

Abstract
Antibody dependent enhancement of infection has been shown to play a major role in Dengue viral pathogenesis. Traditional assays that measure the capacity of antibodies or serum to enhance infection in impermissible cell lines have relied on using viral output in the media followed by plaque assays to quantify infection. More recently, these assays have examined Dengue virus (DENV) infection in the cell lines using fluorescently labeled antibodies. Both these approaches have limitations that restrict the widespread use of these techniques. Here, we describe a simple in vitro assay using Dengue virus reporter viral particles (RVPs) that express green fluorescent protein and K562 cells to examine antibody dependent enhancement (ADE) of DENV infection using serum that was obtained from rhesus macaques 16 weeks after infection with Zika virus (ZIKV). This technique is reliable, involves minimal manipulation of cells, does not involve the use of live replication competent virus, and can be performed in a high throughput format to get a quantitative readout using flow cytometry. Additionally, this assay can be easily adapted to examine antibody dependent enhancement (ADE) of other flavivirus infections such as Yellow Fever virus (YFV), Japanese Equine Encephalitis virus (JEEV), West Nile virus (WNV) etc. where RVPs are available. The ease of setting up the assay, analyzing the data, and interpreting results makes it highly amenable to most laboratory settings.
Keywords: Immunology and Infection, Issue 134, Zika virus, Dengue virus, Antibody dependent enhancement, Reporter viral particles, convalescent serum, Flavivirus
Introduction
Antibody dependent enhancement (ADE) of infection is a process whereby partially cross-reactive antibody responses induced by a serotype of virus enhances uptake of another serotype of virus, leading to increased viral replication and viremia. ADE has been extensively documented in Dengue virus (DENV) infections where four major serotypes are prevalent. In a subset of patients, ADE is associated with Dengue hemorrhagic fever (DHF). We have recently shown that Zika virus (ZIKV) infection induced significantly high levels of DENV cross-reactive antibody responses that caused ADE of DENV in vitro and likely contributed to the enhancement of DENV viremia in vivo1,2. Antibody dependent enhancement assays are a valuable tool to assess the capacity of antibodies to enhance secondary infection with related viruses and provide valuable insights into the pathogenesis of flavivirus infections and inform the development of vaccines.
The assay described here uses DENV RVPs along with K562 cells that are normally impermissible to infection. RVPs are structurally intact replication incompetent DENV viral particles that encode a sub-genomic green fluorescent protein (GFP) replicon that is expressed after a single round of replication3. As such, cells that become infected with RVPs fluoresce green and can be readily detected using flow cytometry or microscopy. The RVPs used in this assay were obtained from commercial sources. They can, however, be generated against other viruses and used in the assay described in this manuscript. Meanwhile, K562 cells are an FcγIII-receptor-expressing leukemia cell line that bind to the Fc region of antibodies and become infected in the presence of sub-neutralizing concentrations of antibody4,5.
ADE assays have been extensively used in studies investigating the risk factors for severe dengue and to delineate the mechanisms of in vitro ADE6,7,8.The ADE assay described here can be quickly and easily used to determine the capacity of serum to enhance in vitro infection using RVPs and flow cytometry, as compared to other assays used currently, which require either the determination of plaque forming units (pfu) in Vero cells or antibody staining of infected cells6,7,8,9,10,11, both of which are time consuming and labor intensive.
Protocol
The serum samples used to demonstrate the protocol described here were obtained from rhesus macaques that were housed and cared for in accordance with local, state and federal policies in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited facility. All animal experiments were reviewed and approved by Institutional Animal Care and Use Committee and samples were acquired through a tissue sharing protocol.
1. Day 1
NOTE: Perform all the steps described below in a sterile laminar flow biosafety cabinet used for tissue culture in a BSL-2 laboratory.
Thaw the serum samples at room temperature and transfer 100 µL of each serum sample to a sterile tube. Heat inactivate for 30 min at 56°C either in a water bath or a temperature-adjustable thermomixer. Make 10-fold serial dilutions of the serum sample ranging from 1:1 to 1:1,000 using cold RPMI-10 (RPMI with 10% fetal bovine serum).
Transfer 10 µL of serially diluted serum sample to each well of a sterile 96-well V-bottom plate. Include two sets of control wells, RPMI-10 with RVPs only and without serum sample, and RPMI-10 only without RVPs and without serum sample. Set up each serum sample and RPMI-10 controls in triplicates.
Remove the Dengue-1, 2, 3 and 4 RVPs from the -80 °C freezer and thaw them in a 37 °C water bath. Transfer the thawed RVPs immediately to ice. Obtain approximately 170 µL RVPs for each serum sample (10 µL of RVPs x 3 wells for each serum dilution (1:1, 1:10, 1:100, 1:1,000) and 10 µL of RVPs x 3 wells for each of the RPMI-10 with RVP only control wells).
Pipette 10 µL of RVPs into each well of the 96-well V-bottom plate containing the serum sample and to the RPMI-10 with RVPs (no serum) control wells. Do not add RVPs to the RPMI-10 only (no serum sample and no RVP) wells. Mix thoroughly by pipetting up and down 5–10 times. Add 10 µL cold RPMI-10 in lieu of RVPs to each cold RPMI-10 only (no serum sample and no RVP) control wells.
Transfer the 96-well V-bottom plate to an incubator and incubate the plate for 1 hour at 37 °C in the presence of 5% CO2. While the 96-well V-bottom plate is incubating, clean the surface of the biosafety cabinet with 70% ethanol and run the UV light for 15 min.
Remove a fully confluent T75 flask of K562 cells from the incubator, mix the cells well using a sterile 5 mL pipet, and transfer 5 mL of cells to a sterile 15 mL conical tube. NOTE: The K562 cells are maintained in RPMI-10 and subcultured at 1 x 106/mL.
Count the number of cells by removing 10 µL cells from the sterile 15 mL conical tube and mix with 10 µL tryphan blue. Count the cells using a hemocytometer to determine the total number of cells in the 15 mL conical tube.
Centrifuge the 15 mL conical with cells at ~1,200 x g for 10 minutes, decant the supernatant, and resuspend the cells in warm RPMI-10 at a concentration of 80,000 cells/30 µL of media (2.66 x 106 cells /mL).
Remove the 96-well V-bottom plate from the incubator (step 1.6), transfer 30 µL of K562 cells to each well of the 96-well V-bottom plate, and mix thoroughly by pipetting up and down 5–10 times. Transfer the 96-well V-bottom plate to an incubator and incubate the plate for 1 hr at 37 °C in the presence of 5% CO2.
After incubating for 1 h, remove the 96-well V-bottom plate from the incubator and centrifuge at ~1,200 x g for 5 minutes. After centrifugation, decant the media from the wells by turning the plate upside down into a container containing 10% bleach.
Wash the cells in each well by resuspending them in 125 µL of warm RPMI-10, mix thoroughly with a pipette, and centrifuge the plate at ~1,200 x g for 5 min. Decant the media by turning the plate upside down into a container containing 10% bleach. Repeat this wash step two times.
After washing, add 100 µL of warm RPMI-10 to each well, mix up and down with the pipet, and incubate the plate for 48 h at 37 °C in the presence of 5% CO2.
2. Day 3
Remove the 96-well V-bottom plate containing 100 µL of cells and media/well from the incubator and move it to a biosafety cabinet . Using a multichannel pipette, mix the contents of each well and transfer the cells and media to a 96 well U-bottom plate or to pre-labeled 5 mL polypropylene tubes.
Rinse each well in the 96-well V-bottom plate with 100 µL of 1% Paraformaldehyde (PFA) in 1x Phosphate Buffered Saline (PBS) and transfer to the respective wells in the 96 well U-bottom plate or labeled 5 mL polypropylene tubes from step 2.1 to yield a final concentration of 0.5% PFA/well or tube. Mix thoroughly using a multichannel pipette, cover the plate with aluminum foil, and let it sit in the incubator for 30 minutes to fix the cells.
Prepare the flow cytometer (see Table of Materials for example) by running unstained K562 cells to calibrate the side and forward scatter along with the fluorescence settings. The only fluorescence channel required is FL1, as GFP is the only fluorescence emitted from the cells. Acquire ~30,000–50,000 cells from each sample. NOTE: Any flow cytometer capable of reading a single fluorescence can be used for acquiring the data, as cells infected with RVPs will only emit green fluorescence due to the presence of GFP, and no other fluorescence channel are needed.
3. Data Analysis
- Analyze data acquired on the flow cytometer using any flow cytometry analysis software (see Table of Materials).
- Set the first gate based on FSC-A (forward scatter – area) vs FSC-H (forward scatter – height) to include single cells and exclude auto-fluorescent doublets from analysis12. Then, gate singlet-gated cells based on SSC-A (side scatter – area) vs FSC-A to exclude any autofluorescent debris.
- Analyze SSC-A vs FSC-A gated cells for the expression of GFP based on SSC-A vs GFP-A. Determine the percentage of GFP+ cells for each dilution of the serum and control samples by setting a gate around GFP+ cells.
Determine the average percentage of GFP+ cells for each serum sample dilution and control wells by dividing the total percentage of GFP+ cells in the triplicate wells by 3.
Calculate fold-enhancement of infection by dividing the average percentage of GFP+ cells in the serum samples for each dilution divided by the average percentage of GFP+ cells in the RPMI-10 with RVPs (no serum sample) control wells.
Graph fold-enhancement (y-axis) versus dilution (x-axis), and perform statistical analysis using ANOVA followed by Tukey's post-hoc test for multiple comparisons.
Representative Results
Sera from 4 rhesus macaques were collected 16 weeks after infection with ZIKV and tested for their potential to enhance DENV-1, 2, 3 and 4 infection in K562 cells. The animals had peak viremia of ~1 x 105 copies of ZIKV RNA/mL of plasma by day 3 after infection that declined to levels that were below the limit of detection by 7 days after infection. No viremia was detected in the plasma at 16 weeks post-infection. Then, the RVP assay was performed and the collected data were analyzed, as described in the above protocol.
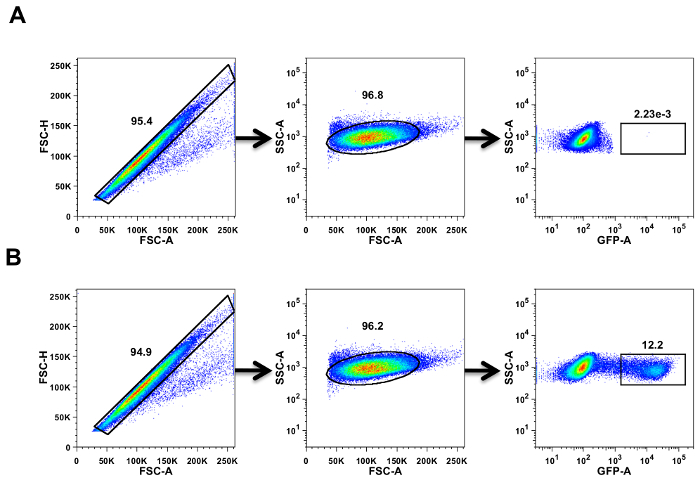
The gating strategy used to identify GFP+ cells are shown in Figure 1. The initial gate was set to exclude autofluorescent doublets and only include single cells based on FSC-A vs FSC-H. The next plot shows only cells from the single cell gate; there, a gate was drawn based on SSC-A vs FSC-A to exclude any autofluorescent debris. These gated cells were then displayed in a third plot based on SSC-A vs FL1-A to identify GFP+ cells. Cells infected with RVPs express GFP, and the percentage of GFP+ cells was determined by setting a gate around these cells. Approximately 12.2% of GFP+ cells are detectable in the ZIKV infected serum sample, as compared to no GFP+ cells in the control sample. This gating strategy is followed for each serum dilution and control sample included in the assay.
The percentage of GFP+ cells was determined for each sample dilution and control sample as described above (Figure 1). Data from a representative animal using DENV-1 RVPs are shown in Figure 2. As compared to the no serum control sample that had 0.253% GFP+ cells, the percentage of GFP+ cells were 2.58% in undiluted sample, 12.8% at 1:10, 0.735% at 1:100, and 0.022% at 1:1,000 dilution. The percentage of GFP+ cells will change as the serum is diluted due to the decreasing concentrations of antibody in the sample.
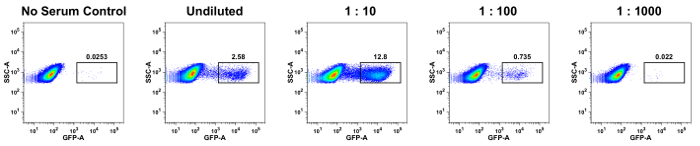
As enhancement of infection is associated with the antibody concentration that is ideal to increase uptake of virus, we observed a dramatic increase in the percentage of GFP+ cells at 1:10 dilution as compared to either undiluted sample or other dilutions, suggesting that the undiluted sample had higher concentrations of antibody whereas the 1:100 and 1:1,000 dilutions had lower levels of antibody that was not sufficient to induce ADE of infection. If antibody concentrations are significantly high, then additional dilutions of serum should be included to determine the dilution at which ADE becomes apparent. In the serum samples we examined, we observed ADE at a dilution of 1:10. On the other hand, ADE may be observed at lower dilutions if the antibody concentration in the serum is quite high.
Next, we calculated the fold-enhancement of infection by dividing the average percentage of GFP+ cells in the triplicate wells for each dilution with the average percentage of GFP+ cells in the triplicate wells for no serum control. The kinetics of ADE against DENV-1, 2, 3 and 4 serotypes were determined by graphing fold-enhancement on the y-axis and serum dilution on the x-axis (Figure 3). The data shows that the serum collected at 16 weeks post infection significantly enhanced infection of K562 cells by DENV-1, 2, 3 and 4 serotypes at a dilution of 1:10.
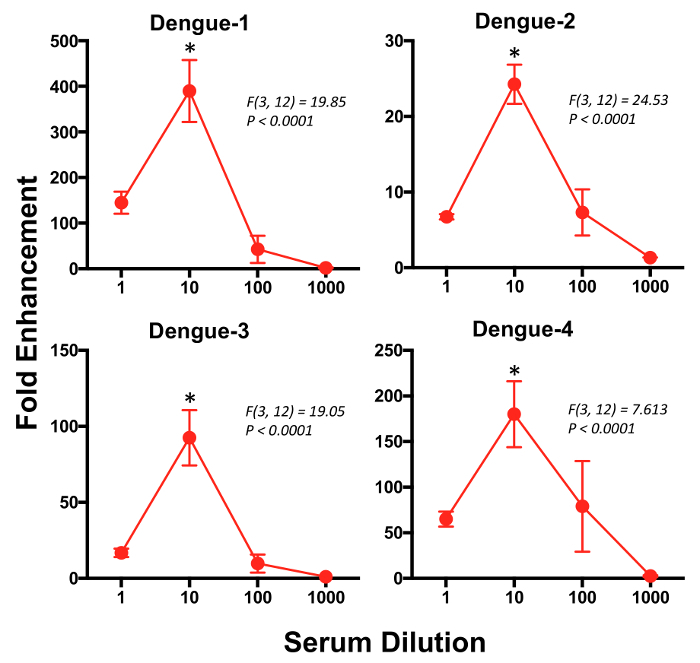
Figure 1: Gating strategy for acquiring and analysis by flow cytometry. K562 cells infected with DENV RVPs express GFP that was analyzed using a flow cytometer. (A) No serum control and (B) 16 week post-ZIKV infected serum. Cells were first gated based on forward scatter area (FSC-A) vs forward scatter height (FSH-H) to exclude doublets followed by a side scatter (SSC-A) vs FSC-A to exclude debris. The SSC-A vs FSC-A gated cells were then gated based on SSC-A vs GFP-A (Green Fluorescence Protein-area). The black rectangles and ovals represent the gates, and the numbers above the gates are the percentage of cells falling within that gate. The black arrow between the FACS plots shows the sequence of gates used. Please click here to view a larger version of this figure.
Figure 2: Representative dot plots showing frequency of DENV RVP+ cells. The percentage of K562 cells that were infected with DENV-1 RVPs was determined by flow cytometry. Serum collected from a representative Rhesus macaque at 16 weeks post infection was incubated with DENV-1 RVPs either undiluted or at a 1:10, 1:100, and 1:1,000 dilutions and compared to no serum control samples. The percentage of GFP+ cells at each concentration of serum is shown. Please click here to view a larger version of this figure.
Figure 3: Significant enhancement of Dengue-1, 2, 3 and 4 infection is observed at 1:10 dilution of serum. Antibody dependent enhancement of Dengue infection was examined for the 4 DENV serotypes by plotting serum dilutions on the x-axis and fold-enhancement (relative to no serum sample controls) on the y-axis. Sera collected from 4 ZIKV immune rhesus macaques at 16 weeks after infection was used in this assay. Error bars represent standard error and * represents p <0.05. Please click here to view a larger version of this figure.
Discussion
Flaviviruses such as DENV and ZIKV share significant evidence of homology in both their structural and non-structural proteins that generate antibodies that cross-react with each other13,14. These cross-reactive antibody responses have been shown to enhance of infection both in vivo and in vitro with either a heterologous serotype or other related flaviviruses1,2. The potential to cross enhance infection has significant implications for management of the disease and also for vaccine development.
Traditional culture based assays use non-permissive cells that are infected with infectious live DENV in the presence of serum. Enhancement of infection is measured by quantifying the amount of virus in the supernatant using standard plaque assays11. This assay requires culture of virus in two cell lines and requires significantly long periods of time to perform. Additionally, not all the DENV serotypes plaque the same way, adding another level of complexity to these assays.
Other assays have modified the plaque based assay protocol to measure infection of cell lines by combining intracellular staining for DENV using fluorescently labeled antibodies and flow cytometry9,10,14. Though this reduces the time required to determine the level of infection as compared to plaque based assays there are a number of optimization steps required for these assays to work consistently. Intracellular staining protocols are time consuming, as they require fixing and permeabilizing of cells followed by intracellular staining and require well-titrated DENV specific antibodies that can clearly discriminate each serotype. Additionally, these assays are not easily amenable to a high throughput format and suffer from high levels of intra-assay variability.
In contrast to the above described assays, the DENV RVPs based assay is easy to set up, does not use infectious virus, and is highly amenable to high throughput that can be readily used to measure the potential of serum to enhance infection. Additionally, this assay is not as labor intensive as other assays, and can be completed within 2 - 3 days, which offers a major advantage over current ADE assay protocols. Though the assay described in the manuscript examined the capacity of ZIKV infected serum to enhance infection of DENV-1, 2, 3 and 4 serotypes, the assay can be readily adapted to examine serum from both experimental and clinical samples obtained from other flavivirus infections such as Yellow Fever virus (YFV), Japanese Equine Encephalitis virus (JEEV), and West Nile virus (WNV) if RVPs are available, and with other cell lines. It is, however, critical to optimize the assay by titrating the volume of RVP, number of cells/well, and the duration of incubation to allow optimal staining of cells without background staining.
Titrations need to be performed by incubating a defined number of target cells (for example, 80,000 K562 cells or a cell line being tested) with variable volumes of RVPs (for example, 2.5 µL, 5 µL, 7.5 µL, 10 µL, 20 µL, etc.). Once the correct titer for each RVPs is obtained, the duration of incubation can be determined using the obtained titer along with the defined number of cells and incubating them as described in the protocol for variable periods of time (for example, 24 h, 48 h, 72 h, etc.).
We observed ADE of infection by all 4 serotypes of DENV at a dilution of 1:10, suggesting that a antibody levels in the serum needed to be diluted 10-fold to detect enhancement of infection. On the other hand, other studies have reported enhancement at lower dilutions, which would indicate that the concentrations of antibody in the serum sample were likely too high, needing lower dilutions to detect ADE in vitro. As ADE depends on the right concentration of antibody in the serum sample, most samples are likely to show ADE at dilutions lower than the undiluted serum.
In conclusion, the protocol described in this manuscript is simple and easy to adopt, and allows for scaling up to enable high throughput screening of a large number of samples in a short period time frame to yield high quality data that can easily be analyzed and interpreted.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The described project was supported by funds from the Uniformed Services University of the Health Sciences to JJM. The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense, the Uniformed Services University of the Health Sciences or any other agency of the U.S. Government.
WGV performed all the experiments and analyzed the data; JJM designed and supervised the study; WGW and JJM wrote the paper.
References
- George J, et al. Prior exposure to zika virus significantly enhances peak dengue-2 viremia in rhesus macaques. Sci Rep. 2017;7(1):10498. doi: 10.1038/s41598-017-10901-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettler K, et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science. 2016;353(6301):823–826. doi: 10.1126/science.aaf8505. [DOI] [PubMed] [Google Scholar]
- Mattia K, et al. Dengue reporter virus particles for measuring neutralizing antibodies against each of the four dengue serotypes. PLoS One. 2011;6(11):e27252. doi: 10.1371/journal.pone.0027252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiofalo MS, Teti G, Goust JM, Trifiletti R, La Via , F M. Subclass specificity of the Fc receptor for human IgG on K562. Cell Immunol. 1988;114(2):272–281. doi: 10.1016/0008-8749(88)90321-8. [DOI] [PubMed] [Google Scholar]
- Klein E, et al. Properties of the K562 cell line, derived from a patient with chronic myeloid leukemia. Int J Cancer. 1976;18(4):421–431. doi: 10.1002/ijc.2910180405. [DOI] [PubMed] [Google Scholar]
- Kliks SC, Nisalak A, Brandt WE, Wahl L, Burke DS. Antibody-dependent enhancement of dengue virus growth in human monocytes as a risk factor for dengue hemorrhagic fever. Am J Trop Med Hyg. 1989;40(4):444–451. doi: 10.4269/ajtmh.1989.40.444. [DOI] [PubMed] [Google Scholar]
- Littaua R, Kurane I, Ennis FA. Human IgG Fc receptor II mediates antibody-dependent enhancement of dengue virus infection. J Immunol. 1990;144(8):3183–3186. [PubMed] [Google Scholar]
- Wang TT, et al. IgG antibodies to dengue enhanced for FcgammaRIIIA binding determine disease severity. Science. 2017;355(6323):395–398. doi: 10.1126/science.aai8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science. 2010;328(5979):745–748. doi: 10.1126/science.1185181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawiecki AB, Christofferson RC. Zika virus-induced antibody response enhances dengue virus serotype 2 replication in vitro. J Infect Dis. 2016;214(9):1357–1360. doi: 10.1093/infdis/jiw377. [DOI] [PubMed] [Google Scholar]
- Morens DM, Halstead SB. Measurement of antibody-dependent infection enhancement of four dengue virus serotypes by monoclonal and polyclonal antibodies. J Gen Virol. 1990;71(Pt 12):2909–2914. doi: 10.1099/0022-1317-71-12-2909. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, et al. Amine reactive dyes: an effective tool to discriminate live and dead cells in polychromatic flow cytometry. J Immunol Methods. 2006;313(1-2):199–208. doi: 10.1016/j.jim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Priyamvada L, et al. B Cell responses during secondary dengue virus infection are dominated by highly cross-reactive, memory-derived plasmablasts. J Virol. 2016;90(12):5574–5585. doi: 10.1128/JVI.03203-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyamvada L, et al. Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc Natl Acad Sci U S A. 2016;113(28):7852–7857. doi: 10.1073/pnas.1607931113. [DOI] [PMC free article] [PubMed] [Google Scholar]