

Abstract
B cells that bind to membrane-bound antigens (e.g., on the surface of an antigen-presenting cell) form an immune synapse, a specialized cellular structure that optimizes B-cell receptor (BCR) signaling and BCR-mediated antigen acquisition. Both the remodeling of the actin cytoskeleton and the reorientation of the microtubule network towards the antigen contact site are essential for immune synapse formation. Remodeling of the actin cytoskeleton into a dense peripheral ring of F-actin is accompanied by polarization of the microtubule-organizing center towards the immune synapse. Microtubule plus-end binding proteins, as well as cortical plus-end capture proteins mediate physical interactions between the actin and microtubule cytoskeletons, which allow them to be reorganized in a coordinated manner. Elucidating the mechanisms that control this cytoskeletal reorganization, as well as understanding how these cytoskeletal structures shape immune synapse formation and BCR signaling, can provide new insights into B cell activation. This has been aided by the development of super-resolution microscopy approaches that reveal new details of cytoskeletal network organization. We describe here a method for using stimulated emission depletion (STED) microscopy to simultaneously image actin structures, microtubules, and transfected GFP-tagged microtubule plus-end binding proteins in B cells. To model the early events in immune synapse formation, we allow B cells to spread on coverslips coated with anti-immunoglobulin (anti-Ig) antibodies, which initiate BCR signaling and cytoskeleton remodeling. We provide step-by-step protocols for expressing GFP fusion proteins in A20 B-lymphoma cells, for anti-Ig-induced cell spreading, and for subsequent cell fixation, Immunostaining, image acquisition, and image deconvolution steps. The high-resolution images obtained using these procedures allow one to simultaneously visualize actin structures, microtubules, and the microtubule plus-end binding proteins that may link these two cytoskeletal networks.
Keywords: Immunology and Infection, Issue 134, Immunology, B cells, immune synapse, actin, microtubules, microtubule-organizing center (MTOC), plus-end tracking proteins, super-resolution microscopy, stimulated emission depletion (STED) microscopy
Introduction
When B cells bind to polarized arrays of antigens (e.g., displayed on the surface of antigen-presenting cells (APCs)), the resulting B-cell receptor (BCR) signaling drives the formation of a classic immune synapse structure, which was first described in T cells1,2,3,4,5,6,7,8,9,10,11,12,13. Initially, microclusters of antigen-bound BCRs form at the periphery of the B cell:APC contact site. These microclusters then move towards the center of the antigen contact site, where they coalesce into a central supramolecular activation cluster (cSMAC) that forms the core of the immune synapse. Immune synapse formation optimizes BCR signaling and facilitates BCR-mediated antigen extraction from the APC membrane14. This antigen acquisition, which is followed by BCR-mediated antigen internalization and subsequent antigen processing, allows B cells to present peptide:MHC II complexes to T cells and elicit T cell help14. Because immune synapse formation promotes B cell activation, elucidating the mechanisms that establish this functional pattern of receptor organization can provide new insights into how humoral immune responses are initiated and regulated.
Reorganization of both the actin and microtubule cytoskeletons is essential for immune synapse formation. Localized BCR signaling stimulated by a spatially-polarized array of antigens induces rapid and dramatic remodeling of the actin cytoskeleton1,15. The formation of dendritic actin structures at the periphery of the B cell exerts pushing forces on the plasma membrane and promotes B cell spreading. This allows the B cell to scan a greater area of the antigen-bearing surface, and increases the number of BCRs that bind antigen and activate BCR signaling pathways. At the same time, the MTOC and the microtubule network are reoriented towards the site of antigen contact. As the MTOC approaches the antigen contact site, microtubules emanating from the MTOC extend along the inner face of the plasma membrane at the interface between the B cell and the antigen-bearing surface16,17. These juxtamembrane microtubules can then act as tracks for the dynein-mediated centripetal movement of antigen-bound BCR microclusters18, leading to the formation of a cSMAC.
The reorientation and polarization of the MTOC towards the immune synapse requires intact actin and microtubule cytoskeletons, and often depends on interactions between the cortical actin network and microtubules16,17,19,20. Cortical actin-binding proteins, such as IQGAP1, can capture microtubules by interacting with protein complexes that decorate the microtubule plus-ends21. These dynamic complexes of plus-end binding proteins include EB1 and CLIP-170, which are collectively referred to as microtubule plus-end tracking proteins (+TIPs)21,22. +TIPs at the ends of microtubules can bind to proteins that are associated with either the plasma membrane or the cortical actin cytoskeleton. This allows force-generating mechanisms (e.g., the minus-end directed movement of cortically-anchored dynein along microtubules) to exert pulling forces on microtubules, and thereby reposition the MTOC. CLIP-170 can bind to the actin-associated scaffolding protein IQGAP123, and we have shown that both of these proteins are required for BCR-induced MTOC polarization towards the immune synapse17. This IQGAP1-CLIP-170 interaction may play a key role in coordinating the remodeling of the actin cytoskeleton with the repositioning of the microtubule network at the B-cell immune synapse.
Conventional fluorescence microscopy has revealed the dramatic reorganization of the actin and microtubule cytoskeletons during B-cell immune synapse formation2. However, this approach cannot resolve small cellular structures in detail due to the diffraction limit of light, which, according to Abbe's law, is dependent on the wavelength of light used to illuminate the sample and the aperture of the objective24. This diffraction limit constrains the resolution of conventional light microscopes to 200-300 nm in the lateral direction and 500-700 nm in the axial direction25. Therefore, smaller subcellular structures, as well as the fine details of the actin and microtubule cytoskeletons, could only be observed using electron microscopy. Electron microscopy imaging of the cytoskeleton is time consuming, requires harsh sample fixation and preparation protocols that can alter biological structures, and is limited to antibody-mediated detection. The ability to immunostain and simultaneously image multiple proteins or cellular structures is a substantial advantage of fluorescence microscopy. Moreover, expressing fluorescent fusion proteins in cells enables real-time imaging and is useful when effective antibodies for immunostaining the protein of interest are not available.
Recent technological advances in super-resolution microscopy have overcome the diffraction limits of light and allowed the visualization of nanoscale cellular structures24. One such super-resolution microscopy technique is called stimulated emission depletion (STED) microscopy. STED employs two lasers, where one laser excites the fluorophore and a second laser with a donut-shaped pattern selectively suppresses the fluorescence emission around the fluorophore. This reduces the point-spread function (apparent area) of a single fluorescent particle and provides a sub-diffraction limit fluorescent image25,26. Ground-state depletion microscopy also employs fluorescence-based techniques to acquire super-resolution images. However, the image acquisition and reconstruction times are long, there are only a limited number of fluorophores that can be used, and the simultaneous high-resolution imaging of multiple cytoskeletal components is technically challenging because maintaining actin and microtubule structures requires different fixation procedures. Therefore, STED has multiple advantages over electron microscopy and other super-resolution microscopy approaches in that it offers rapid image acquisition, has minimal post-processing requirements, and employs the same fluorophores and staining techniques that are used for conventional fluorescence microscopy of fixed samples26.
Super-resolution microscopy has now been used to visualize actin structures at the immune synapse in natural killer (NK) cells and T cells26,27,28,29,30,31. However, super-resolution imaging of the microtubule cytoskeleton, as well as the coordinated reorganization of the actin and microtubule cytoskeletons during immune synapse formation, has only recently been reported17. We used STED microscopy to image B cells that had been allowed to spread on coverslips coated with anti-immunoglobulin (anti-Ig) antibodies, which stimulate BCR signaling and initiate cytoskeleton reorganization. When plated on immobilized anti-Ig antibodies, B cells undergo dramatic actin-dependent spreading, which recapitulates the initial events during immune synapse formation. Importantly, STED microscopy revealed the fine details of the dendritic ring of F-actin that forms at the periphery of the immune synapse and showed that the MTOC, as well as the microtubules attached to it, had moved close to the antigen contact site17. These microtubules extended outward towards the peripheral ring of F-actin. Moreover, multi-color STED imaging of various combinations of F-actin, tubulin, IQGAP1, and GFP-tagged CLIP-170 +TIPs showed that microtubule plus-ends marked by CLIP-170-GFP were closely associated with the peripheral actin meshwork and with IQGAP1, a cortical capture protein17.
Here, we present a detailed protocol for imaging the actin and microtubule cytoskeletons at the immune synapse using STED microscopy. These methods have been optimized using the A20 murine B cell line, which has been widely employed for studying BCR signaling and immune synapse formation17,32,33,34,35,36,37,38,39. Because commercial antibodies to CLIP-170 did not work well for immunostaining in previous experiments, we describe in detail the expression of GFP-tagged CLIP-170 in A20 cells, along with staining protocols for simultaneously visualizing up to three cytoskeletal components or cytoskeleton-associated proteins. Methods for using STED microscopy to image actin at NK cell immune synapses have been described previously40. Here, we extend this to acquiring multi-color super-resolution images of both the actin and microtubule cytoskeletons in B cells.
A critical consideration for super-resolution microscopy is using the appropriate fixation procedures for maintaining cellular structures and preventing damage to fluorescent proteins. The fixation and staining methods presented herein have been optimized to retain GFP fluorescence and provide high-resolution imaging of the actin and microtubule networks. When expressing fluorescent proteins, it should be noted that B cells are usually difficult to transfect. Using this protocol, 20-50% of A20 cells typically express the transfected GFP fusion protein, and among this population the levels of protein expression are variable. Nevertheless, super-resolution imaging of actin and microtubules using the procedures we describe is quite robust and high-quality images are readily obtained. Despite their small size relative to A20 cells, we show that these procedures can also be used to image the microtubule network in primary B cells that have been briefly activated with lipopolysaccharide (LPS). We have shown that LPS-activated primary B cells can be transfected with siRNAs at relatively high efficiency (i.e., such that protein depletion can be detected by immunoblotting), making them a good alternative to the use of B cell lines for some studies17.
Protocol
All animal procedures were approved by the University of British Columbia Animal Care Committee.
1. Expressing GFP-fusion proteins in A20 B-lymphoma cells
Culture A20 cells in complete RPMI medium (RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 µM 2-mercaptoethanol, 2 mM glutamine, 1 mM pyruvate, 50 U/mL penicillin, and 50 µg/mL streptomycin) at 37 °C in a tissue culture incubator with 5% CO2.
Prepare the transfection reagent (see the Table of Materials) according to the manufacturer's instructions. NOTE: This protocol has been optimized for the specific reagents and transfection protocol described in the Table of Materials. Other transfection reagents that we have tried have not yielded sufficient transfection efficiency.
Count the cells using a hemocytometer. Centrifuge 2.5 x 106 A20 cells (for each transfection) for 5 min at 525 x g. Resuspend the cells in 100 µL of transfection reagent containing 1-2.5 µg of plasmid DNA. For DNA encoding CLIP-170-GFP23, use 2.5 µg per transfection. Mix gently.
Transfer the cells to a well of a 6-well plate, and adjust the volume to 2 mL with pre-warmed complete RPMI medium. Culture the cells for 18 h at 37 °C to allow protein expression.
2. Isolating Primary Mouse B Cells and Activating Them with LPS
Use sterilized surgical tools to remove the spleen from a mouse, following protocols approved by the institution's Animal Care Committee.
In the tissue culture hood, place a sterile 70-µm cell strainer into a 35-mm tissue culture dish containing 3 mL of room temperature sterile PBS. Use the rubber part of the plunger from a 5-ml syringe to crush the spleen through the cell strainer.
Pipet the single cell suspension of splenoctyes into a sterile 15-mL tube and centrifuge at 525 x g for 5 min.
Use a magnetic bead-based B cell isolation kit (see Table of Materials) to obtain a highly enriched population of B cells.
Resuspend the B cells to 3 x 106/mL in complete RPMI medium supplemented with 2.5 µg/mL LPS plus 5 ng/mL B cell activating factor (BAFF; a survival cytokine).
Culture the cells for 6 hr at 37 °C.
3. Coating Glass Coverslips with Anti-Ig Antibodies
Prepare the antibody solution for coating the coverslips. Dilute the stock solution of goat-anti-mouse Ig antibodies into room temperature sterile PBS to make a 12.5 µg/mL solution; 400 µL of antibody solution is required for each coverslip. NOTE: Use goat anti-mouse IgG for A20 cells; use goat anti-mouse IgM for primary B cells. Goat IgG antibodies do not bind well to mouse Fc receptors. However, to avoid any potential Fc receptor binding, one can use F(ab) or F(ab')2 fragments of anti-mouse IgG or anti-mouse IgM antibodies.
Dip a #1.5 18-mm round glass coverslip in 100% methanol and let it dry completely (~10 min).
Using forceps, place the dried coverslip into a 12-well tissue culture plate and pipet 400 µL of the 12.5 µg/mL antibody solution (5 µg total; 2 µg/cm2) onto the coverslip so that it forms a bubble at the center of the coverslip and spreads to the edges.
Be careful to cover the entire coverslip, but do not allow the antibody solution to extend past the edge of the glass coverslip and onto the tissue culture plastic. Incubate at room temperature for 30 min.
Wash the coverslips by pipetting 1 mL of sterile PBS into the well and subsequently aspirating out the solution. Repeat two more times to remove unbound antibodies.
Add 1 mL of sterile PBS to the well containing the antibody-coated coverslip until cells are ready to be added. NOTE: The coverslips can be kept at room temperature, covered with PBS, for use on the same day.
4. B Cell Spreading on Anti-Ig-Coated Coverslips
Prepare modified HEPES-buffered saline (mHBS): 25 mM HEPES, pH 7.2, 125 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM Na2HPO4, 0.5 mM MgSO4, 1 mg/mL dextrose, 2 mM glutamine, 1 mM sodium pyruvate, and 50 µM 2-mercaptoethanol). Filter sterilize this solution and store at 4 °C.
On the day of the experiment, make 50 mL of mHBS with 2% FBS (mHBS-FBS) by adding 1 mL of heat-inactivated FBS to 50 mL mHBS. Warm the mHBS-FBS to 37 °C before use.
Centrifuge the transfected A20 cells or the primary B cells that had been cultured with BAFF plus LPS at 525 x g for 5 min.
Resuspend the cells in 1 mL of mHBS-FBS, count the cells using a hemocytometer, and dilute the cells to 2 x 105 cells/mL in mHBS-FBS.
Using forceps, transfer the coverslip from the tissue culture plate to a piece of paraffin film.
Add 250 µL of B cells (5 x 104 cells) to each coverslip.
Incubate the coverslips at 37 °C for 15 min in the dark to allow the B cells to spread over the anti-IgG-coated surface.
5. Fixing and Immunostaining the Cells
Prepare the fixation solution by diluting the 16% paraformaldehyde stock solution and the 50% glutaraldehyde stock solution in room-temperature distilled water to yield final concentrations of 3% paraformaldehyde and 0.1% glutaraldehyde. Prepare the fixation solution on the day it is to be used and discard any excess. CAUTION: The paraformaldehyde and glutaraldehyde stock solutions should only be used in a chemical fume hood. Follow the instructions on the material safety data sheet (MSDS).
Prepare the permeabilization/blocking buffer (3% BSA and 0.1% Triton X-100 diluted in PBS). Filter sterilize this solution and store at 4 °C. On the day of the experiment, warm the required amount to room temperature.
Prepare the staining buffer (1% BSA and 0.1% saponin in PBS). Sterilize this solution using a 0.2 µm filter and store at 4 °C. On the day of the experiment, warm the required amount to room temperature.
Pipet 350 µL of the fixation solution onto each coverslip, ensuring that the surface is completely covered. Incubate at room temperature for 10 min in the dark.
Remove the fixation solution from the coverslip by carefully aspirating from the edge of the coverslip using a micropipet. CAUTION: The fixation solution should be discarded as hazardous chemical waste.
Wash the coverslips once with 500 µL of permeabilization/blocking buffer. Pipet off the liquid.
Permeabilize and block the cells by adding 250 µL of permeabilization/blocking buffer to each coverslip, and incubating for 10 min at room temperature in the dark.
Dilute the rabbit anti-tubulin antibody 1:100 in staining buffer.
Aspirate off the permeabilization/blocking buffer from the coverslips, and add 50 µL of the diluted anti-tubulin antibody solution to each coverslip. Incubate at room temperature for 30 min in the dark.
Prepare the secondary antibody/actin stain solution by diluting both the Alexa Fluor 532-conjugated goat anti-rabbit IgG secondary antibody and Alexa Fluor 568-conjugated phalloidin 1:100 in staining buffer.
Wash the coverslips 3 times to remove excess primary antibody by adding 500 µL of staining buffer and then aspirating it.
Add 50 µL of the secondary antibody/actin stain solution to each coverslip, and incubate for 30 min at room temperature in the dark.
Wash the coverslips 3 times to remove excess secondary antibody by adding 500 µL of staining buffer and then aspirating it.
Add 10 µL of mounting reagent to a microscope slide. Mount the coverslip onto the microscope slide with the cell side down. NOTE: An "anti-fade" mounting medium (see Table of Materials) is strongly recommended to preserve the fluorescence intensity of GFP fusion proteins. Avoid the use of mounting reagents containing DAPI, which can interfere with the imaging of the fluorophores when using the STED laser.
Allow the slides to dry overnight in the dark. Image the slides the next day. NOTE: Imaging the slides as soon as the coverslip is dry is strongly recommended, as the GFP fluorescence will decrease over time.
6. Imaging Using the STED Microscope
NOTE: Please note that all software steps described below are specific to the microscope and software we used (see the Table of Materials). The steps and settings will need to be adjusted if imaging is performed using a different microscope/software.
Turn on the STED microscope, activate the lasers and epifluorescence illumination lamp, and start up the microscope software.
Set the parameters for the STED depletion lasers. NOTE: This will depend on the specific microscope being used and the lasers that it is equipped with. Some recommended settings are white light (WLL) 70%; 592 nm laser 80%; 660 nm laser 80%.
Activate the STED settings and align the STED laser beams using the 100X objective.
Using the eyepiece, focus the sample and select a cell with moderate GFP expression. NOTE: Low GFP expression will not be visible because STED reduces the emission intensity. Conversely, high expression of CLIP-170-GFP induces the spontaneous formation of large clusters, which does not reflect the normal microtubule plus-end protein complex morphology and distribution. Selecting a cell with moderate GFP expression is essential for accurately imaging the cytoskeletal structures at the antigen contact site.
- Zoom into the selected cell and choose the region of interest to be imaged. Adjust the focus so that the x-y plane of the B cell that is closest to the coverslip is in focus.
- To do this, move the objective progressively closer to the coverslip so that the B cell cytoskeletal structures come into focus and then go out of focus. Then gradually move the objective in the opposite direction until the B cell cytoskeletal structures first come back into focus.
Optimize the settings including the laser power, excitation beam, and detector range. Start with the settings recommended by the manufacturer's instructions. Then make adjustments as needed to optimize the signal for the samples. Image all samples to be compared to each other with the same settings.
Under the "acquire" tab of the software, set the image acquisition to sequential frame acquisition in the lower left "Sequential Scan" panel by checking the option "between frames".
Set the acquisition sequence. Note: We acquire the GFP fluorescence first in order to avoid degradation of the GFP signal. Depending on the combination of fluorophores that are used in addition to GFP, imaging the longer wavelength fluorescence signal first may yield better results. However, the order in which the fluorophores or fluorescent proteins are subjected to stimulated emission depletion and imaged should be optimized to obtain the strongest fluorescence signals and best resolution.
Select and set the STED laser power for each fluorophore under the "Acquire" tab of the software, and use the slider bar for either the 592-nm or the 660-nm STED laser to adjust the laser power. Adjust the power for the 592-nm depletion laser for GFP and for Alexa Fluor 532. Adjust the power for the 660-nm depletion laser for Alexa Fluor 568.
To increase the resolution and reduce the background signal, go to the "Acquisition" settings under the "Acquire" tab of the software, increase the line and/or frame averaging by selecting a value greater than 1 using the drop-down menu for each option.
Also, acquire the fluorescent signal using time-gated STED by checking the gating option for the fluorophore under the "Acquire" tab, and specifying values for time-gating (see note below). Increase the STED laser power to enhance the resolution, though this will also increase photobleaching of the fluorophores. NOTE: The gating time will need to be optimized for the microscope, sample, and fluorophores being used. A time gating of 0.3 to 6 ns is recommended. After fixation, GFP is particularly sensitive to high laser power. To reduce the photobleaching of GFP, time gating should be applied and the STED laser power should be reduced.
To capture 3D STED images, use the slider to change the point spread function (PSF) to "3D STED".
Manually acquire multiple images to ensure reproducibility. Deconvolve the images (see Figure 1) using a deconvolution software package41(see Table of Materials).

Representative Results
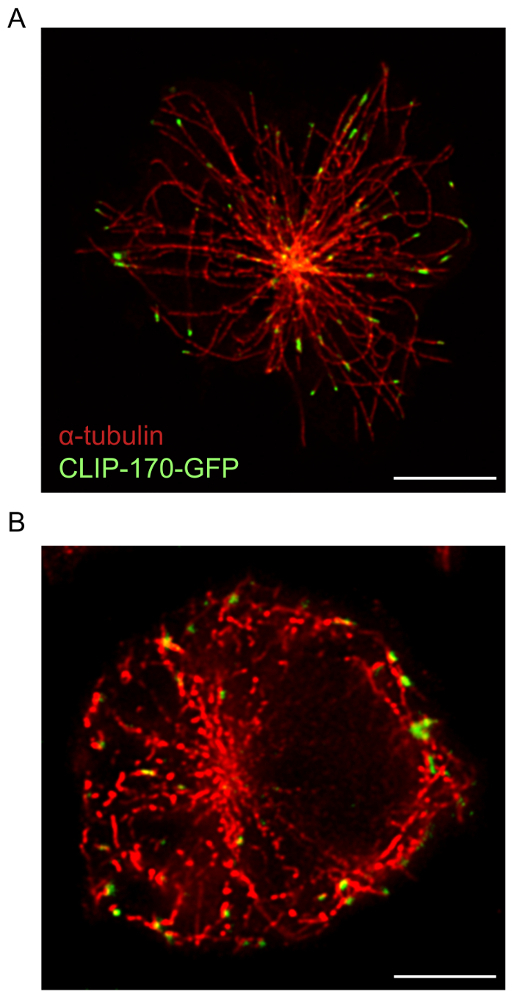
For B cells spreading on immobilized anti-Ig, STED microscopy used in conjunction with deconvolution software provides higher resolution images of cytoskeletal structures than confocal microscopy. This is evident in Figure 1, where the F-actin network was visualized using the protocol described above. A comparison of confocal and STED super-resolution images of the same sample shows that the STED images are of higher resolution and reveal more detailed structures of the actin cytoskeleton (Figure 1). This figure also shows that deconvolution is essential for obtaining high quality STED images in which actin filaments are more clearly defined. Although deconvolution of confocal images yields a substantial improvement in the resolution of the image, deconvolved STED images provide more detailed structural information than deconvolved confocal images. In particular, the dendritic structure of the peripheral F-actin ring is revealed in greater detail by STED microscopy (Figure 1 and Figure 2). The microtubule network at the antigen contact site was also imaged using the sample preparation and imaging protocol described above (Figure 3). Microtubules originate from a central point, which is the MTOC, and emanate outwards towards the periphery of the cell. In this experiment, the B cells were allowed to spread on anti-Ig-coated coverslips for 15 min (Figure 3), a time point at which the MTOC has moved towards the antigen contact site17. CLIP-170-GFP clusters that mark the plus-ends of microtubules can be seen at the ends of the microtubules shown in Figure 3. When the sample preparation and STED imaging of the microtubule network is optimal, continuous and distinct microtubules are observed, with CLIP-170-GFP localized either along the microtubules or at the plus-ends (Figure 3A). Sub-optimal microtubule staining, which was observed when using lower concentrations of staining antibodies or batches of α-tubulin antibodies that are more than one year old, results in microtubules appearing as discontinuous sections that are poorly resolved upon deconvolution (Figure 3B; see also Figure 5C). Although all of the CLIP-170-GFP fluorescence in these images is associated with α-tubulin-immunostained structures, one cannot distinguish whether CLIP-170-GFP is located at the plus ends, or along the length of the microtubules, due to the incomplete staining of the microtubules. Hence it is important that the α-tubulin antibody be stored under the manufacturer recommended storage conditions and used within one year.
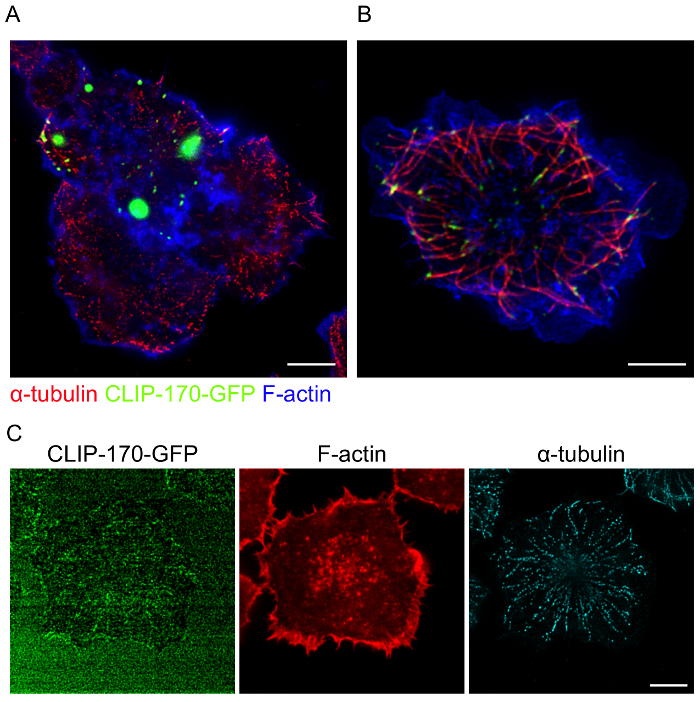
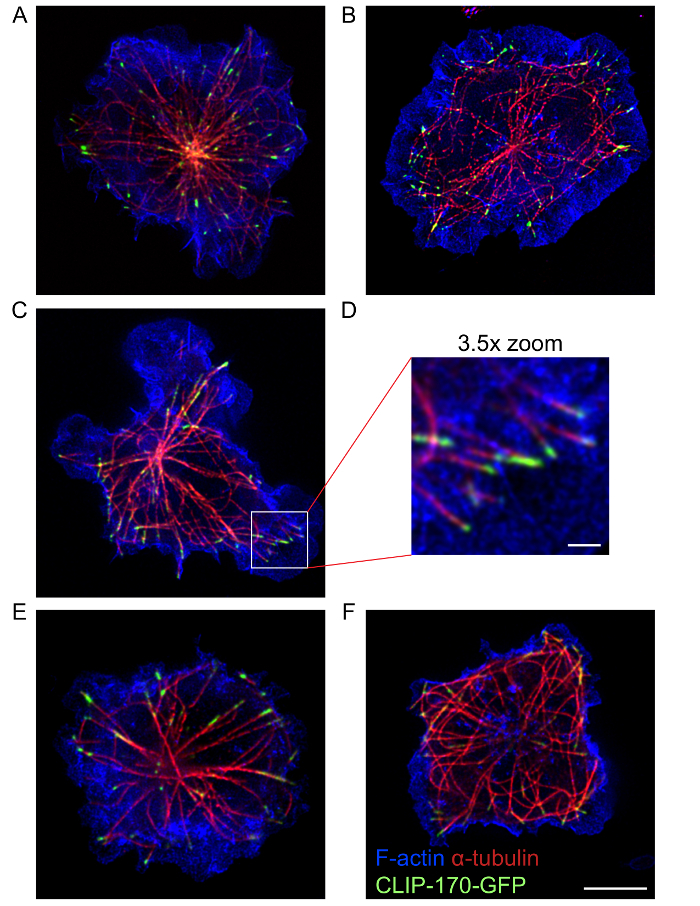
Using this protocol, high quality multi-color STED images that show the organization and structure of the actin cytoskeleton and the microtubule network, as well as proteins such as IQGAP1 and CLIP-170 that associate with these two cytoskeletons17, could be acquired. The STED images in Figure 4 show the peripheral ring of dendritic actin, as well as microtubules that emanate from a central location in the cell where the actin staining is much less dense. CLIP-170-GFP at the ends of these microtubules is closely associated with the peripheral F-actin. This protocol allows one to visualize cytoskeletal structures at the immune synapse using either single-color STED imaging (Figure 2) or multicolor STED imaging (Figure 3 and Figure 4). However, it should be noted that single color STED imaging (Figure 2) may yield better resolution of actin structures and microtubules in B cells than multi-color STED (Figure 4). This may be due to photobleaching caused by sequential STED image acquisition for the different fluorophores. To obtain the best super-resolution images, the combination of fluorophores and fluorescent proteins selected, as well as the sequence in which they are imaged using the excitation and depletion lasers, should be optimized for the sample. Nevertheless, multi-color STED imaging provides higher resolution images of these cytoskeletal structures than conventional confocal microscopy. Additionally, single-color STED can be used to acquire 3-dimensional super-resolution images of the entire B cell actin or microtubule network (Movie 1).
When using cells transfected with fluorescent fusion proteins, achieving optimal expression levels and avoiding artifacts due to overexpression are significant considerations. In cells where CLIP-170-GFP is overexpressed, large aggregates of CLIP-170-GFP are formed (Figure 5A). In addition to this mislocalization of CLIP-170-GFP, only part of the microtubule network in this cell was within the focal plane closest to the coverslip (Figure 5B). This suggests that CLIP-170 overexpression may also impair BCR-induced MTOC polarization. Conversely, because strong fluorescence signals are typically required for acquiring high quality STED images, low expression of fluorescent fusion proteins such as CLIP-170-GFP (Figure 5C) results in poor quality images. Hence, when using cells that have been transfected with fluorescent proteins, it is important to image only those cells with optimal levels of fusion protein expression. It is also important to note that the transfection protocol that was used for A20 cells (see Table of Materials) typically results in 20-50% of the cells expressing the transfected protein. For plasmid DNA (as opposed to siRNAs), transfection frequencies for primary B cells are often much lower than for A20 cells, requiring the use of B cell lines. Nevertheless, high quality STED images of cytoskeletal elements in untransfected primary B cells can be obtained using this protocol (Figure 6).
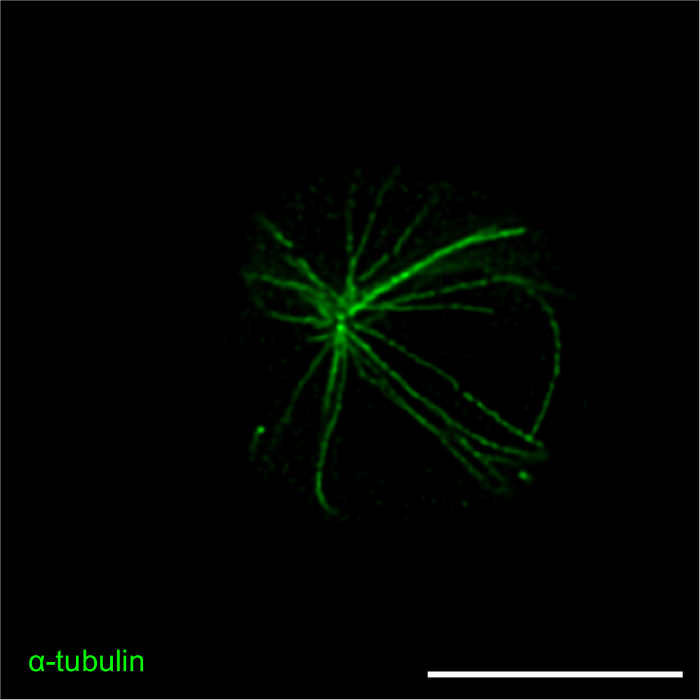
Figure 1: Comparison of confocal and STED imaging of F-actin. Confocal images (top) and STED images (bottom) of an A20 cell that had spread on anti-IgG-coated coverslips for 15 min before being stained with Alexa Fluor 532-conjugated phalloidin. Using the confocal STED microscope, the same cell was imaged first by confocal microscopy and then by STED. The initial confocal and STED images are shown, along with the same confocal and STED images after deconvolution. Scale bar: 5 µm. Please click here to view a larger version of this figure.
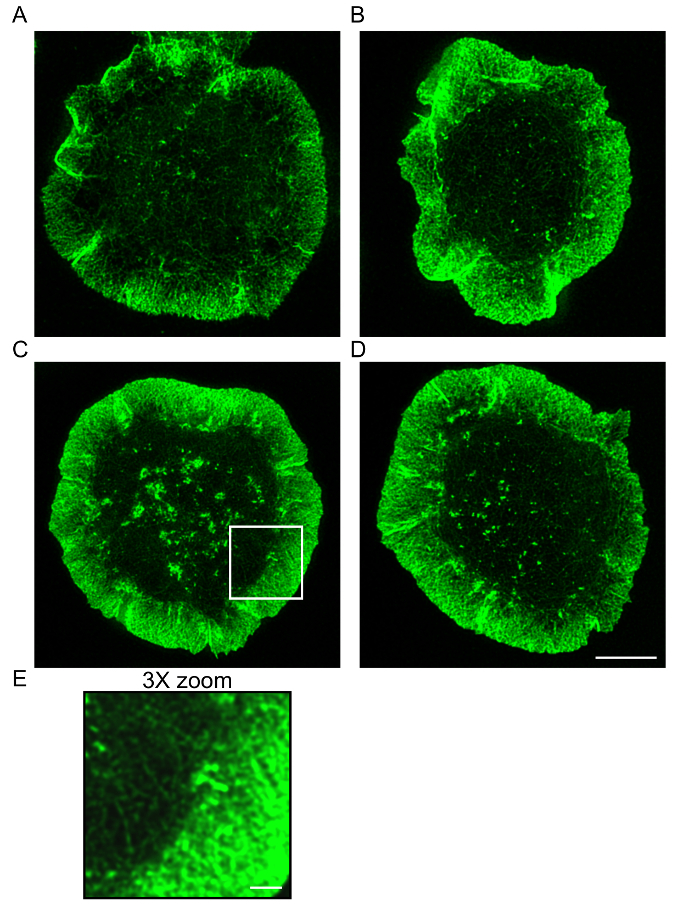
Figure 2: The actin cytoskeleton at the antigen contact site. A20 cells that had been allowed to spread on anti-IgG-coated coverslips for 15 min were stained with Alexa Fluor 568-conjugated phalloidin and imaged by STED microscopy. The initial STED images were deconvolved. Panels A-B and panels C-E show representative images of two different cells. Panel E shows a 3X enlargement of the region in the white box in panel C. Scale bar for panels A-D: 5 µm. Scale bar for panel E: 1 µm. Please click here to view a larger version of this figure.
Figure 3: STED images of the microtubule network and CLIP-170-GFP. A20 cells expressing CLIP-170-GFP were allowed to spread on anti-IgG-coated coverslips for 15 min before being fixed and immunostained with an α-tubulin antibody plus an Alexa Fluor 532-conjugated secondary antibody. (A) Representative image showing immunostaining of the microtubule network, with CLIP-170-GFP located mainly at the plus-ends of microtubules. (B) Sub-optimal staining and resolution of microtubules due to the use of an outdated stock of α-tubulin antibody. Scale bars: 5 µm. Please click here to view a larger version of this figure.
Figure 4: STED images of the actin and microtubule cytoskeletons. A20 cells expressing CLIP-170-GFP were allowed to spread on anti-IgG-coated coverslips for 15 min before being fixed and stained with Alexa Fluor 568-conjugated phalloidin to visualize F-actin, and with an α-tubulin antibody plus an Alexa Fluor 532-conjugated secondary antibody to visualize microtubules. Panels A-D and panels E-F show representative images of two different cells. Panel D is a 3.5X enlargement of the region in the white box in panel C. Scale bars: 5 µm for panels A-C and E-F; 1 µm for panel D. The image in panel A is the same image used in Figure 3A but includes the overlay of the F-actin channel. Please click here to view a larger version of this figure.
Figure 5: Examples of poor quality STED images due to overexpression or insufficient expression of the GFP fusion protein. A20 cells expressing CLIP-170-GFP were allowed to spread on anti-IgG-coated coverslips for 15 min before being fixed and stained as in Figure 4. (A, B) CLIP-170-GFP overexpression results in large, abnormal aggregates of CLIP-170-GFP (A) and impaired MTOC polarization towards the antigen contact site (B). (C) Compensating for insufficient expression of CLIP-170-GFP by increasing the laser power results in poor quality STED images. Scale bars: 5 µm. Please click here to view a larger version of this figure.
Figure 6: STED image of the microtubule cytoskeleton in primary B cells. Primary splenic B cells were cultured for 6 h with 5 ng/µL BAFF plus 2.5 µg/mL LPS, and then allowed to spread for 15 min on coverslips that had been coated with anti-IgM antibodies. The cells were then fixed and stained with an α-tubulin antibody plus an Alexa Fluor 568-conjugated secondary antibody to visualize microtubules. A representative image is shown. Scale bar: 5 µm. Please click here to view a larger version of this figure.
 Movie 1: 3D reconstruction of the B cell microtubule network. A20 cells were allowed to spread on anti-IgG-coated coverslips for 15 min before being fixed and immunostained with an α-tubulin antibody plus an Alexa Fluor 488-conjugated secondary antibody. Z-slices were captured at 0.2 µm step sizes for a total of 37 frames. 3D reconstruction was done using the STED microscope's imaging software. Please click here to view this video. (Right-click to download.)
Movie 1: 3D reconstruction of the B cell microtubule network. A20 cells were allowed to spread on anti-IgG-coated coverslips for 15 min before being fixed and immunostained with an α-tubulin antibody plus an Alexa Fluor 488-conjugated secondary antibody. Z-slices were captured at 0.2 µm step sizes for a total of 37 frames. 3D reconstruction was done using the STED microscope's imaging software. Please click here to view this video. (Right-click to download.)
Discussion
Detailed images of cytoskeletal structures can be obtained using STED super-resolution microscopy, which can theoretically achieve a resolution of 50 nm, compared to conventional confocal microscopy, for which the diffraction-limited resolution is ~200 nm24. The ability to resolve finer structures is further enhanced by using deconvolution software to calculate the most likely position of the original light source from the observed "blurred" fluorescence signal. This protocol describes methods for using STED to image the actin and microtubule cytoskeletons, as well as cytoskeleton-associated proteins.
B cell activation induces remodeling of both the actin cytoskeleton and the microtubule network, with the coordinated regulation of the two cytoskeletons being important for immune synapse formation17,42. The method we present has been optimized for simultaneously imaging the actin and microtubule cytoskeletons at the antigen contact site using multi-color STED, but is equally applicable for single-color STED. Previous studies we conducted on the B cell cytoskeleton highlight that STED can provide new insights into how cellular structures are organized and how they interact with each other. Using confocal and total internal fluorescence (TIRF) microscopy, we had observed that microtubules contact the ring of F-actin at the periphery of the antigen contact site17. Using STED microscopy, we were able to show that the plus ends of microtubules that were marked by the +TIP CLIP-170 associate closely with the dendritic actin network at the cell periphery (see Figure 4).
Multiple factors influence which imaging technique is most suitable for one's specific application. These include the resolution that is required, the structures to be imaged, the labeling technique and its signal-to-noise ratio (i.e., contrast), acquisition time, ease of sample preparation, and reproducibility. Sample preparation for STED is not significantly different than for confocal microscopy, and it combines high resolution with rapid image acquisition. A major advantage of STED is that it is an optical process in which the image is directly acquired from the sample and the resolution can be adjusted by changing the power of the STED laser24. Unlike ground state depletion super-resolution microscopy, which reconstructs images from thousands of successive image captures, extensive computational processing is not required for STED, and the introduction of image reconstruction artifacts is avoided24. However, the contrast in STED images is often low24, in which case post-image processing using software such as ImageJ may be needed to enhance the contrast. This is particularly important for images with dense structures, such as a dendritic actin network. To improve image contrast during image acquisition, one can reduce the depletion laser power and/or apply line or frame averaging. Time-gated STED, which captures photons after a user-set time delay, can increase the resolution by decreasing the area from which photons are collected24,43. We recommend optimizing the STED imaging of cytoskeletal structures at the immune synapse by using a combination of these methods to improve contrast and resolution.
Currently, not all fluorophores are optimal for imaging with STED, and not all fluorophore combinations are suitable for obtaining multi-color STED images. Careful adjustment of the detection ranges is important for ensuring minimal bleed-through of fluorophores into adjacent channels. The combination of fluorophores used in this protocol (i.e., GFP, Alexa Fluor 532, and Alexa Fluor 568) is optimal for multicolor STED super-resolution imaging. Compared to structured illumination microscopy (SIM) and single-molecule localization methods (SMLM), such as photo-activated localization microscopy (PALM), STED is not typically ideal for multi-color imaging. However, we show here that slight over-saturation of fluorophore detection, paired with simple image processing tools, can deliver high-resolution multi-color images of the actin and microtubule cytoskeletons.
This protocol for STED imaging of cytoskeletal structures has revealed new details of cytoskeletal architecture at the B cell immune synapse. Although we optimized this protocol for imaging the actin and microtubule cytoskeletons at the antigen-contact site in B cells, these methods should be applicable to other cell types, especially immune cells (T cells, NK cells, mast cells, etc.) that form immune synapses. Moreover, the utility of this protocol could be extended to coating the coverslips with other ligands or adhesive substrates. However, it is important to optimize the protocol and the image acquisition settings for the cell type and the experimental set-up.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the UBC Life Sciences Institute (LSI) Imaging Facility for supporting and maintaining the STED microscope. This work was funded by grant #68865 from the Canadian Institutes of Health Research (to M.R.G.). We thank Dr. Kozo Kaibuchi (Nagoya University, Nagoya, Japan) for the CLIP-170-GFP plasmid.
References
- Harwood NE, Batista FD. The cytoskeleton coordinates the early events of B-cell activation. Cold Spring Harb Perspect Biol. 2011;3(2):a002360. doi: 10.1101/cshperspect.a002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista FD, Treanor B, Harwood NE. Visualizing a role for the actin cytoskeleton in the regulation of B-cell activation. Immunol Rev. 2010;237(1):191–204. doi: 10.1111/j.1600-065X.2010.00943.x. [DOI] [PubMed] [Google Scholar]
- Harwood NE, Batista FD. Early events in B cell activation. Annu Rev Immunol. 2010;28:185–210. doi: 10.1146/annurev-immunol-030409-101216. [DOI] [PubMed] [Google Scholar]
- Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. 2009;9(1):15–27. doi: 10.1038/nri2454. [DOI] [PubMed] [Google Scholar]
- Kumari S, et al. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta. 2014;1838(2):546–556. doi: 10.1016/j.bbamem.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML. Visualization of Cell-Cell Interaction Contacts: Synapses and Kinapses. Self Nonself. 2011;2(2):85–97. doi: 10.4161/self.2.2.17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Chakraborty AK, Shaw AS. Understanding the Structure and Function of the Immunological Synapse. Cold Spring Harb Perspect Biol. 2010;2(10):a002311. doi: 10.1101/cshperspect.a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML. Modular design of immunological synapses and kinapses. Cold Spring Harb Perspect Biol. 2009;1(1):a002873. doi: 10.1101/cshperspect.a002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML. Visualization of cell-cell interaction contacts-synapses and kinapses. Adv Exp Med Biol. 2008;640:164–182. doi: 10.1007/978-0-387-09789-3_13. [DOI] [PubMed] [Google Scholar]
- Dustin ML. Hunter to gatherer and back: immunological synapses and kinapses as variations on the theme of amoeboid locomotion. Annu Rev Cell Dev Biol. 2008;24:577–596. doi: 10.1146/annurev.cellbio.24.110707.175226. [DOI] [PubMed] [Google Scholar]
- Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285(5425):221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- Monks CR, et al. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395(6697):82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- Yuseff MI, et al. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat Rev Immunol. 2013;13(7):475–486. doi: 10.1038/nri3469. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Batista FD, Treanor B. Dynamics of the actin cytoskeleton mediates receptor cross talk: An emerging concept in tuning receptor signaling. J Cell Biol. 2016;212(3):267–280. doi: 10.1083/jcb.201504137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn JR, Poenie M. Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity. 2002;16(1):111–121. doi: 10.1016/s1074-7613(02)00262-5. [DOI] [PubMed] [Google Scholar]
- Wang JC, et al. The Rap1-cofilin-1 pathway coordinates actin reorganization and MTOC polarization at the B cell immune synapse. J Cell Sci. 2017;130(6):1094–1109. doi: 10.1242/jcs.191858. [DOI] [PubMed] [Google Scholar]
- Schnyder T, et al. B cell receptor-mediated antigen gathering requires ubiquitin ligase Cbl and adaptors Grb2 and Dok-3 to recruit dynein to the signaling microcluster. Immunity. 2011;34(6):905–918. doi: 10.1016/j.immuni.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Nath S, et al. Dynein Separately Partners with NDE1 and Dynactin To Orchestrate T Cell Focused Secretion. J Immunol. 2016;197(6):2090–2101. doi: 10.4049/jimmunol.1600180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs J, et al. Recruitment of dynein to the Jurkat immunological synapse. Proc Natl Acad Sci U S A. 2006;103(40):14883–14888. doi: 10.1073/pnas.0600914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16(12):711–726. doi: 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO. Microtubule +TIPs at a glance. J Cell Sci. 2010;123(20):3415–3419. doi: 10.1242/jcs.062414. [DOI] [PubMed] [Google Scholar]
- Fukata M, et al. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell. 2002;109(7):873–885. doi: 10.1016/s0092-8674(02)00800-0. [DOI] [PubMed] [Google Scholar]
- Wegel E, et al. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Sci Rep. 2016;6:27290. doi: 10.1038/srep27290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu Rev Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace EM, Orange JS. Dual channel STED nanoscopy of lytic granules on actin filaments in natural killer cells. Commun Integr Biol. 2012;5(2):184–186. doi: 10.4161/cib.18818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter AT, et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity. 2015;42(5):864–876. doi: 10.1016/j.immuni.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarit B, et al. Membrane microdomains and cytoskeleton organization shape and regulate the IL-7 receptor signalosome in human CD4 T-cells. J Biol Chem. 2013;288(12):8691–8701. doi: 10.1074/jbc.M113.449918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AC, et al. Super-resolution imaging of remodeled synaptic actin reveals different synergies between NK cell receptors and integrins. Blood. 2012;120(18):3729–3740. doi: 10.1182/blood-2012-05-429977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre L, et al. T Lymphocyte Migration: An Action Movie Starring the Actin and Associated Actors. Front Immunol. 2015;6:586. doi: 10.3389/fimmu.2015.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak GD, et al. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 2011;9(9):e1001151. doi: 10.1371/journal.pbio.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KB, et al. The Rap GTPases regulate the migration, invasiveness and in vivo dissemination of B-cell lymphomas. Oncogene. 2010;29(4):608–615. doi: 10.1038/onc.2009.345. [DOI] [PubMed] [Google Scholar]
- Lin KB, et al. The rap GTPases regulate B cell morphology, immune-synapse formation, and signaling by particulate B cell receptor ligands. Immunity. 2008;28(1):75–87. doi: 10.1016/j.immuni.2007.11.019. [DOI] [PubMed] [Google Scholar]
- McLeod SJ, et al. The Rap GTPases regulate integrin-mediated adhesion, cell spreading, actin polymerization, and Pyk2 tyrosine phosphorylation in B lymphocytes. J. Biol. Chem. 2004;279(13):12009–12019. doi: 10.1074/jbc.M313098200. [DOI] [PubMed] [Google Scholar]
- Christian SL, et al. Activation of the Rap GTPases in B lymphocytes modulates B cell antigen receptor-induced activation of Akt but has no effect on MAPK activation. J Biol Chem. 2003;278(43):41756–41767. doi: 10.1074/jbc.M303180200. [DOI] [PubMed] [Google Scholar]
- Batista FD, Iber D, Neuberger MS. B cells acquire antigen from target cells after synapse formation. Nature. 2001;411(6836):489–494. doi: 10.1038/35078099. [DOI] [PubMed] [Google Scholar]
- Treanor B, et al. Dynamic cortical actin remodeling by ERM proteins controls BCR microcluster organization and integrity. J Exp Med. 2011;208(5):1055–1068. doi: 10.1084/jem.20101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, et al. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity. 2013;38(3):461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Yuseff M-I, et al. Polarized Secretion of Lysosomes at the B Cell Synapse Couples Antigen Extraction to Processing and Presentation. Immunity. 2011;35(3):361–374. doi: 10.1016/j.immuni.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Mace EM, Orange JS. Visualization of the immunological synapse by dual color time-gated stimulated emission depletion (STED) nanoscopy. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]
- Russ JC. The Image Processing Handbook, Sixth Edition. CRC Press; 2011. [Google Scholar]
- Stinchcombe JC, et al. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443(7110):462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- Vicidomini G, et al. Sharper low-power STED nanoscopy by time gating. Nat Methods. 2011;8(7):571–573. doi: 10.1038/nmeth.1624. [DOI] [PubMed] [Google Scholar]