

Abstract
A highly efficient asymmetric synthesis of the key tetrahydropyranol intermediate of DPP-4 inhibitor omarigliptin (1) is described. The successful development of a protecting-group- and precious-metal-free synthesis was achieved via the discovery of a practical asymmetric Henry reaction and the application of a one-pot nitro-Michael–lactolization–dehydration through-process. Other features of the synthesis include a highly efficient MsCl-mediated dehydration and a crystallization-induced dynamic resolution for exceptional ee and dr upgrade. The synthesis of this complex intermediate utilizes simple starting materials and proceeds in four linear steps.
Graphical abstract
INTRODUCTION
Diabetes is a fast-growing, worldwide epidemic known to affect over 415 million people in 2015 and is projected to grow to 642 million patients in 2040. It is estimated that approximately 5 million people died from diabetes and its complications in 2015 alone, representing one of the leading causes of death globally.1,2 Recently, new effective treatment choices have become available for type 2 diabetes patients, including agents such as dipeptidyl peptidase-4 (DPP-4) inhibitors, GLP-1 analogues, and SGLT2 inhibitors.3 Januvia (sitagliptin), a first-in-class DPP-4 inhibitor, taken once-a-day, was approved in 2006.4 Given the clinical success of DPP-4 inhibitors, there has been ongoing interest in the development of drug candidates with longer half-lives that decrease dosing frequency.5 Such therapies would provide advantages to the patient of improved medication compliance and minimized side effects. In this vein, omarigliptin (1, MK-3102), a highly functionalized long-acting DPP-4 inhibitor, was developed and recently approved for the once-weekly treatment of type 2 diabetes in Japan (Figure 1).6
Figure 1.
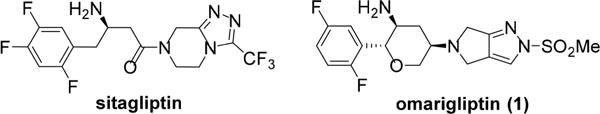
Structures of sitagliptin and omarigliptin (1).
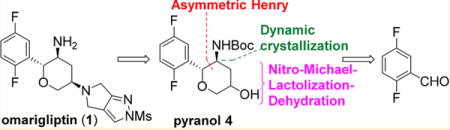
The existing synthesis of omarigliptin is shown in Scheme 1.7 Starting from inexpensive glycine benzophenone imine (9), the current manufacturing route is a highly stereocontrolled synthesis and amenable to produce hundreds of kilograms of 1 with good overall yield.8 While this synthesis is practical and efficient, we identified several specific opportunities for improvement. Specifically, a precious-metal-free process with minimal protecting groups was expected to result in a more sustainable long-term synthesis.9 We targeted pyranol 4, which is an accepted active pharmaceutical ingredient starting material for Japan, and restricted the new approach to include the hydroboration of dihydropyan in order to maintain the alcohol diastereomeric ratio as specified in the regulatory filing. Thus, with this constraint, an alternative approach to pyranol 4 would begin with an asymmetric Henry reaction between benzaldehyde 15 and nitromethane (Scheme 2). We envisioned that the nitro aldol product 14 could undergo a tandem nitro-Michael/lactolization/dehydration with acrolein to afford dihydropyran 12. Subsequent hydroboration/oxidation and nitro reduction/Boc protection should complete the synthesis of pyranol 4. Reducing this approach to practice would result in a more cost-effective and sustainable approach to 4. Herein, we describe a highly efficient alternative pyranol 4 synthesis that features a highly stereoselective Henry reaction and a unique and effective nitro-Michael–lactolization–dehydration sequence.10
Scheme 1.
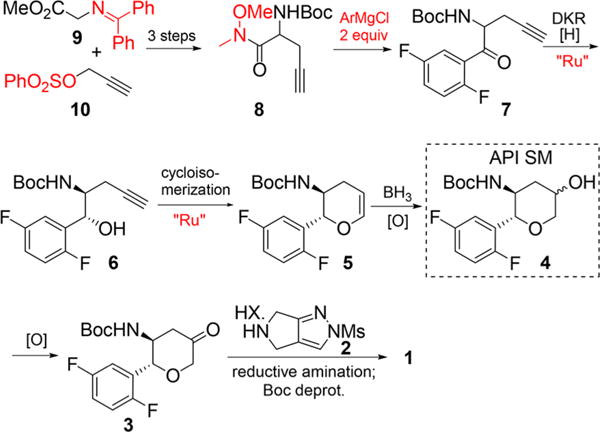
Manufacturing Route of 1
Scheme 2.
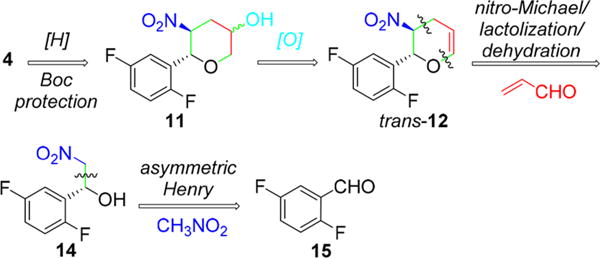
Retrosynthetic Analysis of Pyranol 4
RESULTS AND DISCUSSION
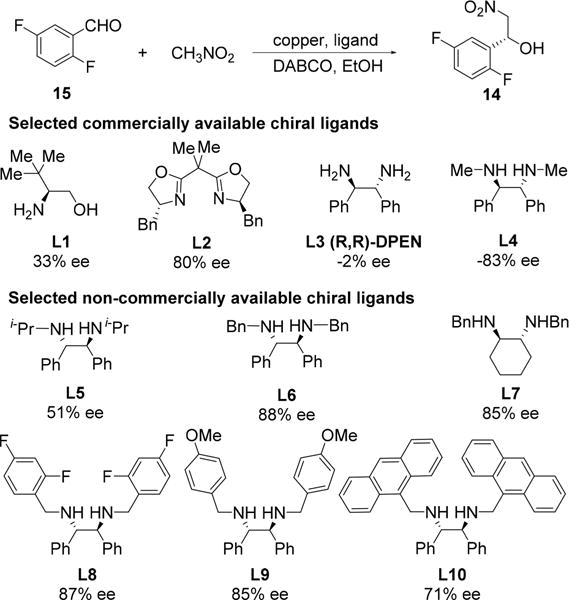
Despite its longstanding recognition as an atom-economical, green approach to chiral 1,2-aminoalcohols, application of the asymmetric Henry reaction to large scale synthesis has remained rare.11 This paucity is likely attributable to the requirement for a high catalyst loading to effect the desired transformation, and/or limited availability of certain chiral ligands.12 Given these known challenges, we focused our initial efforts on identifying a practical ligand for the asymmetric Henry reaction. Using 2,5-difluorobenzaldehyde (15) as substrate, a variety of commercially available chiral ligands were surveyed utilizing high-throughput experimentation tools and techniques.13 Cu(OAc)2, in combination with these ligands, was found to cleanly catalyze formation of the desired arylnitroethanol 14 under mild conditions using ethanol as solvent and DABCO as base (Scheme 3, L1–L4). Diamine L4 was identified as a promising ligand; using (S,S)-L4/Cu(OAc)2 as catalyst, the desired arylnitroethanol intermediate 14 was prepared in 83% ee and 85–90% yields.14
Scheme 3.
Ligand Screening Results
Encouraged by these results with ligand L4, we prepared a series of diamines and evaluated these ligands in our asymmetric Henry reaction (Scheme 3, L5–L10). To our delight, we found that arylnitroethanol 14 could be accessed in enantiomeric excesses as high as 88% with diamine L6 as ligand, which is derived from N,N′-dibenzylation of (S,S)-DPEN. To the best of our knolwedge, this ligand has not been used in an asymmetric Henry reaction to date.
Having identified an efficient ligand, we sought to optimize the reaction conditions to provide the best reactivity and selectivity. A survey of bases revealed that catalytic amounts of DABCO or dimethylpiperazine led to significantly improved reaction rates compared to the reaction without a base and without altering the product enantioselectivity. Extensive optimization studies were then carried out on the copper salt, solvent, concentration, base amount, reagent stoichiometry, catalyst loading, reaction temperature, and mixing mode. The optimized conditions used 0.4 mol % each of CuCl2 and L6, 8 mol % dimethylpiperazine, 5 equiv of nitromethane, and 3 vol EtOH at −16 °C for 15 h, which afforded 14 in 92% yield and 93% ee (Scheme 4). These conditions were successfully demonstrated on kilogram scale.15
Scheme 4.

Optimized Asymmetric Henry Reaction
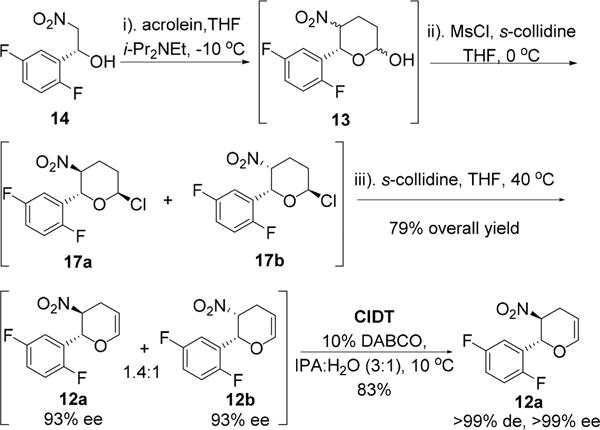
With a reliable synthesis of 14 in hand, we surveyed reaction conditions for the proposed one-pot through-process to dihydropyran 12 by coupling 14 with acrolein (Scheme 5). Our initial nitro-Michael–lactolization sequence reaction conditions were based on literature precedent that utilized a secondary amine (Et2NH or diphenylprolinol trimethylsilyl ether) and acid (formic acid, pivalic acid or boric acid).16 Reaction rates using these conditions were very slow, requiring 4–5 days and 2 equiv of acrolein to reach 95% conversion to lactol 13 in variable yields (60–80%). The major impurities observed were higher molecular weight impurities and aldol byproducts. We hypothesized that the secondary amines may be responsible for promoting acrolein polymerization via iminium activation. Indeed, when the reaction was instead carried out using N,N-diisopropylethylamine (Hünig’s base), a tertiary amine, we observed a faster (10 h) and cleaner Michael–lactolization process (Scheme 5, step (i)). Control studies confirmed the excellent stability of lactol 13 in the presence of Hünig’s base, and the competitive retro-Henry reaction of starting nitroethanol 14 and polymerization of acrolein were both observed to be negligible.17 After optimization of the stoichiometry of base, temperature, concentration, and addition mode, we arrived at a set of simple reaction conditions involving slow addition of 1.15 equiv acrolein to nitroethanol 14 with 1.0 equiv of Hünig’s base in THF at −10 °C to afford lactol 13 in 92% reaction yield (Scheme 5, step (i)).18
Scheme 5.
One-Pot Through-Process from 14 to 12
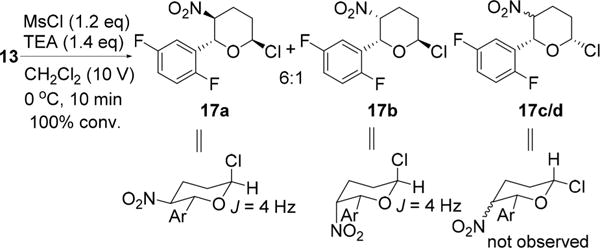
With lactol 13 in hand, we then surveyed reaction conditions for the desired dehydration process. Initial efforts for direct dehydration of 13 were largely unsuccessful, affording a mixture of trans and cis nitro-olefins 12a/b in less than 40% isolated yield.19 Subsequently, we found, to our delight, axial chlorides 17a/b could be prepared cleanly by treatment of 13 with 1 equiv each of MsCl and Et3N as revealed by proton NMR spectroscopic analyses of the reaction mixture (Scheme 6),20 where a small coupling constant (4 Hz) was observed for the anomeric hydrogen. The equatorial chloro anomers 17c/d were not observed. Free energy calculations confirmed a large thermodynamic preference for axial chlorides over the corresponding equatorial chlorides (Table 1). These results are also consistent with the outcomes obtained by NMR and computational methods reported for 2-chlorotetra-hydropyrans.21 The axial chorides are highly suited for E2 dehydrochlorination and should afford a high yield of dihydropyrans.
Scheme 6.
Axial Chlorides Formation
Table 1.
Calculated Relative Free Energies (kcal/mol) of 17
| compound 17
|
M06/6-31G**
|
M062X/6-31G*
|
B3LYP/6-31G**w/D2
|
M062X/6-31+G**
|
||
|---|---|---|---|---|---|---|
| −NO2 | −Cl | ΔG | ΔG | ΔG | ΔG | |
| 17a | equa | axial | 0.0 | 0.0 | 0.0 | 0.0 |
| 17b | axial | axial | 2.5 | 2.4 | 2.5 | 2.7 |
| 17c | equa | equa | 3.5 | 2.6 | 3.1 | 2.6 |
| 17d | axial | equa | 7.7 | 7.0 | 7.5 | 7.3 |
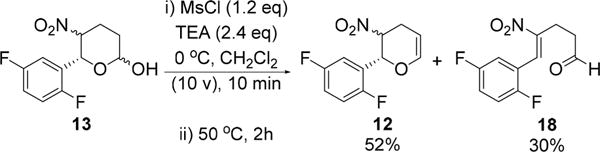
With clean formation of 17 in hand, successful development of a dehydrochlorination process completes the synthesis of nitro-olefins 12 from lactol 13. We identified that the choice of base was critical for dehydrochlorination. For example, when the chlorination reaction mixture was treated with additional Et3N at elevated temperature, a mixture of the desired nitro-olefin 12a/b and a significant amount of aldehyde byproduct 18 were obtained (Scheme 7). Formation of this byproduct is most likely initiated by the α-deprotonation of the nitro compounds 17a/b. We anticipated that use of a weak base should prevent the formation of undesired aldehyde by avoiding this competitive α-deprotonation. Indeed, after screening six mild bases (Table 2, entries 2–7), we were delighted to identify 2,4,6-trimethylpyridine (s-collidine) (pKa ≈ 7) as the optimal base, and the resultant nitro-olefins 12 were obtained in 84% yield (Table 2, entry 3). Furthermore, s-collidine was also demonstrated to be an effective base for the chlorination reaction. Several solvents, including MeCN, MTBE, and THF, provided good results for dehydrochlorination (Table 2, entries 8–10). Therefore, by using THF as a single solvent, and s-collidine for both chlorination and dehydrochlorination, we were able to develop a one-pot through-process that prepared dihydropyrans 12a/b from 14 in 79% overall yield on a 100 g scale (Scheme 5).
Scheme 7.
Dehydration via Chloride Intermediate
Table 2.
Dehydrochlorination Optimization

| |||
|---|---|---|---|
|
| |||
| entry | solvent | 2nd base | assay yield |
| 1 | CH2Cl2 | TEA | 50% |
| 2 | CH2Cl2 | pyridine | 57% |
| 3 | CH2Cl2 | 2,4,6-collidine | 84% |
| 4 | CH2Cl2 | 2,6-lutidine | 80% |
| 5 | CH2Cl2 | Hünig base | 58% |
| 6 | CH2Cl2 | N-methyl morpholine | 72% |
| 7 | CH2Cl2 | dimethylaniline | 69% |
| 8 | MeCN | 2,4,6-collidine | 97% |
| 9 | MTBE | 2,4,6-collidine | 87% |
| 10 | THF | 2,4,6-collidine | 92% |
| 11 | IPAC | 2,4,6-collidine | 50% |
| 12 | 2-MeTHF | 2,4,6-collidine | 75% |
We anticipated that the mixture of diastereomers 12a/b could be converged to a single diastereomer 12a by a base-promoted crystallization-induced diastereomer transformation (CIDT).22,23 After surveying 14 bases24 for the proposed dynamic crystallization, we found that a superior epimerization rate and optimal product stability could be achieved with use of DABCO as base. By careful optimization of the crystallization process, we arrived at conditions that employed 10 mol % DABCO in 3:1 IPA/H2O at 10 °C. This process ultimately afforded 12a in 83% yield, >99% ee, and >99% de from a mixture of 1.4:1 trans/cis 12a/b (93% ee).
The remaining two steps required were hydroboration of the newly formed olefin and subsequent reduction and Boc protection of the nitro group. Using BH3·SMe2 at 0–5 °C, hydroboration of 12a proceeded smoothly within 4 h. After a simple oxidative work up with Na2CO3·1.5(H2O2),25 alcohol 11 was obtained in 94% yield and 99% ee as a 58:42 diastereomeric mixture at the alcohol center (Scheme 8), which is similar to the ratio obtained from the commercial route.
Scheme 8.
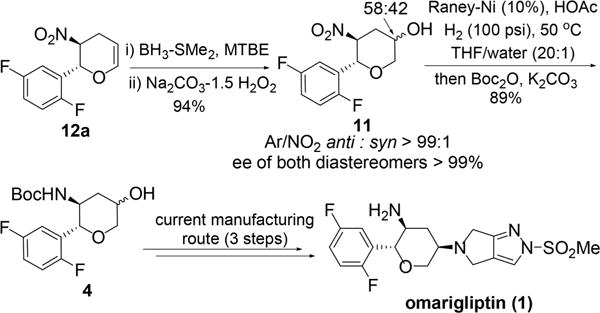
Completion of the Synthesis
Both precious metal and base metal catalysts have been utilized for nitro reductions. For process sustainability considerations, we began surveying the reduction of 11 with a base metal catalyst, Raney-Ni. Our preliminary results indicated that Raney-Ni-catalyzed hydrogenation was very sensitive to the pH of the reaction. The hydrogenation proceeded very slowly under low pH conditions (pH < 2), and a significant amount of the cis-isomer was observed under higher pH conditions (pH > 5) due to epimerization of the nitro group-bearing stereocenter. After examining a variety of additives, we found that optimal results were obtained with acetic acid (pKa ≈ 4.7) as an additive. Therefore, in the presence of 1 equiv of acetic acid in THF, the desired reduction product was prepared in 92% assay yield with 3.7% of the undesired cis-isomer observed. After completion of the hydrogenation and removal of the catalyst, the reaction solution was directly subjected to Boc protection, affording the target product 4 in 96% assay yield. After crystallization from aqueous ethanol, the purity of 4 was upgraded to 99.6% HPLC area percent.26
In summary, a concise, enantioselective synthetic route to the tetrahydropyranol intermediate (4) of omarigliptin (1) has been achieved. The route features a practical, highly enantioselective asymmetric Henry reaction and a CIDT process to establish two contiguous stereogenic centers. Other features of the route include a one-pot nitro-Michael/lactolization/dehydration through-process to assemble the tetrahydropyran ring. The new synthesis requires only four chemical steps for completion and proceeds in 50% overall yield without the need for chromatography.
EXPERIMENTAL SECTION
General Methods
All reagents and solvents were obtained from commercial sources and used as received. All reactions were performed under an inert atmosphere with unpurified reagents and dry solvents. Analytical thin-layer chromatography (TLC) was performed using 0.25 mm silica gel coated plates. Flash chromatography was performed using the indicated solvent and silica gel 60 (230–400 mesh). NMR and NOE spectra were recorded at ambient temperature using 400, 500, and 600 MHz spectrometers. Chemical shifts are reported in parts per million (ppm) scaled from an internal standard. High resolution mass spectra were recorded in ESI mode using a magnetic sector mass analyzer and TOF mass spectrometer.
Preparation of (R)-1-(2,5-Difluorophenyl)-2-nitroethan-1-ol (14)
To a vessel was charged 0.4 mol % (1S,2S)-N1,N2-dibenzyl-1,2-diphenylethane-1,2-diamine (14.03 g, 35.7 mmol), 3.5 vol EtOH (4445 mL; KF 0.03%), and 0.4 mol % CuCl2 (4.81 g, 35.7 mmol). The mixture was stirred at 20–25 °C for 1 h to afford a homogeneous blue solution. To the solution was added 1.0 equiv of 2,5-difluoro-benzaldehyde (1.27 kg, 8.938 mol; contained 0.25% 2,5-difluoro-benzoic acid, KF 0.03%) in one portion with mild exotherm (about 2 °C) followed by 0.08 equiv of N,N′-dimethylpiperizine (82 g, 715 mmol). The resulting mixture was cooled to −16 °C. CH3NO2 (5 equiv, 2728 g, 44.692 mol; KF 0.15%) at 0 °C was slowly added over 90 min with a mild exotherm (about 2 °C). After the mixture stirred at −16 °C for 15 h, HPLC analysis (sample quenched with HOAc) showed 99.1% conversion and 93% ee (sample quenched with 5% EDTA·2Na·2H2O aqueous solution). MTBE (9.4 kg) at 0 °C was slowly added while maintaining the internal temperature at <5 °C. A 5% EDTA·2Na·2H2O aqueous solution (6 kg) was slowly added while keeping the internal temperature at <5 °C, followed by H2O (2.6 kg). The mixture was warmed to 20 °C and stirred for 1 h. The two layers were separated. The organic phase was analyzed for N,N′-dimethyl-piperazine (0.007% vs target ≤0.01%), residual Cu (<10 ppm vs target ≤10 ppm). The organic phase was washed with 10% NaCl aqueous solution (2 × 6.5 kg). Analysis showed the product in the combined aqueous layers was 15 g (about 0.1% loss). The organic phase was concentrated under reduced pressure at <40 °C, which was then azeotropically distilled with a total of 6.6 kg of toluene under the same conditions to a final volume of 2.6 L. The solution was analyzed for residual solvents: nitromethane was 0.06% (vs target ≤0.10%), EtOH was 0.04% (vs target ≤0.10%), and water was 0.06%. THF (2.2 kg) was added and concentrated to a final volume of 3 L at <40 °C. The residual toluene was 11.7% (vs target of 20%), CH3NO2 was 0.02%, MTBE was N.D., EtOH was N.D., and KF was 0.06%. The assay yield of the desired product was 1.68 kg (2.78 kg × 60.3 wt %) for a 92% corrected yield with 93.0% ee and a purity of 94.1% by HPLC. An analytically pure sample was obtained by flash chromatography (5 to 10% MTBE/hexanes); 14: 1H NMR (CDCl3, 500 MHz) δ 7.31 (dddt, 1H, J = 8.5, 5.5, 2.9, 0.6 Hz), 7.09–7.01 (om, 2H), 5.73 (ddd, 1H, J = 8.7, 4.0, 2.4 Hz), 4.64 (dd, 1H, J = 13.8, 2.7 Hz), 4.54 (dd, 1H, J = 13.8, 9.4 Hz), 3.09 (d, 1H, J = 4.6 Hz); 19F NMR (CDCl3, 471 MHz) δ −116.8 (d, J = 17.9 Hz), −124.2 (d, J = 17.9 Hz); 13C NMR (CDCl3, 125 MHz) δ 159.3 (dd, J = 243.9, 2.3 Hz), 155.3 (dd, J = 242.1, 2.6 Hz), 127.1 (dd, J = 16.1, 7.5 Hz), 117.1 (dd, J = 24.2, 1.7 Hz), 116.9 (dd, J = 24.3, 2.1 Hz), 114.6 (dd, J = 25.7, 4.1 Hz), 79.5 (d, J = 1.7 Hz), 65.2 (m); HRMS (ESI-TOF) m/z: [M – H]− calcd for C8H6F2NO3 202.0316; found 202.0316.
Preparation of (1S,2S)-N1,N2-Dibenzyl-1,2-diphenylethane-1,2-diamine (L6)
(1S,2S)-1,2-Diphenylethane-1,2-diamine (30 g, 141 mmol) and dichloromethane (450 mL) was stirred at 25 °C for 1 h until the solid dissolved to give a solution and then cooled to 0 °C. Benzaldehyde (31.5 g, 297 mmol) was added, followed by NaBH-(OAc)3 (90 g, 424 mmol), while being maintained at 0–5 °C. To the resulting mixture was slowly added acetic acid (25.5 g, 424 mmol) while being maintained at <7 °C. The mixture was then warmed to 20–25 °C and aged for 30 h. The mixture was washed with saturated NaHCO3 (3 × 200 mL) to pH 7. The ogranic phase was washed with water (1 × 200 mL) and 25% NaCl (1 × 100 mL), then concentrated. The residue was purified by flash chromatography (10 to 50% EtOAc/n-heptane). The desired fractions were concentrated and the product was crystallized from n-heptane (10 vol) at −10 °C to afford 36.1 g L6 (65% yield). The purity by HPLC was 99.5% and >99.8% ee. L6: 1H NMR (CD3CN, 500 MHz) δ 7.26–7.22 (m, 10H), 7.08–7.16 (m, 10 H), 3.75 (s, 2H), 3.58 (d, 2H, J = 13.4 Hz), 3.45 (d, 2H, J = 13.4 Hz), 2.69 (s, 2H, NH); 13C NMR (CD3CN, 125 MHz,) δ 143.2, 142.5, 129.7, 129.6, 129.5, 129.2, 128.1, 128.0, 69.6, 52.2; HRMS (ESI-TOF) [M + H]+ calcd for C28H28N2 393.2330; found 393.2332.
Preparation of (2R)-2-(2,5-Difluorophenyl)-3-nitro-3,4-dihy-dro-2H-pyran (12a/12b)
To a vessel were charged a solution of 14 (119.1 g × 84 wt % = 100 g, 0.492 mol) in THF/toluene, and THF (200 mL). The solution was cooled to −15 to −10 °C, and then i-Pr2NEt (63.6 g, 0.492 mol) was added over 30 min while maintaing the internal temperature at −15 to −10 °C, followed by 90.4 wt % acrolein (38.8 g, 0.626 mol) over 30 min while maintaing the internal temperature at −15 to −10 °C. After stirring for 18 h at −10 °C, HPLC assay showed 98.3% conversion and 93.7% ee at the benzylic center of 13. S-Collidine (41.7 g, 0.344 mol) was added at < −10 °C, followed by addition of mesyl chloride (90 g, 0.786 mol) over 1 h at −15 to −10 °C. After the mixture stirred for 4 h, HPLC assay showed 99% conversion; THF (1 L), followed by s-collidine (101.4 g, 0.836 mol), was added. The mixture was warmed to 40 °C. After the mixture stirred for 24 h at 40 °C, HPLC showed 96.5% conversion and 93.5% ee at the benzylic center of 12. The mixture was cooled to 15–25 °C; MTBE (740 g), followed by 5 wt % citric acid (500 g), was added (final pH 5–6). The layers were separated. The organic layer was assayed for 12a/12b (5.12 wt % = >80% assay yield), residual Et3N (0.01%), and s-collidine (0.39%). Product loss to aqueous phase was about 0.01%. The organic phase was washed with 5% NaHCO3 solution (400 g) (no loss to the aqueous) and then solvent-switched to MTBE until 3 vol (total 2360 g of MTBE were used). MTBE (1.2 L), n-heptane (0.5 L), and Aquaguard (20 g) were added to the solution, the and mixture was stirred for 18 h at rt. The mixture was filtered through kieselguhr (50 g) and washed with MTBE (200 mL × 2). Product 12 loss to the cake was about 1.7%. The filtrate contained 94.6 g (1521 g × 6.22 wt %) of 12a/12b (79% yield) with 93.9% ee and a purity of 89.5% by HPLC. Analytical samples were obtained by flash chromatography (5 to 100% EtOAc/hexanes). 12a: solid, mp 59–61 °C; 1H NMR (500 MHz, CDCl3) δ 7.18–7.12 (m, 1H), 7.08 (ddd, J = 8.6, 5.9, 1.9 Hz, 2H), 6.57 (dt, J = 6.1, 2.0 Hz, 1H), 5.56 (d, J = 7.3 Hz, 1H), 5.00 (td, J = 7.6, 5.6 Hz, 1H), 4.94 (ddd, J = 6.1, 4.4, 3.4 Hz, 1H), 3.00–2.88 (m, 1H), 2.58 (dddd, J = 17.1, 6.1, 4.5, 1.9 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 158.8 (d, JCF = 244 Hz), 156.1 (d, JCF = 244 Hz), 143.4, 124.7, 117.6, 117.3, 115.3, 97.6, 81.6, 71.6, 24.6; HRMS (ESI−): calcd for C11H8F2NO3 [M – H] 240.0472, found 240.0473. 12b: oil; 1H NMR (500 MHz, CDCl3) δ 7.22 (ddd, J = 8.9, 5.7, 3.1 Hz, 1H), 7.11–7.00 (m, 2H), 6.74–6.53 (m, 1H), 5.33 (s, 1H), 5.24 (dd, J = 6.5, 1.7 Hz, 1H), 5.03 (ddd, J = 4.9, 3.9, 2.2 Hz, 1H), 2.85–2.68 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 159.2 (d, 1JC–F = 242 Hz), 155.2 (d, 1JC–F = 242 Hz), 144.4, 125.5, 116.7, 116.3, 114.7, 98.7, 81.0, 70.4, 24.7; HRMS (ESI-TOF) m/z: [M – H]− calcd for C11H8F2NO3 240.0472; found 240.0463.
Preparation of (2R,3S)-2-(2,5-Difluorophenyl)-3-nitro-3,4-dihydro-2H-pyran (12a)
To a mixture of crude 12a/12b (19.8 g) in 3 vol MTBE from above was added 0.05 equiv of s-collidine (0.5 g), followed by solvent-switching to IPA (total 240 mL IPA used) at <35 °C under reduced pressure until 60 mL (3 vol). The mixture was stirred at 30–35 °C for 1 h until the solid dissolved, seeded with 12a (80 mg), and stirred for 1 h; the mixture was then cooled to 10 °C and aged for 1 h. A solution of 0.1 equiv of DABCO (0.9 g) in 0.5 vol IPA (10 mL) was added at 10 °C, and then the mixture was stirred for 1 h. H2O (1 vol, 19.8 mL) was slowly added over 3 h at 10 °C, and then the mixture was stirred at 10 °C for 1 h. Afterward, residual 12a/12b in the supernatant was 3.9 wt % (cis/trans = 18%/40%) for a total loss equaling an ∼13% yield. The solid was 99.6% trans. The slurry was filtered, and the cake was washed with 2:1 IPA/H2O (2 × 20 mL) at 10 °C. The wet cake was dried under vacuum at 30 °C for 24 h to afford 16.8 g of 12a in 83% assay yield (16.8 g × 98.0 wt % by 1H NMR) with 99.9% ee and 99.4% purity by HPLC. KF and residual IPA of the dried product were 0.07% and 0.03% respectively. See above for NMR and MS data.
Preparation of (5S,6R)-6-(2,5-Difluorophenyl)-5-nitrotetra-hydro-2H-pyran-3-ol (11)
A mixture of 12a (142.7 g × 91.0 wt % = 129.9 g, solid) and MTBE (962.0 g, KF = 0.06%) was stirred until the solid dissolved and then cooled to 0–2 °C. 10 M Borane dimethyl sulfide complex (108.1 g) was added dropwise over 30 min at 0–4 °C, and then the mixture was stirred at 0–4 °C for 3–4 h. Afterward, the conversion was >99.8%. H2O (93.6 g) was slowly added over 60 min at 0–10 °C. H2O (1014 g) was added over 10 min at 0–10 °C. Sodium percarbonate (178.0 g) was added in three portions over 30 min at 0–8 °C. The mixture was stirred at 3–5 °C for 3–4 h. Afterward, the conversion was >99.8%. The aqueous layer was discarded, and the organic phase was washed with 10% NaHSO3 solution (390 g). The organic layer was checked for peroxide with KI-starch paper; if any was present, it washed again with a 10% NaHSO3 solution. The organic phase was washed with 10% NaCl solution (390 g) leading to nondetected levels of residual DMSO in organic phase. The organic phase was solvent-switched to THF until 1.6X at 35–45 °C under vacuum (total 1740 g THF was used) until the residual MTBE and dimethyl sulfide were not detected. The amount of products 11a/b in the solution was 130.7 g (222 g × 58.9 wt %) (93.5% yield) with 88.8% purity and 98.7% ee. The diastereomers were purified by flash chromatography (20 to 100% EtOAc/hexanes): 11a: 1H NMR (500 MHz, CDCl3) δ 7.12–7.08 (m, 1H), 7.06–7.02 (m, 2H), 4.91 (dd, J = 9.7, 0.9 Hz, 1H), 4.77 (ddd, J = 12.2, 9.7, 4.1 Hz, 1H), 4.18 (ddd, J = 11.0, 5.1, 2.0 Hz, 1H), 4.06 (ddt, J = 11.1, 10.0, 4.9 Hz, 1H), 3.43 (t, J = 11.0, 10.2 Hz, 1H), 2.82 (dtd, J = 12.0, 4.4, 2.0 Hz, 1H), 2.31 (td, J = 12.1, 11.1 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 159.9 (dd, JCF = 244.0, 2.4 Hz), 156.7 (dd, JCF = 245.0, 2.6 Hz), 124.9 (dd, JCF = 15.8, 7.5 Hz), 117.8 (dd, JCF = 24.0, 8.8 Hz), 117.4 (dd, JCF = 25.0, 8.5 Hz), 115.4 (dd, JCF = 25.0, 3.9 Hz), 84.6 (d, JCF = 1.9 Hz), 74.9 (t, JCF = 1.6 Hz), 72.9, 64.5, 38.0; HRMS (ESI−) m/z: [M – H]− calcd for C11H10F2NO4 258.0583; found 258.0580. 11b: 1H NMR (500 MHz, CDCl3) δ 7.22–7.16 (m, 1H), 7.07–7.01 (m, 2H), 5.13 (dddd, J = 12.1, 9.8, 4.3, 0.6 Hz, 1H), 4.96 (dd, J = 9.9, 0.9 Hz, 1H), 4.22 (tt, J = 2.8, 1.3 Hz, 1H), 4.08 (dt, J = 12.6, 2.1 Hz, 1H), 3.87 (dd, J = 12.6, 1.4 Hz, 1H), 2.66 (dddd, J = 13.2, 4.3, 3.3, 2.5 Hz, 1H), 2.46 (ddd, J = 13.3, 12.1, 2.9 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 159.0 (dd, JCF = 243.7, 2.3 Hz), 156.6 (dd, JCF = 245.4, 2.5 Hz), 125.3 (dd, JCF = 15.7, 7.5 Hz), 117.8 (dd, JCF = 24.0, 8.8 Hz), 117.4 (dd, JCF = 25.0, 8.5 Hz), 115.6 (dd, JCF = 25.0, 4.0 Hz), 82.5, 84.6 (d, JCF = 1.5 Hz), 75.2 (t, JCF = 1.4 Hz), 72.9, 64.9, 36.2; HRMS (ESI-TOF) m/z: [M – H]− calcd for C11H10F2NO4 258.0583; found 258.0581.
Preparation of Pyranol 4
A 250 mL autoclave was charged with THF (147 mL), a solution of Raney-Ni (0.73 g, 10 wt %, adjust by 0.5 M acetic acid in THF to pH 7) in water (7.4 mL, 1.0 vol.), acetic acid (1.71 g, 28.39 mmol, 1.0 equiv), and nitropyranol 11 (7.36 g, 28.39 mmol, assay 92%). The mixture was purged with argon three times and then with hydrogen three times. The reaction mixture was stirred at 50 °C under 100 psi of H2 for 24 h. After reaching full conversion, the mixture was filtered. The solution (assay yield ∼94%) was diluted with THF to 250 mL. K2CO3 (7.2 g, 52 mmol, 2 equiv) was added in 7 portions over 30 min while being maintained at 0–10 °C. (Boc)2O (6.3 g, 29.0 mmol, 1.1 equiv) was then charged in one portion to the reaction mixture at 0–10 °C. After the mixture stirred at 25 °C for 18 h, full conversion was observed. The reaction mixture was concentrated to about 30 mL, and then H2O (30 mL) and ethyl acetate (48 mL) were added. The two layers were separated, and the organic layer was washed with 5% NaCl (3 × 24 mL) until pH 7 (assay yield ∼96%). The organic layer was solvent-switched to toluene until 45 mL (6 vol). A total of 135 mL of toluene was used to flush the solution until residual ethyl acetate was completely removed. The desired product was crystallized by slow addition of n-heptane (88 mL, 12 vol) over 1 h at 80 °C; the mixture was then cooled to 25 °C over 3 h, and the mixture was stirred for 18 h. The slurry was filtered, and the solid was washed with toluene/n-heptane (1:2). The solid was vacuum-dried for 18 h to afford 7.6 g of crude product (88.6% assay yield). About 2% product was in the mother liquor. The crude product was recrystallized by first dissolving the solid in EtOH (3 vol, 23 mL) at 45 °C, and then water (9 vol, 70 mL) was slowly added at 45 °C over 1 h, followed by aging at 25 °C for 18 h. The slurry was filtered and washed with EtOH/H2O (1:3.5) (about 2% loss in mother liquor). The wet cake was dried under vacuum at 45 °C for 24 h to afford 8.0 g of pyranol 4 (84% yield) with 98.2% purity by HPLC. Characterization data for 4 have been previously reported.7
Supplementary Material
Acknowledgments
We thank Pu Qian, Weijun Guo, Wei Zhu, Xiaoli Liu, Honggui Ouyang, Hui Wu, Honglin Ye, Shebin He, Jianhang Cheng, and Xianghui Wen (WuXi AppTec Co., Ltd.) for assistance with the process development. We also thank Junyong Jo, Ping Zhuang, and Kyle Eckenroad (Merck & Co., Inc., Kenilworth, NJ USA) for assistance with the analytical m ethod development. We also thank Elizabeth Bess (University of Utah) and Professor Matthew Sigman (UOU) for very useful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b01467.
Chiral HPLC methods and chromatograms of 14 and L6, compound characterization and NMR spectral data for all new substrates, HPLC chromatogram of compound 4, Cartesian coordinates for compounds 17a/b/c/d (PDF)
ORCID
Mark McLaughlin: 0000-0003-0595-4754
Edward C. Sherer: 0000-0001-8178-9186
John Y. L. Chung: 0000-0001-6094-5549
Notes
The authors declare no competing financial interest.
References
- 1.IDF Diabetes Atlas. 7th. International Diabetes Federation: Brussels, Belgium; 2015. http://www.diabetesatlas.org. [Google Scholar]; (b) Among three major types of diabetes, type 2 diabetes comprises at least 90% of patient population.
- 2.(a) Adeghate E, Kalasz H, Veress G, Tekes K. Curr Med Chem. 2010;17:517–551. doi: 10.2174/092986710790416281. [DOI] [PubMed] [Google Scholar]; (b) Adeghate E, Feher E, Kalasz H. Expert Opin Invest Drugs. 2015;24:1–15. doi: 10.1517/13543784.2014.954033. [DOI] [PubMed] [Google Scholar]
- 3.Tahrani A, Barnett A, Bailey C. Nat Rev Endocrinol. 2016;12:566–592. doi: 10.1038/nrendo.2016.86. [DOI] [PubMed] [Google Scholar]
- 4.Kim D, Wang L, Beconi M, Eiermann GJ, Fisher MH, He H, Hickey GJ, Kowalchick JE, Leiting B, Lyons K, Marsilio F, McCann M, Patel RA, Petrov A, Scapin G, Patel SB, Sinha-Roy R, Wu JK, Wyvratt MJ, Shang BB, Zhu L, Thornberry NA, Weber AE. J Med Chem. 2005;48:141–151. doi: 10.1021/jm0493156. and references therein. [DOI] [PubMed] [Google Scholar]
- 5.Li S, Xu H, Cui S, Wu F, Zhang Y, Su M, Gong Y, Qiu S, Jiao Q, Qin C, Shan J, Zhang M, Wang J, Yin Q, Xu M, Liu X, Wang R, Zhu L, Li J, Xu Y, Jiang H, Zhao Z, Li J, Li H. J Med Chem. 2016;59:6772–6790. doi: 10.1021/acs.jmedchem.6b00505. and references therein. [DOI] [PubMed] [Google Scholar]
- 6.Biftu T, Sinha-Roy R, Chen P, Qian X, Feng D, Kuethe JT, Scapin G, Gao YD, Yan Y, Krueger D, Bak A, Eiermann G, He J, Cox J, Hicks J, Lyons K, He H, Salituro G, Tong S, Patel S, Doss G, Petrov A, Wu J, Xu SS, Sewall C, Zhang X, Zhang B, Thorn-berry NA, Weber AE. J Med Chem. 2014;57:3205–3212. doi: 10.1021/jm401992e. [DOI] [PubMed] [Google Scholar]
- 7.(a) Xu F, Zacuto MJ, Kohmura Y, Rosen J, Gibb A, Alam M, Scott J, Tschaen D. Org Lett. 2014;16:5422–5425. doi: 10.1021/ol502661g. [DOI] [PubMed] [Google Scholar]; (b) Chung JYL, Scott JP, Anderson C, Bishop B, Bremeyer N, Cao Y, Chen QH, Dunn R, Kassim A, Lieberman D, Moment AJ, Sheen F, Zacuto M. Org Process Res Dev. 2015;19:1760–1768. [Google Scholar]
- 8.Starting from glycine benzophenone imine 9, the overall yield of 1 is 29%.
- 9.A high molecular weight protecting group and leaving group are employed in compounds 9 and 10 respectively. Weinreb amide 8 and 2 equiv of aryl Grignard reagent are used for the preparation of aryl ketone 7.
- 10.Sun G, Wei M, Luo Z, Liu Y, Chen Z, Wang Z. A new synthesis of ketone 3 from alkyne 6 was recently disclosed. Org Process Res Dev. 2016;20:2074–2079. [Google Scholar]
- 11.(a) Ananthi N, Velmathi S. Indian J Chem. 2013;52B(1):87–108. [Google Scholar]; (b) Feng G-f, Gao N, Wang Y-y, Li H-h, Xiao Y-p. Fenzi Cuihua. 2011;25:354–385. [Google Scholar]; (c) Lu G, Zheng F, Wanga L, Guo Y, Li X, Cao X, Wang C, Chi H, Dong Y, Zhang Z. Tetrahedron: Asymmetry. 2016;27:732–739. [Google Scholar]; (d) Arai T, Watanabe M, Yanagisawa A. Org Lett. 2007;9:3595–3597. doi: 10.1021/ol7014362. [DOI] [PubMed] [Google Scholar]; (e) White JD, Shaw S. Org Lett. 2012;14:6270–6273. doi: 10.1021/ol3030023. [DOI] [PubMed] [Google Scholar]; (f) Evans DA, Seidel D, Rueping D, Lam HW, Shaw JT, Downey CW. J Am Chem Soc. 2003;125:12692–12693. doi: 10.1021/ja0373871. [DOI] [PubMed] [Google Scholar]
- 12.Uzarewicz-Baig M, Koppenwallner M, Tabassum S, Wilhelm R. Appl Organomet Chem. 2014;28:552–558. and references therein. [Google Scholar]
- 13.Over 70 commercially available diamines, amino-alcohols, and box-ligands were evaluated.
- 14.Evans DA, Seidel D. The success of ligand L6 in this system is interesting considering that a similar ligand (dibenzyl substituted cyclohexanediamine) was used by Evans and Seidel in their highly selective malonate Michael addition to nitroalkenes. J Am Chem Soc. 2005;127:9958–9959. doi: 10.1021/ja052935r. [DOI] [PubMed] [Google Scholar]
- 15.It is important to note that high levels of residual copper led to significant ee erosion of alcohol 14. With a simple aqueous EDTA solution wash, residual copper was reduced to <10 ppm, and the enantiomeric purity of 14 was maintained for at least a month.
- 16.(a) de la Torre AF, Rivera DG, Concepción O, Echemendía R, Corrêa AG, Paixão MW. J Org Chem. 2016;81:803–809. doi: 10.1021/acs.joc.5b02158. [DOI] [PubMed] [Google Scholar]; (b) Gotoh H, Okamura D, Ishikawa H, Hayashi Y. Org Lett. 2009;11:4056–4059. doi: 10.1021/ol901483x. [DOI] [PubMed] [Google Scholar]; (c) Kaji E, Kohno H, Zen S. Bull Chem Soc Jpn. 1977;50:928–932. [Google Scholar]
- 17.Stronger bases such as DBU and DABCO led to significant acrolein polymerization, whereas weaker bases, piperidine, N-Me-piperidine, morpholine, N-Me-morpholine, and N-Me-pyrrolidine led to poor conversions. Hünig’s base provided the unique balance of basicity and nucleophilicity to successfully promote the desired transformation.
- 18.In the absence of solvent, significant polymerization of acrolein was observed.
- 19.Conditions, including (EtO)2POCl/collidine/0 to 80 °C and pTsOH or MsOH/toluene/0 to 110 °C, were investigated.
- 20.We were surprised to find that the reaction conditions afforded exclusively axial chlorides 17a/b instead of the expected the corresponding mesylate intermediates. This is in sharp contrast to the corresponding NHBoc substrate, which afforded a mixture of anomeric mesylates (unpublished results).
- 21.(a) Anderson CB, Sepp DT. J Org Chem. 1967;32:607–611. [Google Scholar]; (b) Wang C, Ying F, Wu W, Mo Y. J Org Chem. 2014;79:1571–1581. doi: 10.1021/jo402306e. [DOI] [PubMed] [Google Scholar]
- 22.Brands KMJ, Davies AJ. Chem Rev. 2006;106:2711–2733. doi: 10.1021/cr0406864. [DOI] [PubMed] [Google Scholar]
- 23.Compound 12a is a crystalline solid, while 12b is an oil.
- 24.The 14 bases were as follows: triethylamine, N,N-diisopropyl-ethylamine, 4-methylmorpholine, morpholine, 1,4-dimethylpiperazine, 2,4,6-trimethylpyridine, DABCO, DMAP, KOtBu, KOTMS, K2HPO4, NaOH, NaHCO3, and Na2CO3.
- 25.Na2CO3·1.5(H2O2) is a less expensive oxidant than NaBO3·4H2O used in the current route ($1.30/kg vs $14/kg).
- 26.It is worth noting that, by using a superstoichiometric quantity of zinc under acidic conditions, desired pyranol 4 can also be prepared from 11 in 89% yield, >99% ee, and 99.95:0.05 trans/cis ratio. While the zinc process is cost-effective, it was not pursued due to the large amount of waste generated from the process.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.