

Abstract
Embryonic aneuploidy is the major genetic cause of infertility in humans. Most of these events originate during female meiosis, and albeit positively correlated with maternal age, age alone is not always predictive of the risk of generating an aneuploid embryo. Therefore, gene variants might account for incorrect chromosome segregation during oogenesis. Given that access to human oocytes is limited for research purposes, a series of assays were developed to study human gene function during meiosis I using mouse oocytes. First, messenger RNA (mRNA) of the gene and gene variant of interest are microinjected into prophase I-arrested mouse oocytes. After allowing time for expression, oocytes are synchronously released into meiotic maturation to complete meiosis I. By tagging the mRNA with a sequence of a fluorescent reporter, such as green fluorescent protein (Gfp), the localization of the human protein can be assessed in addition to the phenotypic alterations. For example, gain or loss of function can be investigated by establishing experimental conditions that challenge the gene product to fix meiotic errors. Although this system is advantageous in investigating human protein function during oogenesis, adequate interpretation of results should be undertaken given that protein expression is not at endogenous levels and, unless controlled for (i.e. knocked out or down), murine homologs are also present in the system.
Keywords: Genetics, Issue 134, meiosis, oocyte, aneuploidy, gene variants, chromosome segregation, fertility
Introduction
Infertility is a condition that affects 10-15% of the human population of reproductive age 1, from which nearly one-half seek medical treatment 2. Although the etiology of infertility is diverse and in many cases multifactorial, the most common genetic abnormality in humans is embryonic aneuploidy 3. Aneuploidy is defined as the deviation (either gain or loss) of the correct number of chromosomes in a cell. The phenomenon of aneuploidy in human embryos is common and increases with advanced maternal age 4,5. Four randomized controlled trials have highlighted the benefit of selecting only chromosomally normal (euploid) embryos for uterine transfer because this strategy resulted in increased implantation rates, lower miscarriage rates and a shorter time for achieving pregnancy 6,7,8,9. Therefore, understanding the etiology of human aneuploidy can have important implications in assisted reproduction.
Although pre-implantation genetic testing for aneuploidies is beneficial in infertility treatments, a thorough understanding of how aneuploidies originate is still lacking. It is widely accepted that there is a positive correlation of meiotic aneuploidies (originated during gamete production) and maternal age, however, some women present embryonic aneuploidy rates that deviate from the mean rate for their given age 4. These cases suggest that age alone is not always predictive of the risk of generating an aneuploid embryo. Other factors may play a role in increasing the risk of embryonic aneuploidy, such as gene variants.
A key aspect of investigating the potential contribution of a gene variant to aneuploidy during oocyte meiosis is to design a system to rapidly evaluate meiotic gene function. Due to ethical constraints and limited access, it is impractical to perform these experiments using human eggs. These issues can be circumvented by using mouse oocytes, and here a series of assays to assess human gene function during meiosis I are described. By microinjecting the messenger RNA (mRNA) coding for the gene variant of interest, the localization of the human protein in the mouse egg can be visualized and used to determine if the ectopic expression of the wild-type and mutated human protein results in any phenotypic alterations that could lead to aneuploidy. These phenotypes include an increase in microtubules that attach to the improper to sister kinetochore and the inability to support chromosome alignment at metaphase of meiosis I. Importantly, this protocol can be used to investigate both gain and loss of function genetic variants by establishing specific experimental conditions to challenge key events in oocyte meiosis such as spindle building and chromosome alignment 10.
Protocol
1. Molecular Cloning
Obtain full-length coding sequence of the gene of interest and the plasmid in vitro transcription (pIVT) vector11. NOTE: Full-length cDNA clones are commercially available from various vendors or can be generated via reverse transcription polymerase chain reaction (RT-PCR). The National Center for Biotechnology Information (NCBI) online resource provides transcript sequences for genes from the Nucleotide and Expressed sequence tagged (EST) databases. For ease of protein visualization and analysis of expression levels, fusion to green fluorescent protein (Gfp) or another suitable fluorescent tag may be desired in the cloning strategy.
Using a validated cloning strategy, insert the coding sequence of the gene of interest into the multiple cloning region of pIVT. A detailed description of molecular cloning and the required steps have previously been described 12. NOTE: If a gene-fusion product is to be generated, be sure that the cloning is in the proper reading frame.
Sequence the construct using Globin primers to ensure correct gene insertion, proper in-frame fusion, and that no polymerase-induced point mutations were created. NOTE: If Sanger DNA sequencing equipment is not available, many companies offer low cost DNA sequencing options.
Mutagenize DNA if generating single nucleotide polymorphisms (SNPs) or insertions/deletions (INDELS). DNA mutagenesis is described in steps 1.5-1.7 below. For wild-type constructs, proceed with the linearization step 1.7.
Design mutagenesis primers and complete the mutagenesis PCR reaction to generate mutant construct per manufacturer's protocol. PCR-mediated site-directed mutagenesis has been previously described 13. Additionally, many companies offer site-directed mutagenesis kits which include protocols.
Transform mutagenized DNA into bacterial host. Isolate and purify plasmid. NOTE: Many companies make kits that can be used to easily isolate and purify plasmid DNA. Follow manufacturers protocol accordingly. If using a gram-negative bacterial strain such as E. coli, it is recommended to use a kit that removes endotoxins.
Sequence the isolated DNA from bacterial transformants using standard Sanger DNA sequencing to confirm introduction of the desired SNP or INDEL.
Linearize final products using single restriction enzyme digestion of the purified DNA. NOTE: The pIVT vector contains multiple single-enzyme cut sites listed in the plasmid map on Addgene 14.
Ensure that the product is linear via agarose gel electrophoresis. The methodology for agarose gel electrophoresis has previously been defined 14. NOTE: For all remaining steps, use barrier (filter) pipet tips and RNase-free materials to ensure production of stable, high quality RNA.
Purify the digested product using a validated DNA clean-up protocol or kit, and elute in 30 µL RNase/DNase-free water. NOTE: Silica-matrix spin column-based kits are recommended for high quality, purified DNA. Follow manufacturers protocol accordingly.
In vitro transcribe DNA using T7 polymerase following the manufacturer's protocol.
Purify and elute RNA in 30 µL of RNase/DNase free water using a silica-matrix spin column-based nucleic acid purification kit following the manufacturer's protocol.
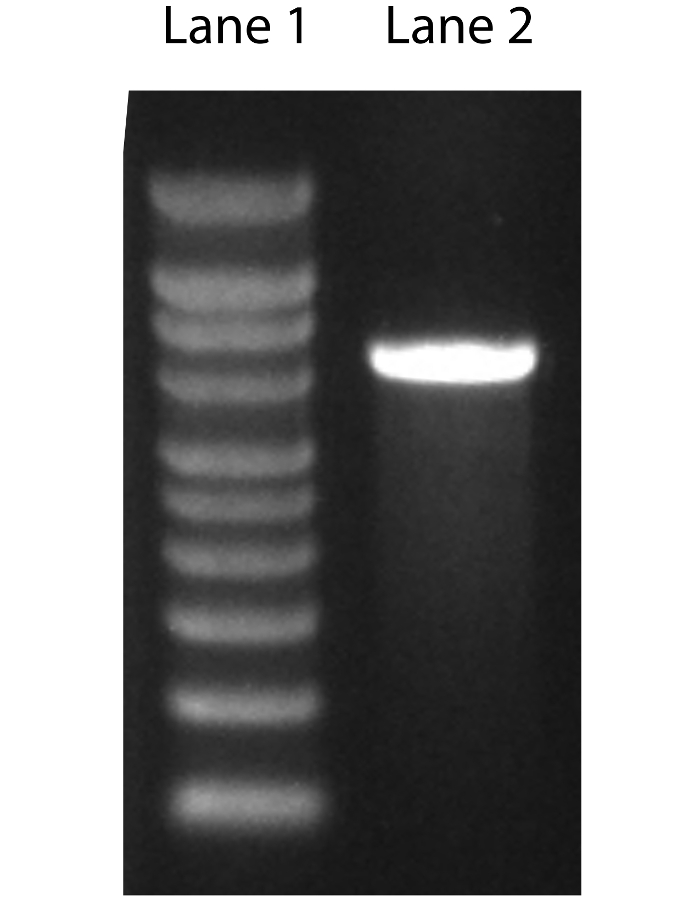
Perform denaturing agarose gel electrophoresis using formaldehyde to confirm the final RNA product size. See steps 1.13-1.22 below 14. (Figure 1).
Prepare gel by heating 1 g agarose in 72 mL water until dissolved, then cool to 60 °C.
Prepare 10X MOPS running buffer by adding 0.4 M MOPS (pH 7.0), 0.1 M sodium acetate, and 0.01 M EDTA.
Add 10 mL of 10X MOPS running buffer, and 18 mL of 37% formaldehyde (12.3 M) to the cooled agarose solution.
Caste the gel by pouring the agarose solution into a gel-forming container. Use a comb large enough to accommodate 10 µL. After gel has set and is firm to the touch, gently remove the comb.
Assemble the gel in the electrophoresis tank, and add enough 1X MOPS running buffer, ensuring the gel is completely covered.
Prepare RNA sample by adding 1 µg of RNA to 0.5X volumes of formaldehyde-containing loading dye.
Add ethidium bromide to RNA solution at a concentration of 10 µg/mL.
Denature RNA sample solution at 70 °C for 5 min.
Using sterile, 10 µL filter tips, load 5 µL of molecular weight marker and 5 µL of RNA sample(s) into separate wells of the gel and electrophorese at 5 V/cm until the dye has migrated at least two-thirds of the length of the gel.
Visualize the RNA in the gel on a UV trans illuminator. Ensure the RNA is the correct size by comparing to the molecular weight marker.
To measure the concentration and purity of the RNA, transfer 1.2 µL of purified RNA using sterile 10 µL filter tips onto a UV-spectrophotometer. Note that high quality RNA should have absorbance ratios of A260/280 (1.8-2.2) and 260/230 (>1.7) respectively.
Using 10 µL filter tips, aliquot the remaining purified RNA into sterile 0.5 µL centrifuge tubes (3-5 µL/tube). Store tubes at -80 °C until ready for microinjection. NOTE: A final injection concentration of 500 ng/µL is an optimal starting point. It is recommended to dilute RNA 1:2 in RNase free H20 prior to microinjection to reduce stickiness of RNA in microinjection pipette.
2. Kinetochore-Microtubule Attachment Assay
Divide oocytes into equal groups for microinjection. NOTE: Oocyte collection, transfer, microinjection, and meiotic maturation has been described in detail in JoVE previously 15.
Microinject one group with experimental mRNA and the other group(s) with the WT control and PBS or Gfp mRNA. NOTE: 500 ng/µL is the recommended injection concentration to begin with 10. A picoinjector can be used to control injection amounts. An injection volume of 5-10 pL is recommended 15.
Using a hand or mouth operated pipetting apparatus where the glass pipette opening is slightly larger than the diameter of an oocyte (100-150 µm), transfer oocytes into a 100 µL drop of milrinone-free culture medium in a Petri dish covered in embryo quality mineral oil and incubate oocytes for 7 h at 37 °C in 5% C02 to allow sufficient maturation to metaphase of meiosis I (Met I) 15.
After incubation, using the same pipette apparatus as above, transfer oocytes into a center-well organ culture dish containing 700 µL of pre-chilled minimum essential media (MEM) collection medium in the center well and 500 µL H20 in the outside ring and place dish on ice for 7 min. NOTE: It is recommended to pre-chill MEM media and center well organ culture dishes at -20 °C for at least 20 min prior to treatment of oocytes.
Fix the oocytes by transferring them using the pipette apparatus into a well of a clear glass spot plate containing 400 µL of 2% paraformaldehyde in 1X PBS for 20 min at room temperature.
Using same pipette apparatus, transfer the oocytes into another well on a spot plate containing 400 µL blocking solution (PBS + 0.3% BSA + 0.01% Tween-20 + 0.02% NaN3) and store at 4 °C until ready to process for immunostaining. NOTE: For storing oocytes at 4 °C, cover glass spot plate with parafilm to prevent evaporation.
Using same pipette apparatus, transfer oocytes to a new well on a spot plate containing 400 µL permeabilization solution (PBS + 0.3% BSA + 0.1% TritonX-100 + 0.02% NaN3) and incubate at room temperature for 20 min. Transfer oocytes quickly through three drops of 400 µL blocking solution to remove any residual detergent. NOTE: A 9-well clear glass spot plate works well for moving groups through multiple treatments on a single dish (steps 2.4-2.5).
Perform the remaining immunocytochemistry procedures in a humidified chamber, protected from light at room temperature. Use a medium-sized plastic container lined with damp paper towels and cover with aluminum foil.
Using the pipette apparatus, transfer oocytes to 30 µL blocking solution on the lid of a 96-well plate and place within the humidified chamber for 10 min. NOTE: Drops are placed in the circular indentions on the plate.
To label centromeres and spindle, using the pipette apparatus, transfer oocytes to 30 µL blocking solution containing anti-centromeric antigen (ACA, Crest) (1:30) and anti-acetylated tubulin (1:1000) and incubate for at least 1 h.
Rinse oocytes by transferring through three 30 µL drops of blocking solution for 10 min each.
Using the pipette apparatus, transfer oocytes to a 30 µL drop of blocking solution containing secondary antibodies complimentary to the antibodies used above (anti-human IgG 1:200, anti-mouse IgG 1:200) and incubate for 1 h.
Repeat the 3, 10-min rinsing steps in 30 µL blocking solution as described in step 2.11.
Transfer the oocytes, using the pipette apparatus, into 3-5 µL mounting medium containing DAPI (5.9 µg/µL) on a glass microscope slide.
Place 4 small drops (size of a pin head) of petroleum jelly on the corners of the cover slip to prevent crushing of the oocytes.
Gently place the cover slip petroleum-jelly side down onto the slide.
Seal coverslip edges with clear nail polish and allow to dry for 5 min.
Store slides at 4 °C, protected from light, until processing via confocal microscopy. NOTE: Ensure the refractive index of mounting medium, and coverslip, matches the microscope objectives being utilized. Recommended mounting materials have previously been described 15.
Image oocytes using a 40-63x objective on a confocal microscope, capturing small steps (≤ 1 µm) in the z-plane, optimized for the working objective, and image the entire region of the metaphase spindle. NOTE: Make sure to image all 3 channels based on specific secondary antibodies used in step 2.11. Image averaging ≥ 4 and a zoom of 3 are ideal for adequate resolution. A 1 µm step size provides clear visualization of microtubules and kinetochores in mouse oocytes. The procedure for image capture will vary from microscope to microscope. Please refer to the manufacturers confocal user guide for specific steps on how to configure microscope settings.
Identify kinetochore-microtubule attachment status using imaging software following steps 2.21-2.23 below. NOTE: The following steps describe this procedure using Image J (NIH) free downloadable software, however, alternate imaging software can be used. Attachment status types have previously been defined 16 .
Open image by dragging file into Image J tool bar.
In the opened z-series, make all channels visible (DNA, kinetochore, and spindle) by clicking "merge channels", available in the "Image" drop-down menu, under the "color" sub tab.
Using the toggle at the bottom of the image, move through each z-slice and determine kinetochore microtubule attachment. Detailed attachment descriptions have been previously described 16
3. Chromosome Alignment Challenge Assay
Using the pipette apparatus as described in step 2.3, transfer oocytes into a 100 µL drop of milrinone-free culture medium under oil and incubate oocytes for 7 h to allow sufficient maturation to metaphase of meiosis I (Met I) as previously described in step 2.3 17. NOTE: Oocyte collection, microinjection, and maturation procedures were outlined previously 15. Refer to the attachment assay protocol in steps 2.1-2.2 for controls and for RNA microinjection concentrations.
Using same pipette apparatus, transfer oocytes to a center-well organ culture dish containing 700 µL culture medium containing monastrol [100 µM] in the center well and 400 µL H20 in the surrounding ring and incubate at 37 °C for 2 h.
Rinse out monastrol by transferring oocytes, using the pipette apparatus as above, through three drops of 100 µL CZB culture medium, and then transfer the oocytes to a new center-well organ culture dish containing 700 µL culture medium + MG132 [5 µM] for 3 h in the center ring. Add 400 µL H20 to the outside ring.
Fix the oocytes by transferring them, using the pipette apparatus, into a well of a clear glass spot plate containing 400 µL 2% paraformaldehyde in PBS for 20 min at room temperature.
After fixation, transfer the oocytes, using the pipette apparatus, into a new well of a clear glass spot plate containing 400 µL blocking solution and store at 4 °C until processed for immunostaining. NOTE: For storing oocytes at 4 °C, cover glass spot plate with parafilm to prevent evaporation.
Permeabilize and label oocytes via immunocytochemistry as described in steps 2.6-2.16.
Image oocytes using a 40-63x objective on a confocal microscope, capturing <1 µm steps in the z-plane making sure to image the entire region of the metaphase spindle.
To identify chromosome alignment status open image files using imaging software. NOTE: The following steps describe this procedure using Image J (NIH) free downloadable software, however, alternate imaging software can be used.
Drag image into image J control panel.
In the opened z-series, use the point tool (available by clicking the point tool icon on the Image J control bar) to mark points at the end of each spindle pole in their respective z-slice by placing the tool over the spindle pole in the image and clicking on the image. Add these points to the Region of interest (ROI) manager by pressing Command+t on the keyboard.
Determine the specific coordinates for each of the positions set in step 3.10 by highlighting the specific point in the ROI manager and clicking the measure button. This will provide a results table-containing x and y coordinates for each point. These will be the X1, Y1, and X1, Y2, points respectively.
Next, determine the true Z coordinate. This is done by multiplying the slice number of the specific z-slice in the z-stack in which the spindle pole was identified (i.e. slice #2 of 6) that each individual point is in by the stack thickness (i.e. 1 µm) (example: slice 2 x 1 µm = 2). These will be the Z1 and Z2 points respectively.
Determine spindle length using the Pythagorean theorem equation (a2 + b2 = c2) using the previously defined x, y, and z coordinates from steps 3.11-3.13. For each spindle the final length is equal to: √((X1-X2)2+(Y1-Y2)2+(Z1-Z2)2)
Determine the spindle midzone. This is calculated as one half the length of the spindle. Using the line tool in Image J, by clicking the line icon in the Image J tool bar, draw a line that is from one spindle pole to the spindle midzone. The length will be displayed on the Image J tool bar.
Determine chromosome alignment status by assessing chromosome distance from the spindle midzone using the line measurement tool. Using the line tool available by clicking the line icon in the Image J tool bar, draw a line from the spindle midzone to the chromosome. The length will be displayed on the Image J tool bar. Chromosomes greater than 4 µm from the spindle midzone are considered unaligned 18.
Representative Results
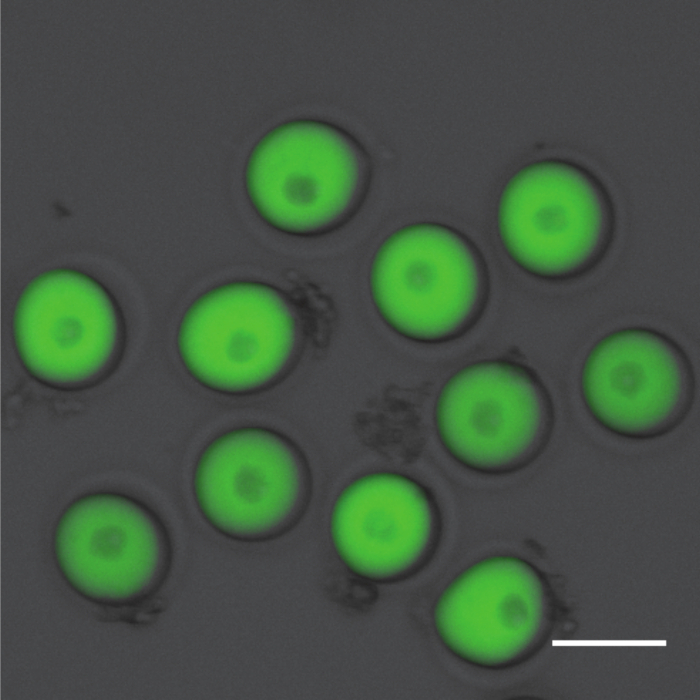
After in vitro transcription high quality RNA will have an A260/A280 ratio of (1.8-2.2) and an A260/A230 ratio ≥1.7 when measured using a spectrophotometer. The picture in Figure 1 shows the migration of in vitro-produced RNA on a denaturing agarose gel after electrophoresis. A band that is smeared in pattern or a sample that has multiple sized bands can indicate contamination or degradation of the sample. Alternatively, multiple bands may indicate that the mRNA is not completely denatured, or the starting plasmid was not completely linearized. The mRNA will run larger than predicted because of the poly-adenylated tail built into pIVT, which allows stable expression of mRNA in vitro. After microinjection it is recommended to ensure uniform injection volumes and expression levels via fluorescent microscopy. The picture in Figure 2 shows prophase I-arrested oocytes uniformly injected with Gfp. Consistent microinjection technique is critical for this protocol. Image analysis software, such as Image J, can be used to validate expression levels to ensure uniformity and mastery of the technique. Note that subcellular localization information can be obtained while doing the imaging for both assays. For all microinjection experiments assessing the effects of a gene variant, the wild-type gene should be injected into a subset of oocytes to serve as a control. This allows one to control for potential overexpression phenotypes of the human gene in a mouse oocyte. PBS, or Gfp for fluorophore-labeled constructs, should be also be injected into a subgroup of oocytes to control for microinjection-induced phenotypes. For constructs without a fluorescent tag, Gfp mRNA can be co-injected to ensure sufficient microinjection. If co-injecting two or more constructs simultaneously be sure to increase the mRNA concentration such that the final solution contains 500 ng/µL of each mRNA.
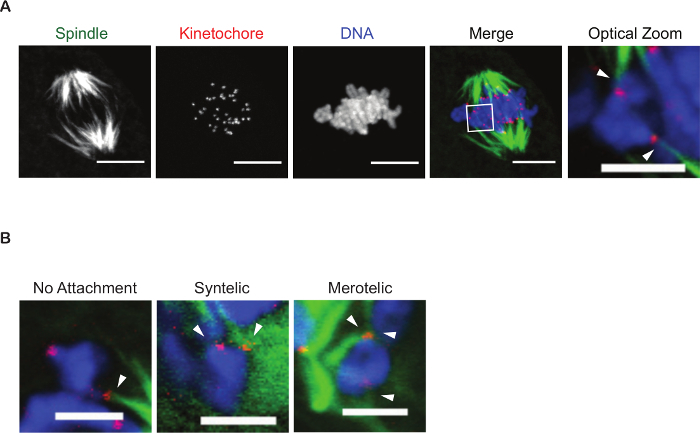
The cold-stable microtubule assay provides a valuable tool for assessing the attachment status of kinetochores to microtubules. Figure 3 is a z-projection of a confocal microscope image of a metaphase I oocyte. This oocyte was subjected to cold media treatment to depolymerize all microtubules that had not attached to kinetochores. Oocytes that have been treated for an insufficient time will contain microtubule fibers not attached to chromosomes (too short results in too many fibers) or lack fibers all together (too long results in no spindle structure) and it is recommended that these oocytes not be used in the analysis. At metaphase of meiosis I it is common for oocytes to contain unattached or laterally attached kinetochores, therefore it is recommended that a control group be used in any attachment study. On average, wild-type oocytes will contain 5-10% of kinetochores at MI will not yet be stably attached 19. It is essential that all oocytes be at the same stage for analysis of kinetochore-microtubule attachment status. To ensure that experimental and control groups mature with similar time scales it is recommend to assess cell-cycle kinetics. To perform this assay, a subset of control and experimental oocytes should be matured and visualized live under time-lapse microcopy. The use of milrinone allows oocytes to be synchronized in prophase of meiosis I, such that release into media lacking milrinone with allow resumption of meiosis at the same time 20. Oocytes from both control and experimental groups should extrude a polar body, indicative of reaching metaphase of meiosis II (Met II), at similar time points. If live-cell microscopy is not available, oocytes can be matured to Met I (7 h) and to Met II (16 h), fixed, and stained to detect DNA and microtubules as described earlier. At Met I and Met II a barrel-shaped, bipolar spindle should be visible. In control oocytes, chromosomes should be aligned at the metaphase plate. The alignment of chromosomes will differ in experimental groups depending on variant affects, however, chromosomes should be condensed and attached to microtubules with the majority of chromosomes centrally located on the spindle. A similar number of oocytes should have reached meiosis II in control and experimental groups. To reduce difficulties in determining attachment status it is recommended imaging oocytes using small steps in the z-plane, optimized for the specific working objective. Analysis will involve viewing the z-slices individually and in merged settings to determine which pole that attached microtubules emanate from. Figure 3B illustrates examples of abnormal kinetochore-microtubule attachments. Shown is a bivalent in which only one sister kinetochore pair is attached to a spindle microtubule, defined as a no attachment. Next, a syntelic attachment is shown in which both sister kinetochore pairs are attached to the same spindle pole. Finally, a merotelic attachment is shown in which one sister kinetochore is attached to both spindle poles while the other sister kinetochore of the pair is attached to a single spindle pole.
The images in Figure 4 illustrate the progressive expected DNA and spindle configurations as one moves through the steps of the chromosome alignment challenge assay. In this assay it is critical that different experimental groups progress through meiosis with similar kinetics. As described previously it is recommend to complete cell-cycle kinetics analyses prior to conducting the challenge assay. In this assay, the ability for oocytes to successfully correct abnormal kinetochore-microtubule attachments is assessed. To test this, oocytes are first matured to Met I to allow kinetochore-microtubule attachments to be established. Next, oocytes are incubated in monastrol, and Kinesin 5 (EG5) inhibitor, that collapses the bipolar spindle into a monopolar spindle 21. The generation of a monopolar spindle induces 100% incorrect microtubule-kinetochore attachments. The monastrol inhibitor is then washed out to allow recovery. During the recovery period the oocytes regenerate a bipolar spindle and attempt to correct microtubule attachments. Addition of the proteasome inhibitor MG132 prevents anaphase onset. As a control, a small subset of oocytes from each experimental group can be fixed at each stage in the protocol (Met I, monastrol treated, post recovery) to ensure all groups are responding to treatments, and to the same extent. Metaphase I oocytes should contain a barrel-shaped bipolar spindle with chromosomes aligned at the metaphase plate. Oocytes treated with monastrol should contain a monopolar spindle with chromosomes oriented around the single pole. Recovered oocytes should once again contain a barrel-shaped bipolar spindle. Chromosome alignment is defined as any chromosomes more than 4 μM from the spindle midzone 22. It is helpful to stain oocytes to detect DNA and kinetochores to establish if both kinetochores are outside this central zone as this can be difficult to determine using only DNA detection.
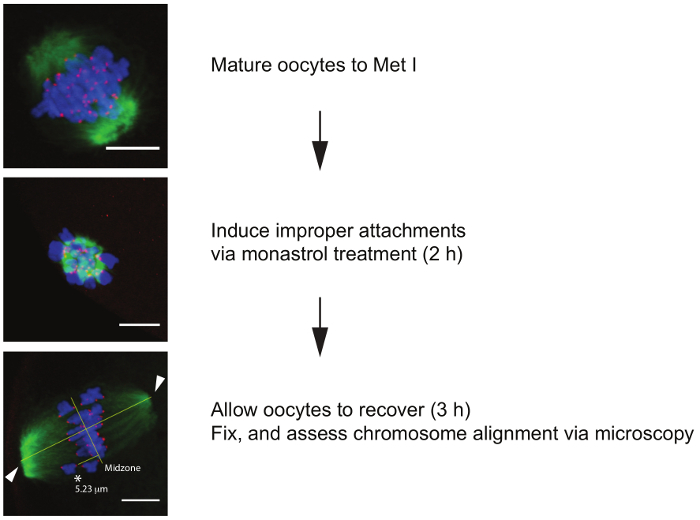
Figure 1. Denaturingagarose gel electrophoresis of RNA. RNA quality and size can be assessed by electrophoresis on a denaturing agarose gel. A denaturing gel is recommended to prevent secondary structure formation. Lane 1 contains a RNA molecular weight marker. Lane 2 is an RNA sample. Note that the RNA will run larger than calculated because of the built-in polyA tail. Please click here to view a larger version of this figure.
Figure 2. Validating oocyte microinjection. Fluorescent microscopy can be used to validate successful oocyte microinjection and expression. Prophase I-arrested oocytes microinjected with Gfp are shown after overnight expression of the construct. This picture was obtained using the Invitrogen EVOS FL Auto imaging system equipped with GFP light cube. Scale bar is 100 µm. Please click here to view a larger version of this figure.
Figure 3. Assessing kinetochore-microtubule attachments. (A) A representative z-projection of a cold-treated Met I oocyte. Spindle (green), kinetochores (red), and DNA (blue) are shown. The box indicates the region of the optical zoom. Arrowheads denote end-on attachments of kinetochores to microtubules of a single bivalent. Scale bar is 10 µm for spindle, kinetochore, DNA, and merge channels. Scale bar for optical zoom is 5 µm. (B) Examples of abnormal kinetochore-microtubule attachment types in mouse oocytes. Arrows indicate kinetochore-microtubule attachment sites. Scale bar is 5 µm. Please click here to view a larger version of this figure.
Figure 4. Chromosome alignment challenge assay. Flow chart of the procedure with representative z-projections of oocytes at each step. Oocytes are first matured to metaphase of meiosis I (Met I), then transferred to a new dish containing monastrol for 2 h to induce monopolar spindle formation. Next oocytes are allowed to recover in maturation medium supplemented with a proteasome inhibitor (MG132) to prevent anaphase onset for 3 h. Once recovered oocytes are fixed, labeled for spindle (green), kinetochores (red), and DNA (blue), and imaged via confocal microscopy to determine alignment status. Lines indicate length of spindle (40 µm) and spindle midzone (20 µm) determined using Pythagorean theorem equation. Arrows indicate point tool positions at each pole. Any kinetochores >4 µm from the spindle midzone are considered unaligned. Asterisk indicates unaligned chromosome (5.6 µm from midzone). Scale bar is 10 µm. Please click here to view a larger version of this figure.
Discussion
Because of the rapid and increasing identification of human genetic variants associated with disease, it is essential that systems are established to evaluate their biological significance. Understanding protein function in human meiosis poses particular challenges because human oocytes are precious and rare and human sperm are not amenable to genetic manipulation. Mouse oocytes are a mammalian model system valuable for evaluating human meiotic gene function 10,23. This model bypasses the impracticalities of using human eggs while providing a readily available source of low-cost, manipulable oocytes that undergo meiosis similar to human germ cells while being useful for understanding how genetic variants may impact female fertility.
Because these methods employ ectopic expression of the human protein in mouse oocytes via microinjection, it is critical to first establish a concentration of control, wild-type RNA that does not alter meiotic maturation. It is recommended that meiotic maturation kinetics are monitored (nuclear envelope breakdown, polar body extrusion) in control oocytes to establish baseline parameters, and assessing spindle and chromosome morphology. In addition, ensuring uniform microinjection volumes is critical to comparing groups. Including a fluorescent tag on the gene of interest can simplify this technical challenge. Techniques for mastering oocyte microinjection have been previously demonstrated 15. Visualization of injected oocytes under a fluorescence microscope or determining protein expression via western blot are also valuable tools for confirming consistent injection concentrations.
Because errors in chromosome segregation in mammalian oocytes are a leading cause of embryonic aneuploidy and pregnancy loss5, analysis of kinetochore-microtubule attachments provide a valuable tool for evaluating whether a gene of interest affects chromosome segregation (i.e. improper attachments will cause mis-segregation). A 10 min exposure to chilled media is sufficient to depolymerize microtubules that have not made a kinetochore-microtubule attachment, allowing visualization of stable attachments via confocal microscopy 24. For this assay, it is important to not over-chill the oocytes as this will lead to the depolymerization of stable microtubules. In addition, optimization of staining procedures is key for visualization of kinetochore and microtubule structures. To ensure adequate image resolution for analysis, small steps (≤ 1 μm) in the z-plane using a 40x-63x objective should be captured, and obtaining images through the entire spindle structure to ensure all attachments are visible. Note that it is common to have unattached kinetochores during prometaphase of meiosis I 25.
The chromosome alignment challenge assay is useful for the evaluation of gain or loss-of-function variants. Gain-of-function variants can be difficult to assess because mouse oocytes from young females have low levels of aneuploidy. This assay overcomes this hurdle by generating a situation in which the oocyte must correct experimentally induced incorrect kinetochore-microtubule attachments. Establishing a time point in which control oocytes contain 1-2 misaligned chromosomes after recovery provides a quantifiable scenario for determining whether a mutant provides increased alignment efficiency (i.e. better alignment capabilities will protect euploidy). For quantification of chromosome misalignment, a previously established protocol in which chromosomes greater than 4 μm from the spindle midzone are characterized as unaligned can be used 18.
To study loss-of-function variants, additional experimental conditions are necessary because of the endogenous homolog within the study system. One way to alleviate this issue is to use or generate a knockout mouse model of the variant of interest. The advent of the Crisper/Cas9 system could provide a quick way to generate such knockouts 26. Alternatively, if a knockout model is not an option, classical knockdown techniques such as co-injecting antisense morpholino oligonucleotides or small interfering RNAs could be employed. However, attention to the sequence design is required to avoid any potential interference with the microinjected variant under study. For example, the morpholino oligonucleotide could be designed to bind to the 5' untranslated region (5' UTR) of the mouse gene that is not included in the human mRNA variant introduced via microinjection. If using small interfering RNAs one could target a non-homologous sequence region or introduce silent point mutations in the human gene that would evade targeting. However, having the endogenous protein copy still present upon expression of the variant can be informative as many variants are found in the heterozygous state in the human genome.
Finally, although not described in this protocol, chromosome aneuploidy assessment can be readily assessed in Met II eggs as the final assay in the pipeline for human variant analysis. A detailed description of this in situ chromosome spread protocol has previously described 15. Briefly, microinjected oocytes are matured in vitro until they reach the Met II stage, following treatment with a meiotic spindle-depolymerizing agent prior to fixation. Kinetochore and chromosome staining as described here allow for assessment of chromosome content.
While mouse oocytes provide a valuable tool for the study of proteins critical to female meiosis some limitations to the model exist. Overexpression of proteins can perturb meiotic maturation and therefore an RNA concentration may not exist that does not induce a phenotype. In addition, the endogenous mouse homolog may interfere with localization of exogenous human protein. Generation of knockout mouse models provides a solution to this problem; however, this may not be feasible in all situations. An alternative approach would be to deplete the mouse protein by co-injecting RNAi or morpholino oligonucleotides designed to not target the exogenous RNA and protein. Finally, it is important to note that while many proteins are homologous between human and mouse, differences may exist, leading to differences in localization and function that could preclude this type of cross-species analysis. As genetic manipulation tools like Cas9-genome editing27 become cheaper and more common-place, this protocol could evolve into making knock-in, humanized mouse lines instead of relying on microinjection.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by a Research Grant from the American Society for Reproductive Medicine and from the Charles and Johanna Busch Memorial Fund at Rutgers, The State University of NJ to K.S. A.L.N. was supported by a grant from the N.I.H. (F31 HD0989597).
References
- Thoma ME, et al. Prevalence of infertility in the United States as estimated by the current duration approach and a traditional constructed approach. Fertil Steril. 2013;99(5):1324–1331. doi: 10.1016/j.fertnstert.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin J, Bunting L, Collins JA, Nygren KG. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Human Reproduction. 2007;22(6):1506–1512. doi: 10.1093/humrep/dem046. [DOI] [PubMed] [Google Scholar]
- Treff NR, Zimmerman RS. Advances in Preimplantation Genetic Testing for Monogenic Disease and Aneuploidy. Annu Rev Genomics Hum Genet. 2017. [DOI] [PubMed]
- Franasiak JM, et al. The nature of aneuploidy with increasing age of the female partner: a review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil Steril. 2014;101(3):656–663. doi: 10.1016/j.fertnstert.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2(4):280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- Scott RT., Jr Blastocyst biopsy with comprehensive chromosome screening and fresh embryo transfer significantly increases in vitro fertilization implantation and delivery rates: a randomized controlled trial. Fertil Steril. 2013;100(3):697–703. doi: 10.1016/j.fertnstert.2013.04.035. [DOI] [PubMed] [Google Scholar]
- Scott RT, Jr, Upham KM, Forman EJ, Zhao T, Treff NR. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: a randomized and paired clinical trial. Fertil Steril. 2013;100(3):624–630. doi: 10.1016/j.fertnstert.2013.04.039. [DOI] [PubMed] [Google Scholar]
- Yang Z, et al. Selection of single blastocysts for fresh transfer via standard morphology assessment alone and with array CGH for good prognosis IVF patients: results from a randomized pilot study. Mol Cytogenet. 2012;5(1):24. doi: 10.1186/1755-8166-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio C, et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: a randomized, controlled study. Fertil Steril. 2017;107(5):1122–1129. doi: 10.1016/j.fertnstert.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Nguyen AL, et al. Identification and characterization of Aurora Kinase B and C variants associated with maternal aneuploidy. Mol Hum Reprod. 2017. [DOI] [PMC free article] [PubMed]
- Igarashi H, Knott JG, Schultz RM, Williams CJ. Alterations of PLCbeta1 in mouse eggs change calcium oscillatory behavior following fertilization. Dev Biol. 2007;312(1):321–330. doi: 10.1016/j.ydbio.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Database JSE. Basic Methods in Cellular and Molecular Biology. Molecular Cloning. JoVE. 2017.
- Carey MF, Peterson CL, Smale ST. PCR-mediated site-directed mutagenesis. Cold Spring Harb Protoc. 2013;2013(8):738–742. doi: 10.1101/pdb.prot076505. [DOI] [PubMed] [Google Scholar]
- Armstrong JA, Schulz JR. Current Protocols Essential Laboratory Techniques. John Wiley & Sons, Inc; 2008. [Google Scholar]
- Stein P, Schindler K. Mouse oocyte microinjection, maturation and ploidy assessment. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Watanabe Y. Geometry and force behind kinetochore orientation: lessons from meiosis. Nat Rev Mol Cell Biol. 2012;13(6):370–382. doi: 10.1038/nrm3349. [DOI] [PubMed] [Google Scholar]
- Shuda K, Schindler K, Ma J, Schultz RM, Donovan PJ. Aurora kinase B modulates chromosome alignment in mouse oocytes. Mol Reprod Dev. 2009;76(11):1094–1105. doi: 10.1002/mrd.21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SI, Yun Y, Jones KT. Timing of anaphase-promoting complex activation in mouse oocytes is predicted by microtubule-kinetochore attachment but not by bivalent alignment or tension. Development. 2012;139(11):1947–1955. doi: 10.1242/dev.077040. [DOI] [PubMed] [Google Scholar]
- Nguyen AL, et al. Phosphorylation of threonine 3 on histone H3 by haspin kinase is required for meiosis I in mouse oocytes. J Cell Sci. 2014;127(Pt 23):5066–5078. doi: 10.1242/jcs.158840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsafriri A, Chun SY, Zhang R, Hsueh AJ, Conti M. Oocyte maturation involves compartmentalization and opposing changes of cAMP levels in follicular somatic and germ cells: studies using selective phosphodiesterase inhibitors. Dev Biol. 1996;178(2):393–402. doi: 10.1006/dbio.1996.0226. [DOI] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. Eg5. J Cell Biol. 2000;150(5):975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KT, Lane SI. Molecular causes of aneuploidy in mammalian eggs. Development. 2013;140(18):3719–3730. doi: 10.1242/dev.090589. [DOI] [PubMed] [Google Scholar]
- Fellmeth JE, et al. Expression and characterization of three Aurora kinase C splice variants found in human oocytes. Mol Hum Reprod. 2015;21(8):633–644. doi: 10.1093/molehr/gav026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL. The structure of the cold-stable kinetochore fiber in metaphase PtK1 cells. Chromosoma. 1981;84(1):145–158. doi: 10.1007/BF00293368. [DOI] [PubMed] [Google Scholar]
- Brunet S, et al. Kinetochore fibers are not involved in the formation of the first meiotic spindle in mouse oocytes, but control the exit from the first meiotic M phase. J Cell Biol. 1999;146(1):1–12. doi: 10.1083/jcb.146.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12(4):828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]