7,10,11-hexabromotriphenylene (HBT), accordingly. Caution: Solid CH3SNa has a strong smell and should be handled in a fume hood.
Add 0.216 g of HBT (0.30 mmol) to the flask under a counter flow of N2, and seal the flask with a septum.
Transfer 10 mL of anhydrous and airless 1,3-dimethyl-2-imidazolidinone (DMEU) into the flask via cannula under N2 protection from the Schlenk line.
Replace the septum with a regular glass stopper, and stir the reaction mixture and use a salt bath to heat it to 240 °C for 48 h under N2.
TLC monitoring of the progress of the reaction: Under N2, use a glass dropper to withdraw a small aliquot of the reaction mixture (about 0.1 mL) and immediately inject it into a neat liquid sample of valeroyl chloride (about 0.1 mL) in a plastic microcentrifuge vial, and then shake for 1 min; the mixture should immediately turn turbid with a grey-white color. Add 0.4 mL of deionized water and 0.1 mL of ethyl acetate, close the cap, and shake it for a few seconds. NOTE: If the reaction has completed already, there will not be anything that appears between the layers of water and ethyl acetate. If not, a white insoluble substance will appear between the two layers.
Pipet out the upper portion and use it for spotting the TLC plates. Use 1:4 ethyl acetate/petroleum ether to develop the TLC plate. For a complete reaction, the target molecule shows up as a regular spot around Rf = 0.4 under a UV lamp.
Turn off the heating and take the reaction flask out of the salt bath to cool it to room temperature, and then use an ice bath to cool the flask down to 0 °C.
Inject 1.5 mL of valeroyl chloride (12.6 mmol) into the flask dropwise (over a period of about 2 min) with a syringe under N2. Keep stirring at 0 °C for 2 h. Note: adding valeroyl chloride slowly and at low temperature helps to reduce the generation of by-products.
Pour the mixture into 50 mL of ice water and extract using ethyl acetate (3 30 mL). Then wash the combined organic layer with water (6 60 mL), dry over anhydrous MgSO4, and remove the volatiles by a rotary evaporator.
Isolate the oily crude product by column chromatography using 1:10 ethyl acetate/petroleum ether as the eluent, which affords a light-yellow oily product after rotary evaporation of the solvents. Use 1:4 ethyl acetate/petroleum ether to develop the TLC plate, Rf = 0.4. Further purify the oily product using the trituration step below.
Add 5 mL of methanolto the oily product, and sonicate for 2 min. Collect the resultant off-white solid using suction filtration (yield: 59%).
3. Preparation of Single Crystals of the HTT-Pb Framework Material
Mix 11.4 mg of PbOAc·3H2O (0.030 mmol) in 1.0 mL of ethylene diamine in a vial to make a clear solution.
Load 9.1 mg of HVaTT (0.010 mmol), 2.0 mL of pre-degassed methanol solution of NaOH (70 mmol/L), and 1.0 mL of ethylene diamine into an empty 10-mL vial and sonicate for 5 min.
Add the PbOAc solution to the sonicated HVaTT mixture. Then bubble the reaction mixture with N2 for 1 min, and put on the screw cap to seal the vial.
Heat the vial at 90 °C in an oven for 48 h, followed by natural cooling to room temperature, during which yellow-orange, octahedral single crystals suitable for single-crystal X-ray diffraction studies were formed (yield: 45%).
Suction filtration off the crystals and quickly wash with MeOH. Transfer the crystals with a glass dropper into a vial containing airless MeOH (5 mL) for storage at room temperature.
4. Interaction of Paraquat Diiodide with HTT-Pb Crystals
Use a glass Pasteur pipet to withdraw a few grains of the HTT-Pb single crystal from the MeOH stock and drop it onto a Petri dish (diameter 35 mm and depth 10 mm). Soak up the methanol liquid by tissue or filter paper (or let the MeOH dry up naturally), and then drop onto the crystals a few drops of an aqueous solution of paraquat diiodide (0.1%, w/w).
Observe the color change of the crystals by eye or under a microscope.
Representative Results
The IR spectrum of the HVaTT molecule (collected by the KBr pellet method) features its strongest absorption at 1,700 cm-1, in accordance with the carbonyl stretching of the thioester functional group. The 1H-NMR spectrum of HVaTT (400 MHz, CDCl3) reveals a singlet at δ 8.47 from the aromatic hydrogens, together with 4 multiplets from the aliphatic protons: δ 8.47 (s, 6H, CHAr), 2.75 -2.72 (t, J = 7.4, 12H, CH2), 1.81-1.77 (m, 12H, CH2), 1.50-1.45 (dd, J = 7.4, 12H, CH2), 1.00-0.97 (t, J =7.3, 18H, CH3). The 13C-NMR spectrum of HVaTT (100 MHz, CDCl3) features a peak at δ 196.52 from the carbonyl, three peaks (δ 132.71, 132,35, 130.02) from the triphenylene part, and 4 peaks in correspondence with the aliphatic chains: δ 196.52, 132.71, 132.35, 130.02, 77.35, 77.04, 76.72, 43.56, 27.66, 22.27, and 13.83.
The IR spectrum of the HTT-Pb crystalline product (collected by the KBr pellet method) indicates the absence of carbonyl stretching (around 1,700 cm-1) and the aliphatic C-H (2,800-3,000 cm-1) stretching as seen in the HVaTT reactant. The powder X-ray diffraction (PXRD) pattern of the crystalline HTT-Pb sample thus obtained is consistent with the single crystal structure reported29, indicating the crystalline phase purity of the HTT-Pb product. The single crystals of HTT-Pb are also shown to be highly responsive to the paraquat diiodide in water. Namely, upon contact with a paraquat diiodide solution (e.g., 0.1% w/w), the crystals turned black within one or two minutes, pointing to strong charge transfer interactions between the electron-rich HTT-Pb framework and the highly electron-deficient paraquat guest.
Figure 1: Synthesis of the HVaTT molecule. Synthesisstarts from the hexabromotriphenylene HBT and sodium thiomethoxide, which generates the HTT hexaanion that is consequently reacted with valeroyl chloride to form the thioester product, HVaTT. Please click here to view a larger version of this figure.
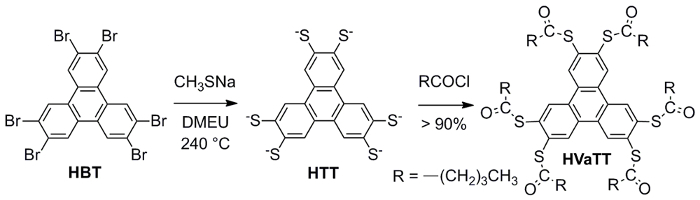
Figure 2: The IR spectra of the thioester precursor molecule of HVaTT (a) and an as-made sample of HTT-Pb crystals. Reproduced from reference 29 with permission from The Royal Society of Chemistry.
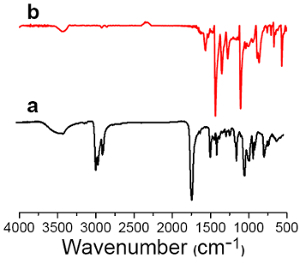
Figure 3:1H-NMR spectrum of HVaTT. 400 MHz, using CDCl3 as the solvent.
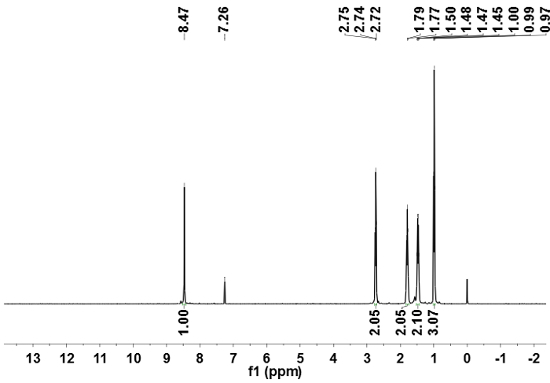
Figure 4: 13C-NMR spectrum of HVaTT. 100 MHz, using CDCl3 as the solvent.
Figure 5: X-ray diffraction patterns (Cu Kα, λ= 1.5418 Å) of HTT-Pb: (a) calculated from the single-crystal structure; (b) freshly prepared bulk sample of crystals. Adapted from reference 29 with permission from The Royal Society of Chemistry.
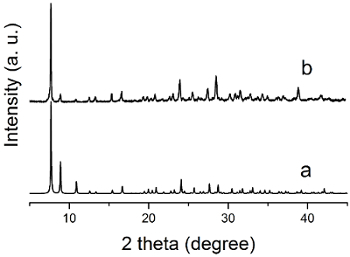
Figure 6. Panel (a): an octahedral single crystal of HTT-Pb before and after treatment with a paraquat diiodide solution (PDI, 0.1% w/w). The edge of the octahedron is about 0.4 mm. Panel (b): an overview of the bulk sample of the PDI-treated HTT-Pb crystals.

Discussion
The reaction between the bromo group and the thiomethoxide anion apparently first produced the methyl thioether, which was then demethylated by the excess thiomethoxide to provide the thiolate anion product. To ensure complete conversion to the desired thiolate anion (especially for a polybromide substrate like HBT), the vigorous conditions of prolonged heating (e.g., 240 °C over 48 h) with a large excess of sodium thiomethoxide (e.g., over three times the moles of the bromo groups) are essential. Nitrogen protection is also needed for preventing the oxidation of the thiolate species of the reactants and products. The acylation step (e.g., injecting valeryl chloride into the reaction mixture cooled by an ice bath) not only provides the thioester as a stable and easily isolatable product, but also offers convenience in monitoring the progress of the reaction. For example, a small aliquot of the reaction mixture can be withdrawn (under N2 protection) and immediately injected into a valeryl chloride solution, which can then be used for TLC monitoring of the progress of the reaction.
Notice that the quality of the sodium thiomethoxide reagent (NaSMe) is also crucial for the making of the HVaTT molecule in the above procedure. Even though the commercial NaSMe reagent has repeatedly proven reliable, highly efficacious NaSMe samples can also be economically prepared in-house on a gram scale (as described in the protocol).
In the solvothermal reaction for preparing single crystals of the HTT-Pb network, which features an anionic framework of the formula [Pb3OH0.5(HTT)]1.5-, it is crucial to use the ester form, i.e., 2,3,6,7,10,11-hexakis(pentanoylthio)triphenylene (HVaTT), as a starting material. For example, if the free thiol of HTT and the Pb(OAc)2 solution was directly mixed together, an amorphous powder was observed to precipitate immediately, with no distinct enhancement of crystallinity effected even with prolonged heating treatment of the reaction mixture. Under the reaction conditions, the HVaTT reactant underwent in situ hydrolysis to generate the HTT thiol species, and thus functioned as a masked version of the highly reactive thiol building block of HTT. As a result, HVaTT effectively serves to mitigate the network formation process, i.e., slowing down the reaction between the sulfur units and the Pb2+ ions, and allowing for more ordered crystalline product to be accessed. The formation of large single crystals (yellow-orange octahedral, about 0.3 mm) of HTT-Pb is also brought about by its structural features. Namely, the individual Pb(II)-dithiolene units in HTT-Pb are integrated through a µ3-oxo atom; thus, the labile Pb3O hinge (as compared with the more intractable Pb-S bonds) as the weakest link in the 3D net offers more reversibility for achieving the highly ordered single crystalline state. Nevertheless, we expect that the crystallinity of other metal-thiolate nets in general should also benefit from the use of protected thiols (like the thioester HVaTT) for in situ supply of the thiol linker.
Besides the benefits of the safe and low-cost reagents (e.g., compared with sodium metal in liquid ammonia), this protocol offers more compatibility with other functional groups crucial for framework construction. For example, our ongoing exploration demonstrates that carbonitrile and pyridinyl groups are generally tolerant of the reaction conditions, and thioester-equipped carbonitrile and N-heterocycles can be efficiently prepared by a similar protocol. Under alkaline conditions, both the carbonitrile and thioester groups can be readily hydrolysed into carboxyl and thiol groups, respectively, thereby generating thiol-equipped carboxylic linkers that have been proven to be important for MOF construction10,30,31,32,33,34. The thiol-equipped N-heterocycle molecules, on the other hand, can be directly used for coordinating with metal ions for network construction, a research area that also features a noted lierature record as well as ongoing intense studies2,4.
The compatibility with ester and carboxylate groups, however, remains problematic at times. For example, ester groups are generally turned into carboxylates under these vigorously nucleophilic conditions (e.g., via dealkylation of the ester group). The carboxylate group, with its negative charge, tends to hinder the thiomethoxide anion in its attack on the methylthio ether; moreover, decarboxylation is often observed under the condition of prolonged heating used in the protocol. Research efforts to ameliorate this situation are therefore truly warranted.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21471037), Guangdong Natural Science Funds for Distinguished Young Scholars (15ZK0307), Science and Technology Planning Project of Guangdong Province (2017A050506051), and the Research Grants Council of HKSAR [GRF 11303414].
References
- Zhao Y, et al. A paramagnetic lamellar polymer with a high semiconductivity. Chem Commun. 2001;0(11):1020–1021. [Google Scholar]
- Su W, Hong M, Weng J, Cao R, Lu S. A semiconducting lamella polymer [{Ag(C5H4NS)}n] with a graphite-like array of silver(I) ions and its analogue with a layered structure. Angew Chem Int Ed. 2000;39(16):2911–2914. doi: 10.1002/1521-3773(20000818)39:16<2911::aid-anie2911>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Tang X-Y, Li H-X, Chen J-X, Ren Z-G, Lang J-P. Synthetic and structural chemistry of groups 11 and 12 metal complexes of the zwitterionic ammonium thiolate ligands. Coord Chem Rev. 2008;252(18-20):2026–2049. [Google Scholar]
- Takaishi S, et al. Electroconductive porous coordination polymer Cu[Cu(pdt)2] composed of donor and acceptor building units. Inorg Chem. 2009;48(19):9048–9050. doi: 10.1021/ic802117q. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Jacobs B, Allendorf MD, Long JR. Conductivity, Doping, and Redox Chemistry of a Microporous Dithiolene-Based Metal-Organic Framework. Chem Mater. 2010;22(14):4120–4122. [Google Scholar]
- Low K-H, Roy VAL, Chui SS-Y, Chan SL-F, Che C-M. Highly conducting two-dimensional copper(I) 4-hydroxythiophenolate network. Chem Commun. 2010;46(39):7328–7330. doi: 10.1039/c0cc02348e. [DOI] [PubMed] [Google Scholar]
- Kambe T, et al. π-Conjugated Nickel Bis(dithiolene) Complex Nanosheet. J Am Chem Soc. 2013;135(7):2462–2465. doi: 10.1021/ja312380b. [DOI] [PubMed] [Google Scholar]
- Mensforth EJ, Hill MR, Batten SR. Coordination polymers of sulphur-donor ligands. Inorg Chim Acta. 2013;403:9–24. [Google Scholar]
- Cui J, Xu Z. An electroactive porous network from covalent metal-dithiolene links. Chem Commun. 2014;50(30):3986–3988. doi: 10.1039/c4cc00408f. [DOI] [PubMed] [Google Scholar]
- Sun L, Miyakai T, Seki S, Dincă M. Mn-2(2,5-disulfhydrylbenzene-1,4-dicarboxylate): A Microporous Metal-Organic Framework with Infinite (-Mn-S-)∞ Chains and High Intrinsic Charge Mobility. J Am Chem Soc. 2013;135(22):8185–8188. doi: 10.1021/ja4037516. [DOI] [PubMed] [Google Scholar]
- Dong R, et al. Large-Area, Free-Standing, Two-Dimensional Supramolecular Polymer Single-Layer Sheets for Highly Efficient Electrocatalytic Hydrogen Evolution. Angew Chem Int Ed. 2015;54(41):12058–12063. doi: 10.1002/anie.201506048. [DOI] [PubMed] [Google Scholar]
- Clough AJ, Yoo JW, Mecklenburg MH, Marinescu SC. Two-Dimensional Metal-Organic Surfaces for Efficient Hydrogen Evolution from Water. J Am Chem Soc. 2015;137(1):118–121. doi: 10.1021/ja5116937. [DOI] [PubMed] [Google Scholar]
- Xu Z, Li K, Fettinger JC, Li J, King MM. A Semiconductive Coordination Network Based on 2,3,6,7,10,11-Hexakis(methylthio)triphenylene and BiCl3. Cryst Growth Des. 2005;5(2):423–425. [Google Scholar]
- Sheberla D, et al. High Electrical Conductivity in Ni3(2,3,6,7,10,11-hexaiminotriphenylene)2, a Semiconducting Metal-Organic Graphene Analogue. J Am Chem Soc. 2014;136(25):8859–8862. doi: 10.1021/ja502765n. [DOI] [PubMed] [Google Scholar]
- Dirk CW, et al. Metal poly(benzodithiolenes) Macromolecules. 1986;19(2):266–269. [Google Scholar]
- Edwards JD, Pianka M. 1346. Isomerisation of 2-butyl-4,6-dinitrophenyl thiocarbamates. J. Chem. Soc. 1965. p. 7338.
- Kwart H, Evans ER. The Vapor Phase Rearrangement of Thioncarbonates and Thioncarbamates. J Org Chem. 1966;31(2):410–413. [Google Scholar]
- Newman MS, Karnes HA. The Conversion of Phenols to Thiophenols via Dialkylthiocarbamates1. J Org Chem. 1966;31(12):3980–3984. [Google Scholar]
- Wolman Y. The Thiol Group (1974) John Wiley & Sons, Ltd; 2010. pp. 669–684. [Google Scholar]
- Harnisch JA, Angelici RJ. Gold and platinum benzenehexathiolate complexes as large templates for the synthesis of 12-coordinate polyphosphine macrocycles. Inorg Chim Acta. 2000;300:273–279. [Google Scholar]
- Yip HK, Schier A, Riede J, Schmidbaur H. Benzenehexathiol as a template rim for a golden wheel: synthesis and structure of [CSAu(PPh3)]6. J Chem Soc Dalton Trans. 1994. pp. 2333–2334.
- Sakamoto R, et al. pi-Conjugated Trinuclear Group-9 Metalladithiolenes with a Triphenylene Backbone. Inorg Chem. 2013;52(13):7411–7416. doi: 10.1021/ic400110z. [DOI] [PubMed] [Google Scholar]
- Testaferri L, Tiecco M, Tingoli M, Chianelli D, Montanucci M. Simple Syntheses of Aryl Alkyl Thioethers and of Aromatic Thiols from Unactivated Aryl Halides and Efficient Methods for Selective Dealkylation of Aryl Alkyl Ethers and Thioethers. Synthesis. 1983. pp. 751–755.
- Testaferri L, Tingoli M, Tiecco M. Reactions of polychlorobenzenes with alkanethiol anions in HMPA. A simple, high-yield synthesis of poly(alkylthio)benzenes. J Org Chem. 1980;45(22):4376–4380. [Google Scholar]
- Tiecco M. Selective dealkylations of aryl alkyl ethers, thioethers, and selenoethers. Synthesis. 1988. pp. 749–759.
- Tiecco M, Tingoli M, Testaferri L, Chianelli D, Maiolo F. Selective dealkylation of bis[alkylthio]benzenes: elimination-substitution competition with methoxide and methanethiolate ions in hexamethylphosphoric triamide. Synthesis. 1982. pp. 478–480.
- Wark TA, Stephan DW. Early metal thiolato species as metalloligands in the formation of early/late heterobimetallic complexes: syntheses and molecular structures of Cp2Ti(SMe)2, Cp2V(SMe)2, (Cp2Ti(µ-SMe)2)2Ni and (Ni(µ-SMe)2)6. Organometallics. 1989;8(12):2836–2843. [Google Scholar]
- Chakraborty P. Sodium methanethiolate. e-EROS Encycl Reagents Org Synth. 2014. pp. 1–5.
- Huang J, et al. A semiconducting gyroidal metal-sulfur framework for chemiresistive sensing. J Mater Chem A. 2017;5(31):16139–16143. [Google Scholar]
- He J, et al. Building thiol and metal-thiolate functions into coordination nets: Clues from a simple molecule. J Solid State Chem. 2009;182(7):1821–1826. [Google Scholar]
- Yee K-K, et al. Effective Mercury Sorption by Thiol-Laced Metal-Organic Frameworks: in Strong Acid and the Vapor Phase. J Am Chem Soc. 2013;135(21):7795–7798. doi: 10.1021/ja400212k. [DOI] [PubMed] [Google Scholar]
- Yee K-K, et al. Room-temperature acetylene hydration by a Hg(II)-laced metal-organic framework. Chem Commun. 2015;51(54):10941–10944. doi: 10.1039/c5cc03943f. [DOI] [PubMed] [Google Scholar]
- Gui B, et al. Tackling poison and leach: catalysis by dangling thiol-palladium functions within a porous metal-organic solid. Chem Commun. 2015;51(32):6917–6920. doi: 10.1039/c5cc00140d. [DOI] [PubMed] [Google Scholar]
- He J, Zeller M, Hunter AD, Xu Z. Functional shakeup of metal-organic frameworks: the rise of the sidekick. CrystEngComm. 2015;17(48):9254–9263. [Google Scholar]