

Abstract
METLIN originated as a database to characterize known metabolites and has since expanded into a technology platform for the identification of known and unknown metabolites and other chemical entities. Through this effort it has become a comprehensive resource containing over 1 million molecules including lipids, amino acids, carbohydrates, toxins, small peptides, and natural products, among other classes. METLIN’s high-resolution tandem mass spectrometry (MS/MS) database, which plays a key role in the identification process, has data generated from both reference standards and their labeled stable isotope analogues, facilitated by METLIN-guided analysis of isotope-labeled microorganisms. The MS/MS data, coupled with the fragment similarity search function, expand the tool’s capabilities into the identification of unknowns. Fragment similarity search is performed independent of the precursor mass, relying solely on the fragment ions to identify similar structures within the database. Stable isotope data also facilitate characterization by coupling the similarity search output with the isotopic m/z shifts. Examples of both are demonstrated here with the characterization of four previously unknown metabolites. METLIN also now features in silico MS/MS data, which has been made possible through the creation of algorithms trained on METLIN’s MS/MS data from both standards and their isotope analogues. With these informatic and experimental data features, METLIN is being designed to address the characterization of known and unknown molecules.
Graphical abstract

The METLIN tandem mass spectrometry (MS/MS) database was created in 2003 and made publicly available in 20051 to help identify metabolites; at that time, no such database existed for identifying metabolites or any other chemical entities. METLIN, a freely accessible cloud-based technology platform and metabolite database, has since grown from a small collection of MS/MS spectra on 100 metabolites in its first iteration1 to more than 10 000 metabolites in 2012,2 with an additional 12 000 metabolites and compounds having been analyzed in the last 5 years. METLIN data are broadly useful across multiple tandem mass spectrometry instrument types, with the data collected in both positive and negative ionization modes at multiple collision energies, providing high-resolution spectra, systematically acquired and manually curated directly from standards and their stable isotope analogues. These data complement the data from other databases, which have been collected for electron impact (EI) or nuclear magnetic resonance (NMR) instrumentation.3–5 Recently, to improve the coverage of metabolites and aid with its annotation, in silico MS/MS spectra have now been generated on METLIN’s additional molecules (that currently have no experimental data). These data are based on advanced machine learning algorithms,6–8 the growing METLIN database, and the unique fragmentation information provided by stable isotopes.
Since the introduction of METLIN, numerous other databases have followed, with over 20 different databases currently available.5 Their impact has been profound, essentially bringing metabolomics from the fringes to what is now a mainstream technology, offering valuable insight into areas as diverse as therapeutic drug discovery, clinical diagnostics, pharmacology, food safety, sports medicine, toxicology, forensics, environmental analyses, and microbiology.9–11 For example, these databases serve to identify metabolites as indicators of a microorganism’s activity,11 disease onset,11–13 and disease progression14,15 or as responsive elements to therapeutics,16,17 and they provide mechanistic insights into biological systems, extending in some cases to the prioritization and identification of endogenous metabolites for the modulation of phenotype.18–20 The increasing ability to obtain and process complex data sets has been pivotal to these achievements, through the identification of metabolites and other chemicals represented by these dysregulated features. However, as addressed in this paper, the primary obstacle facing the field has now shifted from identifying molecules with known MS/MS spectra to identifying the unknowns that are not present in the databases or that are present yet do not have experimental MS/MS data. METLIN is being designed to meet this challenge.
EXPERIMENTAL SECTION
Metabolite Data Acquisition and Analysis
Pichia pastoris extracts corresponding to 2 × 109 unlabeled or 13C-labeled cells were generated by growing cells on natural and 13C-glucose, respectively, as previously reported.21 Extracts were reconstituted with 1 mL of acetonitrile/H2O (1:1, v/v) and aliquots (8 μL) were injected into an Agilent 1200 series high-performance liquid chromatography (HPLC) system (Agilent Technologies, Santa Clara, CA) coupled to a Bruker Impact II quadrupole/time-of-flight mass spectrometer (Q-TOF MS; Bruker, Billerica, MA). The mass spectrometer was set to auto MS/MS mode, selecting the 10 most intense precursor ions in the MS scan to fragment in each cycle and acquiring data over the m/z range 50–1000. Cycle time was set to 3 s. The electrospray source conditions were set as follows: end plate offset = 500 V, dry gas temperature = 220 °C, drying gas = 6 L/min, nebulizer = 1.6 bar, capillary voltage = 3500 V. Samples were analyzed at four different collision energies: 0, 10, 20, and 40 eV. Samples were run in reversed phase and hydrophilic interaction liquid chromatography (HILIC) in both positive and negative ion modes to cover the widest range of the metabolome, as previously described.22
Raw .d data files were converted to .mzXML format by use of ProteoWizard MS Converter version 3.0.7529.23 Peaks were first detected, integrated, and aligned by use of XCMS Online (https://xcmsonline.scripps.edu).11,24 Afterward, isotopically labeled samples were analyzed to identify isotope labeling patterns, by use of the X13CMS software package.25,26 The output was composed of a table where putative molecules were sorted by isotopologues. The grouped putative isotopologues should have a mass shift compared to the unlabeled ion that represents an integer multiple of the mass defect introduced by the isotopic atom (1.0034 Da) within the error of the mass spectrometer. To consider a pair of unlabeled and labeled metabolites, the signal of the 12C-ion in the 13C-glucose-fed P. pastoris extract should not be detectable (or negligible compared to its 13C analogue), and conversely for the 13C-molecule in the 12C-glucose-fed yeast extract. Once this refinement was accomplished, the MS/MS spectra of natural and isotope-labeled putative metabolites were manually compared by use of METLIN functions, as described in the Results and Discussion section.
METLIN Data Curation
METLIN database entries are curated by use of both automated scripts and manual inspection of the data. Briefly, a script reads the MS files determining charge state (positive or negative) and precursor m/z. These are linked with the METLIN entry, and a new entry for MS/MS data is initialized in the database. Once this is confirmed, the script collects the mass and intensity values for each collision energy (i.e., 0, 10, 20, and 40 eV). A signal filter is then employed to identify and remove signal that is due to noise. The largest MS1 peak is chosen that is closest to the precursor mass; the resulting values are normalized and inserted into a database. Normalization is done by equating the maximum MS/MS peak to 100%. Finally, the resulting MS/MS spectrum is manually checked before committing it to the database to be viewed on the METLIN site.
RESULTS AND DISCUSSION
METLIN Search Functions
Simple and Advanced Searching
In addition to more than 1 million metabolites and other small molecules in the database, METLIN has incorporated tools to automate the identification process of known and unknown molecules by use of experimental MS/MS data (Figure 1). For example, once the m/z of a feature of interest is defined, the Simple Search menu allows users to perform an exact mass search and thus obtain putative molecules within a user-defined mass tolerance window. This search menu also offers the possibility to take into consideration different adducts of the molecule that could match the selected m/z (Figure 1A). The Advanced Search tool allows a more general search of metabolites based on different parameters, such as name, m/z range, chemical formula, common names, simplified molecular-input line-entry system (SMILES), Kyoto Encyclopedia of Genes and Genomes (KEGG), and METLIN identification number (MID), among others (Figure 1A).
Figure 1.
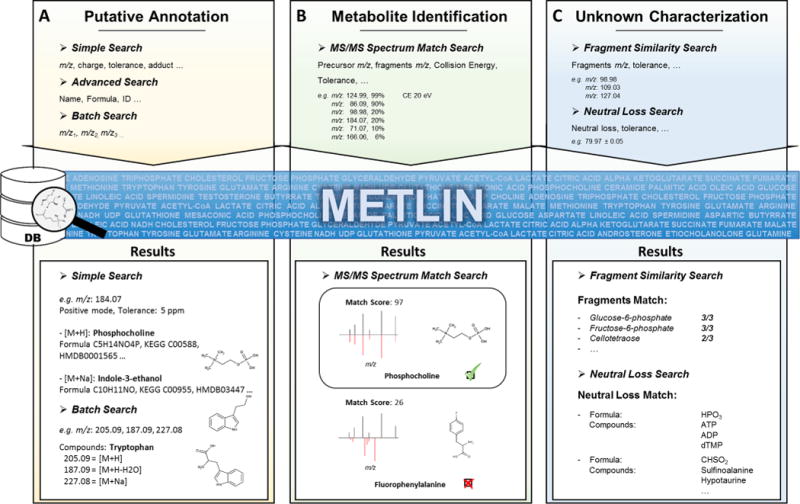
METLIN search functions for metabolite identification. (A) Simple Search and Advanced Search allow the user to search small molecules against a database of 1 million compounds attending to different criteria and retrieve their chemical, spectral and other information of interest. Batch Search facilitates the search of many m/z of interest simultaneously, helping to identify different m/z values as distinct adducts or water losses of the same molecule. (B) With the MS/MS Spectrum Match Search, experimental and library MS/MS spectra can be searched, matched, and scored in an automatic way. (C) Fragment Similarity Search and Neutral Loss Search aid the identification of metabolites or chemical structures by searching m/z values of the fragments or neutral losses, respectively, regardless of the precursor mass.
The output of both search engines consists of a list of molecules with specific identification information. This information includes MID, exact mass, name, formula, CAS number, a link to its KEGG record, its structure, and the availability of experimental or in silico MS/MS spectra. Since experimental MS/MS data provide a higher level of identification confidence compared to in silico MS/MS data, the listing of metabolites has been configured to prioritize molecules with experimental MS/MS spectra first. By clicking on each molecule, users can access detailed information, including links of interest for identification, chemical properties, commercial availability, and biological activity. In the MS/MS spectra section, most fragment structures can be visualized by hovering the cursor over the fragment of interest. This information can be useful during the identification of unknown molecules, as will be explained in greater detail.
Finally, the Batch Search permits searching for multiple m/z values simultaneously, facilitating the identification of different adducts and water losses possibly from the same metabolite. Similarly, ions with a different molecular origin can be easily distinguished and linked to other putative candidates with this search feature (Figure 1A).
Autonomous Identification Tools
The MS/MS Spectrum Match Search automatically matches and scores experimental MS/MS spectra with METLIN MS/MS data to efficiently annotate compounds more rapidly, relying on a modified X-Rank similarity algorithm2,27 (Figure 1B). In this tool, three different collision energies (10, 20, and 40 eV) can be selected to match against the database spectra, thereby allowing users to select the most suitable conditions for their experimental settings and render better scores. Also, this tool has a feature to perform an analysis of each experimental MS spectrum with the METLIN spectrum at 0 eV, to take into account possible in-source fragmentation during the analysis. This is especially useful with molecules that are easily fragmented within the ionization source, producing characteristic in-source fragment ions, capable of aiding in the identification of those molecules by reducing the number of putative metabolites. Nevertheless, this tool expressly requires the experimental MS/MS spectrum of the putative compound to be in METLIN. Alternatively, since most of the metabolites can be accurately defined by a low number of substructures, complementary tools such as Fragment Similarity Search and Neutral Loss Search have been implemented into METLIN. These functions are best suited for the search of compounds or families of compounds with characteristic fragments and thus help to classify compounds within a chemical group of molecules and narrow the number of putative metabolites/identifications (Figure 1C). Examples of the use of these tools in the identification of several compounds can be found in Figures 2, 4, and 5.
Figure 2.
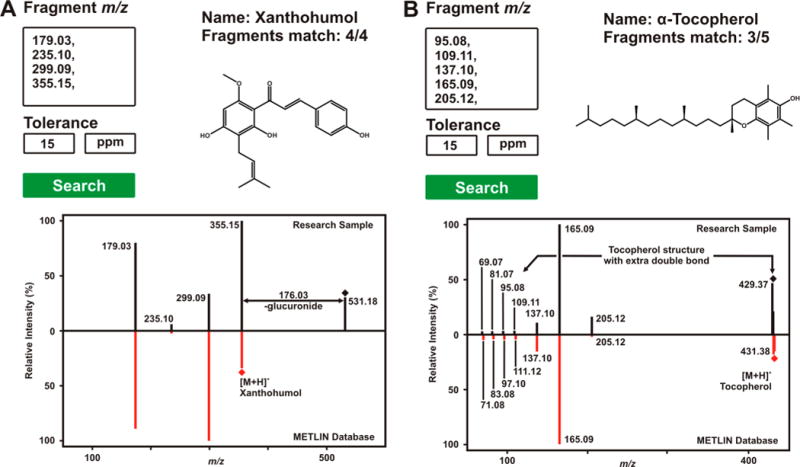
Fragment Similarity Search facilitates the identification of unknown metabolites where no MS/MS spectral data are available. Two examples are shown where an unknown metabolite is characterized by use of Fragment Similarity Search: (A) a glucuronide of xanthohumol and (B) a desaturation variation of α-tocopherol. (A) The fragments of an unknown metabolite were searched against METLIN and all of the four fragments were found to match with xanthohumol. The comparison between experimental and library MS/MS spectra implies high structural similarities. Furthermore, the 176.03 Da difference between the precursor of the experimental spectra and the protonated species of xanthohumol can be attributed to glucuronidation. This mass difference represents the protonated species of xanthohumol + glucuronic acid − H2O (condensation product). (B) Five selected fragments of an unknown metabolite matched three fragments of α-tocopherol; however, the mass difference for nonmatching fragments as well as the precursor is 2.01 Da. This could be attributed to an extra double bond within the structure of α-tocopherol, presumably on the long aliphatic chain.
Figure 4.
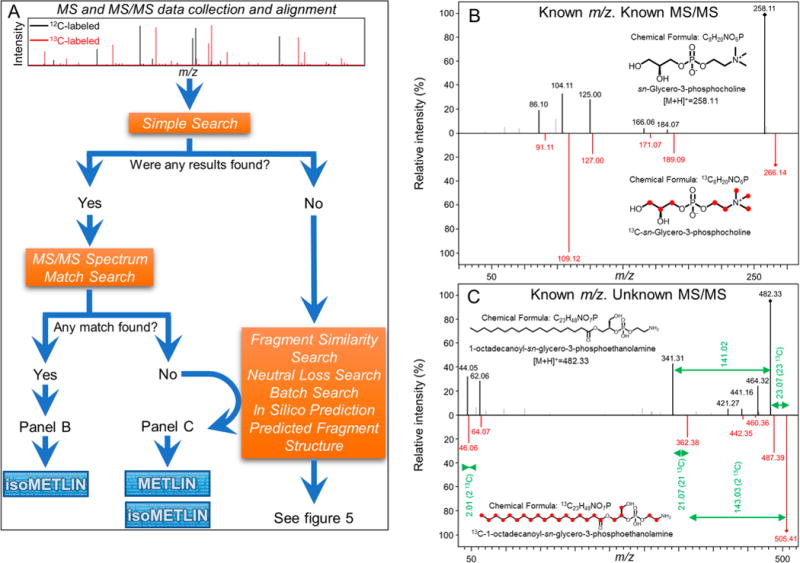
Isotope-labeled microorganisms as a source of MS/MS spectra to populate spectral repositories. (A) An untargeted metabolomics analysis of two extracts of 12C- and 13C-labeled yeast was carried out to collect MS/MS spectra for METLIN and isoMETLIN. (B) If the putative metabolite MS/MS spectrum is recorded in METLIN, the fragmentation spectrum of its 13C-labeled analogue is easily identified for inclusion into isoMETLIN. (C) If the putative metabolite MS/MS spectrum is not displayed in METLIN, it is possible to obtain both 12C- and 13C-labeled spectra for their inclusion into METLIN and isoMETLIN, respectively, through the use of METLIN search functions, together with the in silico prediction and fragment predicted structure of structurally related molecules. Even if the parent m/z of the candidate molecule is not found in METLIN, it is likely that one will obtain structural information leading to its identification by use of METLIN tools. With this workflow, spectral databases are used to self-populate, by using their tools and current spectra to identify new MS/MS spectra.
Figure 5.
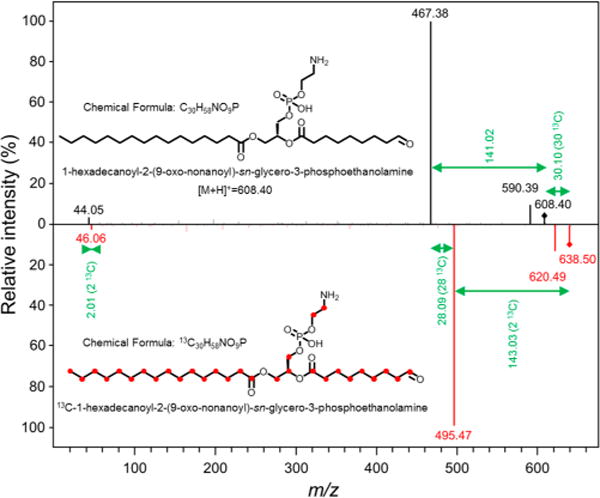
Use of isotope-labeled microorganisms and METLIN to determine the structure of unknown molecules. Starting from the unlabeled and 13C-labeled MS/MS spectra of an unknown metabolite, it is possible to obtain structural information with the use of METLIN tools. The m/z shift of 30.10 Da in the parent ions points out the presence of 30 carbons in this metabolite. The neutral loss of 141.02 Da in the unlabeled molecule, together with the neutral loss of 143.03 Da in the 13C-labeled molecule, indicates the presence of a phosphoethanolamine group (C2H8NO4P). Fragments of 44.05 and 46.06 Da represents the main fragments of the phosphoethanolamine group in unlabeled and labeled molecules, respectively. Given that the glycerophosphoethanolamine group is composed of 5 carbons, the rest of the molecule must have 25 carbons. The most likely biomolecule fitting those requirements and with a parent m/z instrument error within 10 ppm is 1-hexadecanoyl-2-(9-oxononanoyl)-sn-glycero-3-phosphoethanolamine.
Fragment Similarity Search for Unknowns
One of the major challenges in metabolomics is the limited availability of experimental MS/MS spectra in databases. METLIN alone has over 1 million molecules, including metabolites, drugs, xenobiotics, and toxicants, yet only a small percentage have experimental MS/MS data, and this does not include currently undiscovered metabolites and other chemical entities. To overcome this limitation, several algorithms have been developed to assign chemical substructures to unknown molecules on the basis of database queries. These efforts were originally applied to the interpretation of electron impact (EI) ionization mass spectrometry data through the development of algorithms such as STIRS and SISCOM.28,29 These original algorithms were further refined by including neutral losses, peak intensity weighing, and similarity of mass spectra.30,31 Since EI fundamentally differs from the ESI MS/MS fragment ion generation, extrapolating these algorithms to ESI MS/MS data was not immediately possible. The first effort to accomplish this by use of tandem mass spectrometric data was the Fragment Similarity Search algorithm, originally implemented into METLIN and XCMS to facilitate the autonomous identification of small molecules relying on a shared peak count method.32 The algorithm was developed to detect possible structural motifs in unknown metabolites, which may produce characteristic fragment ions and neutral losses to related reference compounds contained in METLIN, independent of their chemical formula and mass.
Algorithms for the structural characterization of unknowns, essentially based on in silico simulated data, have been applied to other biological molecules like peptides and proteins with significant success. However, extrapolation of these algorithms to metabolites and other small molecules still constitutes a major challenge, due to their chemical heterogeneity and the computational challenges in calculating energetically favorable losses. This complexity makes fragment similarity searching algorithms based on experimental MS/MS data a viable alternative for the identification of unknown metabolites, as we will demonstrate.
METLIN’s Fragment Similarity Search in combination with the growing database evolved to facilitate the identification of metabolites and other small molecules that have no library MS/MS data. This is accomplished through the search of common fragments across the METLIN MS/MS library. To illustrate the power of this tool, two examples of unknown metabolite characterization from an extract of mice fecal matter are provided. In Figure 2A, the four main fragments of an unknown compound with a mass-to-charge ratio of 531.18 were investigated by use of the Fragment Similarity Search tool. The search yielded similarities to more than 100 compounds; however, only one, the anticarcinogenic natural product xanthohumol, which has been observed in hops,33 shared all four fragments (Figure 2A). The primary difference between the MS/MS spectral data of the known and unknown molecules was the precursor peak of the unknown metabolite. The m/z shift between the protonated species of xanthohumol with the precursor of the unknown metabolite represents a difference of 176.03 Da. This mass shift can be assigned to glucuronidation, a common metabolic pathway by which the organism makes molecules more water-soluble and thus prone to excretion. This reaction involves the condensation of glucuronic acid (194.04 Da) with xanthohumol (354.15 Da) with the corresponding loss of water (18.01 Da), yielding a molecule with a molecular mass of 530.18 Da (531.18 as its protonated species) (Figure 2A). This putative identification was further confirmed via a bibliographic search.34
The Fragment Similarity Search feature can also be used for the identification of molecules even if only a few fragments of the unknown molecules match MS/MS data in the database. In Figure 2B, five fragments of an unknown molecule were searched with the Fragment Similarity Search tool. In this case, no candidates containing all fragments were found, and only two molecules showed three hits matching the input fragments. Among those molecules was α-tocopherol, the main component of vitamin E.35 When the fragmentation data of α-tocopherol are compared to the experimental data, a m/z shift of 2.01 Da is observed between both precursor ions and other low mass fragments (Figure 2B). This could be attributed to an extra double bond within the α-tocopherol structure, likely in the aliphatic chain, since the METLIN predicted structure for those nonmatching fragments contains that section of the molecule. Moreover, the three matching fragments include the chromanol structure, indicating that the configuration of that structure for the unknown molecule is likely to be the same double ring as α-tocopherol. To the best of our knowledge, only one α-tocopherol desaturation product has been reported, α-tocomonoenol, another component of vitamin E.35
In Figure 2, the utility of the Fragment Similarity Search tool was demonstrated for the identification of molecules whose MS/MS data are not present in the spectral databases and also for metabolites that are not listed in any database or have not been reported previously. Several efforts are currently being carried out to automate the use of this tool within METLIN, allowing the user to upload the MS/MS data and have a reduced number of putative candidates with similarities to the MS/MS spectra via a one-click procedure.
Uniformly 13C-Labeled Metabolites
METLIN and iso-METLIN Data To Facilitate Absolute Quantification
In recent years, uniformly 13C-labeled organisms have been generated by growing different organisms, such as bacteria, yeast, or grains, with 13C-labeled substrates to create 13C-labeled endogenous metabolites.36,37 To take advantage of this trend, metabolite extracts from Escherichia coli or P. pastoris have been used as a source of 13C-labeled molecules as internal standards in metabolomics36,38,39 and lipidomics studies21 (Figure 3), where labeling efficiencies above 99% have been achieved. These extracts show a high dynamic range for use in quantitative experiments, and more importantly, when added as internal standards, more than 100 labeled compounds are spiked into the samples simultaneously, allowing the absolute quantitation of many compounds in one experiment.21,38 Even though the launch of these isotope-labeled internal standards is a step forward to the simultaneous quantitation of multiple compounds in metabolomics, the generation of accurate MS/MS spectra of the 13C-labeled molecules is necessary for generating a quantitative multiple reaction monitoring (MRM) workflow (Figure 3).
Figure 3.
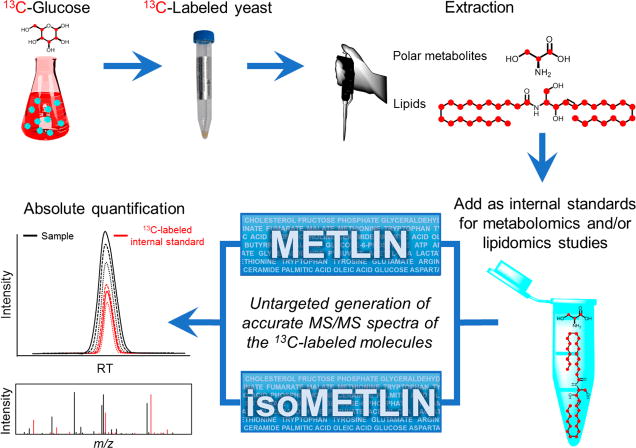
METLIN-guided use of 13C-labeled microorganism extracts as internal standards in mass spectrometry. Yeast are grown in the presence of 13C-glucose, yielding a labeling efficiency of 99% for their metabolites. After the extraction of the compounds of interest to use as internal standards, samples are spiked with those extracts to quantify many metabolites at the same time, using the MS/MS data provided by the spectral databases. The generation of MS/MS spectra to populate databases is a limiting step in this workflow.
In the last three years, the METLIN version for isotope-labeled compounds, isoMETLIN, has been populated with MS/MS spectra of several metabolite isotopologues of analytical standard quality.40 Although isoMETLIN has facilitated untargeted global isotope-tracer experiments,26 the limited number of commercially available stable isotope-labeled molecules makes this approach insufficient for the absolute quantitation of many compounds.5 To address this limitation, we have developed an approach to add metabolites from uniformly 13C-labeled microorganism extracts. To accomplish this, an untargeted analysis of P, pastoris cell extracts was used to generate MS/MS spectra of 13C-labeled metabolites for incorporation into isoMETLIN, which facilitates the absolute quantitation of hundreds of metabolites using the same internal standard mixture. It is worth noting that this approach for MS/MS data generation of isotopically labeled metabolites is guided by METLIN’s database functions and pre-existing data (Figure 4A). This creates a positive feedback loop within the database, which in turn facilitates the generation of additional data.
MS/MS Data from Isotope-Labeled Microorganisms
After RAW MS and MS/MS spectra are acquired, data curation (see Experimental Section) allows for the creation of a list of metabolites that include the unlabeled base metabolite and all possible 13C-labeled isotopologues25 (Figure 4A). With this approach, hundreds of putative isotopologues can be sorted in each analysis. The first step to identify the labeled metabolites is to search their corresponding unlabeled m/z by use of the METLIN Simple Search menu (Figure 4A). Depending upon whether a match is identified in the search, the next step is to compare the MS/MS data of the unlabeled metabolite with all candidates retrieved by the database by use of the autonomous MS/MS Spectrum Match Search tool in METLIN (Figure 4A). If a match is found, the final step compares the MS/MS spectra of both the unlabeled and the candidate 13C-labeled molecule, followed by verification of the analogue fragments in the isotopically labeled MS/MS data (Figure 4A,B). Given that METLIN provides the chemical formula of the metabolites and a predicted structure for most of their fragments, this facilitates the confirmation that the MS/MS spectrum corresponding to the 13C analogue of the previously identified naturally occurring metabolite (Figure 4B). In summary, starting from the extracts of uniformly labeled microorganisms, the use of METLIN throughout the identification process can lead to the generation of MS/MS spectra of an unknown 13C-labeled molecule and its inclusion into isoMETLIN.
Interestingly, this approach is also useful for collecting MS/MS data for unlabeled metabolites that are recorded in METLIN but whose experimental MS/MS spectra have not been added to the database (Figure 4A). For example, the experimental MS/MS data of both the naturally occurring lysoPE(18:0) and its uniformly labeled isotopomer 13C-lysoPE(18:0) were identified for their incorporation into METLIN and isoMETLIN, respectively (Figure 4C). To do so, the experimental MS/MS data of lysoPE(18:0) are compared against the MS/MS spectra of chemically related molecules included in the database [e.g., lysoPE(14:1(9Z)) or lysoPE(15:0), among others], the in silico prediction of MS/MS spectra (Figure 6B), and the m/z shift of each pair of analogous fragments. These complementary data suffice to unequivocally assign the experimental MS/MS spectrum to the candidate molecule, and subsequently, by the approach previously detailed, the related MS/MS spectrum is defined for the corresponding 13C-labeled isotopologue (Figure 4C). In this example, the precursor ion m/z shift is 23.07 Da, which corresponds to a molecule containing 23 carbon atoms. The neutral loss of 141.02 Da as the main fragment peak indicates the presence of a phosphoethanolamine polar head-group. This is further confirmed by the m/z fragment of 44.05, which is characteristic of phosphoethanolamine. In the 13C-labeled isotopologue MS/MS data, both fragments are clearly observed; however, a mass shift of 2.01 Da is present, indicating the presence of two 13C in each of those fragments, matching with the atomic composition of the phosphoethanolamine group (C2H8NO4P). Finally, a mass difference of 21.07 Da between the fragments results from the phosphoethanolamine neutral loss, which corresponds to the 21 carbons of that fragment (23 carbons from the intact metabolite minus 2 carbons from the phosphoethanolamine group) (Figure 4C). Other less prominent fragments further validate the identification and characterization of this lipid species by comparing their m/z shifts with the predicted structures of chemically related molecules. All in all, even when MS/MS spectra for putative metabolites are not available, we were able to generate the fragmentation spectra of those compounds, not only for isoMETLIN but also for METLIN (Figure 4C). It is worth noting that other METLIN informatic tools, such as Neutral Loss and Fragment Similarity Search, were used to identify the fragments described above, resulting in METLIN being capable of self-populating the database by generating more MS/MS spectra.
Figure 6.
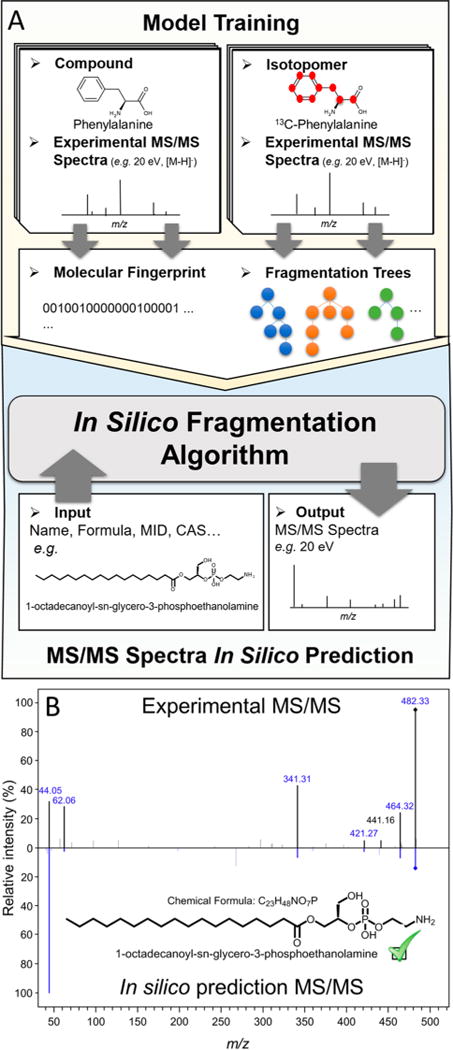
In silico data generation. (A) Workflow for in silico data simulation. A generalization of the input–output kernel regression model, especially designed to predict fragments of known molecules, is used to generate in silico data. Both unlabeled and isotope-labeled compounds are used for model training, providing additional information through the number of isotope-labeled atoms of each fragment. (B) Comparison between experimental MS/MS spectrum generated by lysoPE(18:0) with its in silico prediction in METLIN, at a collision energy of 10 eV. It is worth noting that 6 out of 7 main fragments of the experimental spectrum match with the in silico simulated data (highlighted in blue).
Isotope-Labeled Metabolites to Assist in the Identication of Unknowns
Finally, with this approach, it is possible to facilitate the identification of an endogenous metabolite that is not present in METLIN, starting from its experimental MS/MS data (Figures 4A and 5). Here, the unlabeled molecule shows a neutral loss of 141.02 Da as the main fragment and another fragment of 44.05 Da. Again, its 13C-labeled analogue shows those fragments with a difference of two 13C atoms; hence, it is likely to contain a phosphoethanolamine group (Figure 5). In addition, the precursor ion shift corresponds to a molecule containing 30 carbons. Given that the glycerophosphoethanolamine group contains 5 carbons, the rest of the molecule contains another 25 carbon atoms. Together with that information and high-resolution MS/MS data, the most likely molecule within an error lower than 10 ppm would be the oxidized phospholipid 1-hexadecanoyl-2-(9-oxononanoyl)-sn-glycero-3-phosphoethanolamine (Figure 5). Its phosphatidylcholine analogue has been reported as a product of lung surfactant phospholipid oxidation in smokers,41 and some oxidized ethanolamine phospholipids have also been described as ozonolysis products in bronchoalveolar lavage.42 Although in this case the MS/MS data of the natural occurring metabolite and its isotopologue were not added to the databases due to the lack of complementary information to accurately define the position of the carbonyl within the fatty acid chain, the use of isotope-labeled microorganisms, together with other METLIN tools available, provides a good estimation for the characterization of this unknown natural product synthesized by these microorganisms.
The overall use of 13C-labeled microorganisms is valuable for populating the METLIN MS/MS spectral library. With this approach, MS/MS data for uniformly labeled metabolites and unlabeled molecules with only in silico fragmentation spectra have been detected, identified, manually curated at four different collision energies, and incorporated into isoMETLIN and METLIN, respectively. Furthermore, mass shifts between the endogenous and labeled metabolites provide useful information about the chemical structure of molecules, which is of high interest in fields such as drug design and natural products discovery.
In Silico Data Generation
Experimental MS/MS spectra of more than 20 000 molecules in METLIN, together with some of their isotopologues contained within isoMETLIN, were used for development of the in silico library (Figure 6). One of the strengths of using isotopic fragmentation data is the additional information provided by the number of labeled atoms in each fragment (typically 13C, 2H, or 15N) compared to the endogenous isotopologue. Our in silico algorithm was trained by use of METLIN experimental MS/MS spectra at three discrete collision energies (10, 20, or 40 eV). Accordingly, in silico fragmentation data were generated at collision energies of 10, 20, and 40 eV.
For the computational prediction of MS/MS spectra, many methods have been proposed in the last five years, including CFM-ID,6,43 MetFrag,44 and MyCompoundID,45,46 among others. However, in the latest report of the Critical Assessment of Small Molecule Identification (CASMI) contest,47 held in 2016, CSI:FingerID7 and an input–output kernel regression (IOKR) machine learning approach ranked better than the other tested methods in terms of metabolite structure prediction and computational time efficiency.8,47 A detailed description of the IOKR model can be found in refs 48 and 49. The principle behind our approach is based on the assumption that the IOKR logic is reversible, allowing us to generalize its functionality in the opposite direction: to generate MS/MS data from known molecular structures. IOKR principle is to learn from the similarities among molecules and mass spectral data to identify molecules from MS/MS data, yielding an in silico model. Therefore, given the MS/MS data of an unknown compound, it can predict molecular identities by taking into account these similarities.8 In our approach, we reversed the logic and generalized it to predict in silico MS/MS data from known molecular structures contained in METLIN. Briefly, natural and isotope-labeled compounds are transferred to molecular fingerprints that represent the structure of the molecule encoded into a binary vector. These fingerprints are used as inputs into regression models that describe the relationship between the molecules and their spectra as described by fragmentation trees. This information is employed to train a model by use of the modified IOKR-based approach, finally predicting in silico MS/MS spectral data from known molecules (Figure 6A). Details of the in silico fragmentation model will be published elsewhere. One example of the performance of the in silico algorithm performance is provided for the lipid species lysoPE(18:0), whose experimental MS/MS spectrum was identified in Figure 4C. It is observed that in silico-generated data predicted 6 out of 7 characteristic fragments of the molecule, although intensity correlation is still an aspect of the algorithm that requires improvement (Figure 6B).
CONCLUSION
In summary, the combination of MS/MS experimental data and informatic features within METLIN now make it possible to autonomously identify known molecules and, more importantly, to characterize unknowns. The ultimate goal of METLIN is to help overcome challenges in areas such as global metabolomics, isotope-tracer experiments, and metabolomics activity screening and to facilitate the use of metabolomics to guide systems biology data interpretation. Among its most used features is the Fragment Similarity Search for characterizing unknowns, the development of which takes advantage of the growing number of compounds with MS/MS data that have been recently incorporated. Equally important to the conventional database is the incorporation of data from stable isotopes, which are key to the development of in silico algorithms for MS/MS data prediction on the molecules without experimental data. Together with METLIN’s integration in the cloud-based global metabolomics XCMS Online platform, METLIN is constantly evolving and expanding to facilitate the analysis of known molecules and to identify unknowns.
Acknowledgments
We thank the National Institutes of Health for Grants R01 GM114368 and PO1 A1043376-02S1, and Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov), a Scientific Focus Area Program at Lawrence Berkeley Laboratory for the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Contract DE-AC02-05CH11231.
Footnotes
ORCID
Carlos Guijas: 0000-0001-7993-3388
J. Rafael Montenegro-Burke: 0000-0001-7787-3414
Xavier Domingo-Almenara: 0000-0002-0133-6863
Benedikt Warth: 0000-0002-6104-0706
Tao Huan: 0000-0001-6295-2435
Dennis W. Wolan: 0000-0001-9879-8353
Gary Siuzdak: 0000-0002-4749-0014
Notes
The authors declare no competing financial interest.
References
- 1.Smith CA, O’Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G. Ther Drug Monit. 2005;27:747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 2.Tautenhahn R, Cho K, Uritboonthai W, Zhu Z, Patti GJ, Siuzdak G. Nat Biotechnol. 2012;30:826–8. doi: 10.1038/nbt.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kind T, Tsugawa H, Cajka T, Ma Y, Lai Z, Mehta SS, Wohlgemuth G, Barupal DK, Showalter MR, Arita M, Fiehn O. Mass Spectrom Rev. 2017;9999:1–20. doi: 10.1002/mas.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L. Nucleic Acids Res. 2007;35:D521–6. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, Yanes O. TrAC, Trends Anal Chem. 2016;78:23–35. [Google Scholar]
- 6.Allen F, Greiner R, Wishart D. Metabolomics. 2015;11:98–110. [Google Scholar]
- 7.Duhrkop K, Shen H, Meusel M, Rousu J, Bocker S. Proc Natl Acad Sci U S A. 2015;112:12580–5. doi: 10.1073/pnas.1509788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brouard C, Shen H, Duhrkop K, d’Alche-Buc F, Bocker S, Rousu J. Bioinformatics. 2016;32:i28–i36. doi: 10.1093/bioinformatics/btw246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patti GJ, Yanes O, Siuzdak G. Nat Rev Mol Cell Biol. 2012;13:263–9. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warth B, Spangler S, Fang M, Johnson CH, Forsberg EM, Granados A, Martin RL, Domingo-Almenara X, Huan T, Rinehart D, Montenegro-Burke JR, Hilmers B, Aisporna A, Hoang LT, Uritboonthai W, Benton HP, Richardson SD, Williams AJ, Siuzdak G. Anal Chem. 2017;89:11505–11513. doi: 10.1021/acs.analchem.7b02759. [DOI] [PubMed] [Google Scholar]
- 11.Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, Ivanisevic J, Cho K, Wick EC, Hechenbleikner EM, Uritboonthai W, Goetz L, Casero RA, Jr, Pardoll DM, White JR, Patti GJ, Sears CL, Siuzdak G. Cell Metab. 2015;21:891–7. doi: 10.1016/j.cmet.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Priolo C, Pyne S, Rose J, Regan ER, Zadra G, Photopoulos C, Cacciatore S, Schultz D, Scaglia N, McDunn J, De Marzo AM, Loda M. Cancer Res. 2014;74:7198–204. doi: 10.1158/0008-5472.CAN-14-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim CK, Bilgin A, Lovejoy DB, Tan V, Bustamante S, Taylor BV, Bessede A, Brew BJ, Guillemin GJ. Sci Rep. 2017;7:41473. doi: 10.1038/srep41473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hocher B, Adamski J. Nat Rev Nephrol. 2017;13:269–284. doi: 10.1038/nrneph.2017.30. [DOI] [PubMed] [Google Scholar]
- 15.Roberts LD, Koulman A, Griffin JL. Lancet Diabetes Endocrinol. 2014;2:65–75. doi: 10.1016/S2213-8587(13)70143-8. [DOI] [PubMed] [Google Scholar]
- 16.Armitage EG, Southam AD. Metabolomics. 2016;12:146. doi: 10.1007/s11306-016-1093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warth B, Raffeiner P, Granados A, Huan T, Fang M, Forsberg EM, Benton HP, Goetz L, Johnson CH, Siuzdak G. Cell Chem Biol. 2018 doi: 10.1016/j.chembiol.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yanes O, Clark J, Wong DM, Patti GJ, Sanchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Nat Chem Biol. 2010;6:411–7. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beyer BA, Fang M, Sadrian B, Montenegro-Burke JR, Plaisted WC, Kok BPC, Saez E, Kondo T, Siuzdak G, Lairson LL. Nat Chem Biol. 2018;14:22–28. doi: 10.1038/nchembio.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G. Nat Biotechnol. 2018 doi: 10.1038/nbt.4101. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rampler E, Coman C, Hermann G, Sickmann A, Ahrends R, Koellensperger G. Analyst. 2017;142:1891–1899. doi: 10.1039/c7an00107j. [DOI] [PubMed] [Google Scholar]
- 22.Ivanisevic J, Zhu ZJ, Plate L, Tautenhahn R, Chen S, O’Brien PJ, Johnson CH, Marletta MA, Patti GJ, Siuzdak G. Anal Chem. 2013;85:6876–84. doi: 10.1021/ac401140h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessner D, Chambers M, Burke R, Agus D, Mallick P. Bioinformatics. 2008;24:2534–6. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. Anal Chem. 2012;84:5035–9. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Chen YJ, Cho K, Nikolskiy I, Crawford PA, Patti GJ. Anal Chem. 2014;86:1632–9. doi: 10.1021/ac403384n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurczy ME, Forsberg EM, Thorgersen MP, Poole FL, 2nd, Benton HP, Ivanisevic J, Tran ML, Wall JD, Elias DA, Adams MW, Siuzdak G. ACS Chem Biol. 2016;11:1677–85. doi: 10.1021/acschembio.6b00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mylonas R, Mauron Y, Masselot A, Binz PA, Budin N, Fathi M, Viette V, Hochstrasser DF, Lisacek F. Anal Chem. 2009;81:7604–10. doi: 10.1021/ac900954d. [DOI] [PubMed] [Google Scholar]
- 28.Damen H, Henneberg D, Weimann B. Anal Chim Acta. 1978;103:289–302. [Google Scholar]
- 29.McLafferty FW, Stauffer DB. J Chem Inf Model. 1985;25:245–252. [Google Scholar]
- 30.Stein SE. J Am Soc Mass Spectrom. 1995;6:644–55. doi: 10.1016/1044-0305(95)00291-K. [DOI] [PubMed] [Google Scholar]
- 31.Demuth W, Karlovits M, Varmuza K. Anal Chim Acta. 2004;516:75–85. [Google Scholar]
- 32.Benton HP, Wong DM, Trauger SA, Siuzdak G. Anal Chem. 2008;80:6382–9. doi: 10.1021/ac800795f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Chen Y, Wang J, Chen J, Aggarwal BB, Pang X, Liu M. Curr Mol Med. 2012;12:153–62. doi: 10.2174/156652412798889072. [DOI] [PubMed] [Google Scholar]
- 34.Yilmazer M, Stevens JF, Buhler DR. FEBS Lett. 2001;491:252–6. doi: 10.1016/s0014-5793(01)02210-4. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto Y, Fujisawa A, Hara A, Dunlap WC. Proc Natl Acad Sci U S A. 2001;98:13144–8. doi: 10.1073/pnas.241024298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiner M, Trondle J, Schmideder A, Albermann C, Binder K, Sprenger GA, Weuster-Botz D. Anal Biochem. 2015;478:134–40. doi: 10.1016/j.ab.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Bueschl C, Kluger B, Lemmens M, Adam G, Wiesenberger G, Maschietto V, Marocco A, Strauss J, Bodi S, Thallinger GG, Krska R, Schuhmacher R. Metabolomics. 2014;10:754–769. doi: 10.1007/s11306-013-0611-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neubauer S, Haberhauer-Troyer C, Klavins K, Russmayer H, Steiger MG, Gasser B, Sauer M, Mattanovich D, Hann S, Koellensperger G. J Sep Sci. 2012;35:3091–105. doi: 10.1002/jssc.201200447. [DOI] [PubMed] [Google Scholar]
- 39.Schwaiger M, Rampler E, Hermann G, Miklos W, Berger W, Koellensperger G. Anal Chem. 2017;89:7667–7674. doi: 10.1021/acs.analchem.7b01624. [DOI] [PubMed] [Google Scholar]
- 40.Cho K, Mahieu N, Ivanisevic J, Uritboonthai W, Chen YJ, Siuzdak G, Patti GJ. Anal Chem. 2014;86:9358–61. doi: 10.1021/ac5029177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimura T, Shibata Y, Yamauchi K, Igarashi A, Inoue S, Abe S, Fujita K, Uosaki Y, Kubota I. Lung. 2012;190:169–82. doi: 10.1007/s00408-011-9331-2. [DOI] [PubMed] [Google Scholar]
- 42.Wynalda KM, Murphy RC. Chem Res Toxicol. 2010;23:108–17. doi: 10.1021/tx900306p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen F, Pon A, Wilson M, Greiner R, Wishart D. Nucleic Acids Res. 2014;42:W94–9. doi: 10.1093/nar/gku436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruttkies C, Schymanski EL, Wolf S, Hollender J, Neumann S. J Cheminf. 2016;8:3. doi: 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huan T, Tang C, Li R, Shi Y, Lin G, Li L. Anal Chem. 2015;87:10619–26. doi: 10.1021/acs.analchem.5b03126. [DOI] [PubMed] [Google Scholar]
- 46.Shen H, Duhrkop K, Bocker S, Rousu J. Bioinformatics. 2014;30:i157–64. doi: 10.1093/bioinformatics/btu275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schymanski EL, Ruttkies C, Krauss M, Brouard C, Kind T, Duhrkop K, Allen F, Vaniya A, Verdegem D, Bocker S, Rousu J, Shen H, Tsugawa H, Sajed T, Fiehn O, Ghesquiere B, Neumann S. J Cheminf. 2017;9:22. doi: 10.1186/s13321-017-0207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brouard C, Szafranski M, d’Alche-Buc F. J Mach Learn Res. 2016;17:1–48. [Google Scholar]
- 49.Brouard C, D’Alché-Buc F, Szafranski M. Semi-supervised Penalized Output Kernel Regression for Link Prediction; 28th International Conference on Machine Learning (ICML 2011); Bellevue, WA. June 28, 2011; pp. 593–600. http://www.icml-2011.org/papers/367_icmlpaper.pdf. [Google Scholar]