

Abstract
While the introduction of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulator drugs has revolutionized care in Cystic Fibrosis (CF), the genotype-directed therapy model currently in use has several limitations. First, rare or understudied mutation groups are excluded from definitive clinical trials. Moreover, as additional modulator drugs enter the market, it will become difficult to optimize the modulator choices for an individual subject. Both of these issues are addressed with the use of patient-derived, individualized preclinical model systems of CFTR function and modulation. Human nasal epithelial cells (HNEs) are an easily accessible source of respiratory tissue for such a model. Herein, we describe the generation of a three-dimensional spheroid model of CFTR function and modulation using primary HNEs. HNEs are isolated from subjects in a minimally invasive fashion, expanded in conditional reprogramming conditions, and seeded into the spheroid culture. Within 2 weeks of seeding, spheroid cultures generate HNE spheroids that can be stimulated with 3',5'-cyclic adenosine monophosphate (cAMP)-generating agonists to activate CFTR function. Spheroid swelling is then quantified as a proxy of CFTR activity. HNE spheroids capitalize on the minimally invasive, yet respiratory origin of nasal cells to generate an accessible, personalized model relevant to an epithelium reflecting disease morbidity and mortality. Compared to the air-liquid interface HNE cultures, spheroids are relatively quick to mature, which reduces the overall contamination rate. In its current form, the model is limited by low throughput, though this is offset by the relative ease of tissue acquisition. HNE spheroids can be used to reliably quantify and characterize CFTR activity at the individual level. An ongoing study to tie this quantification to in vivo drug response will determine if HNE spheroids are a true preclinical predictor of patient response to CFTR modulation.
Keywords: Medicine, Issue 134, Cystic Fibrosis, CFTR, Precision Medicine, Organoid, Spheroid, Nasal Cell, Cell Culture
Introduction
Cystic Fibrosis (CF) is a fatal, autosomal recessive disease affecting over 70,000 people worldwide1. This life-shortening genetic disease is caused by mutations in the Cystic Fibrosis Transmembrane conductance Regulator protein (CFTR)2. CFTR is a member of the adenosine triphosphate-binding cassette family, and functions as an ion channel allowing movement of chloride and bicarbonate across the apical membranes of multiple polarized epithelia including the gastrointestinal tract, sweat gland, and respiratory tree, among others3,4. As such, dysfunctional CFTR leads to multisystem epithelial dysfunction, with most mortality stemming from the respiratory disease1. In the CF lung, loss of CFTR-driven airway surface liquid (ASL) regulation and mucus release leads to thickened mucus, airway obstruction, chronic infection, and progressive airway remodeling and loss of lung function1,5.
Despite the identification of CFTR dysfunction as the cause of disease, therapies in CF traditionally focused on mitigation of symptoms (e.g., pancreatic enzyme replacement therapy, airway clearance therapies)1. This approach was recently revolutionized by the advent of novel therapeutics, termed "CFTR modulators," that directly target dysfunctional CFTR. This approach has shifted the clinical landscape from symptom management to disease-modifying care but carries several limitations6,7,8,9,10. Modulator activity is specific to the protein defect accompanying each CFTR mutation and limited to select genetic populations11. This limitation is driven by the heterogeneous nature of protein defects, but also by the impracticality of clinical trials in rare mutation groups. In addition, among subjects with well-studied genotypes (e.g., F508del/F508del CFTR, the most common CFTR mutation), there is wide variability in disease burden and modulator response6,7,8,9,11.
To overcome both of these issues, investigators have proposed the use of personalized models for preclinical testing12. This concept utilizes patient-specific tissue to generate an individualized ex vivo model system for compound testing, predicting in vivo subject response to therapies in a personalized fashion. Once validated, such a model could be used by clinicians to drive precision therapy regardless of the patient's underlying CFTR genotype.
Human bronchial epithelial (HBE) cells obtained from explant tissue at the time of lung transplant established the possibility of such a model for CF13,14. HBEs grown at an air-liquid interface (ALI) allow for functional CFTR quantification directly through electrophysiologic testing or indirectly through measures of ASL homeostasis13,15. This model has been critical to understanding CFTR biology and was a key driver in the development of CFTR modulators16. Unfortunately, HBE models are not tenable as a personalized model due to the invasive nature of acquisition (lung transplant or bronchial brushing) and lack of samples for those with rare mutations or mild disease. Conversely, intestinal tissue, obtained from rectal or duodenal biopsy specimens, can be used for intestinal current measurement (ICM) or a swelling-based organoid assay to study individualized CFTR function17,18,19. Organoid assays, in particular, are very sensitive models of CFTR activity20,21,22. Both models are limited by the tissue source (intestinal tissue, while most disease pathology is respiratory) and the semi-invasive method of acquisition. Alternatively, several investigators have studied human nasal epithelial (HNE) cells to model CFTR restoration23,24,25. HNEs can be safely harvested by brush or curettage in subjects of any age and, when cultured in ALI, recapitulate many characteristics of HBEs25,26,27,28. HNE monolayer cultures have traditionally been limited by squamous transformation and a long time to maturity29. Moreover, reported short-circuit current measurements in HNEs are smaller than those in HBEs, suggesting a smaller window to detect therapeutic efficacy25.
Given the need for a personalized, non-invasive, respiratory tissue culture model of CFTR function, we sought to merge the sensitivity of a swelling-based, organoid assay with the non-invasive and respiratory nature of HNEs. Here, we describe our method of generating 3-dimensional "spheroid" cultures of HNEs for individualized CFTR study in a swelling-based assay30. HNE spheroids polarize reliably with the epithelial apex towards the sphere center, or lumen. This model recapitulates numerous characteristics of a lower respiratory epithelium and matures more quickly than ALI cultures. As a functional assay, HNE spheroids reliably quantify a range of CFTR function, as well as modulation in well-studied mutation groups (e.g., F508del CFTR). This swelling-based assay capitalizes on the ion/salt transport properties of CFTR, indirectly measuring fluid influx into the spheroid as water follows apical salt efflux. In this fashion, stimulated spheroids with fully functional CFTR swell robustly, while those with dysfunctional CFTR swell less or shrink. This is quantified through image analysis of pre- and 1 h post-stimulation spheroids, measuring luminal area and determining the percent change. This measure can then be compared across experimental groups to screen for drug bioactivity in a patient-specific fashion.
Protocol
HNE samples were procured from subjects recruited through the Cincinnati Children's Hospital Medical Center CF Research Center. All methods described here have been approved by the Institutional Review Board (IRB) of Cincinnati Children's Hospital Medical Center. Written consent was obtained from all subjects prior to testing.
1. Prepare Expansion Media and Antibiotic Media
Gather media components listed in Table 1. Thaw frozen materials at room temperature and make necessary stock solutions as indicated in Table 1 under "Expansion Media". NOTE: Use care when handling cholera toxin, which is toxic if ingested, and is a skin, eye, and respiratory irritant. Make and combine all solutions and media under clean conditions in the biosafety cabinet.
Using a 50 mL serologic pipette, remove and discard 50 mL of base medium (Dulbecco Modified Eagle Medium/F12) from one container to compensate for the addition of other materials. Pour all base media by hand into an autoclaved 1 L glass bottle.
Using a 50 mL serologic pipette or 1 mL pipette as appropriate, transfer all remaining components from the "Expansion Media" section of Table 1 into the autoclaved glass bottle containing the media. Gently swirl the bottle by hand to mix.
Filter media into a fresh, sterile 1 L, 0.22 µm polyethersulfone (PES) filter flask using the manufacturer's instructions and annotate with "Expansion Media" and date.
- Prepare Antibiotic Media. Make necessary antibiotic stock solutions as indicated in Table 1 under "Antibiotic Media."
- Using a 50 mL serological pipette, transfer 150 mL of Expansion Media to a fresh 250 mL autoclaved glass bottle.
- Using a 1 mL pipette, add all components from the "Antibiotic Media" section of Table 1 into the autoclaved glass bottle containing the media. Swirl by hand to mix until the media is uniform in appearance.
- Filter media into a fresh, sterile 250 mL, 0.22 µm PES filter flask using the manufacturer's instructions and annotate with "Antibiotic Media" and date.
Store media at 4 °C for up to one month, discarding if cloudy, suggesting contamination.
2. Prepare Differentiation Media
Gather media components listed in Table 2. Thaw frozen materials at room temperature and make necessary stock solutions as indicated in Table 2. NOTE: Use caution when handling retinoic acid, which is a reproductive toxin, can be toxic if swallowed, and causes skin irritation. Make, aliquot, and combine all solutions and media under clean conditions in the biosafety cabinet.
Remove and discard 52 mL of the base medium from one container to compensate for the addition of other materials. Pour all base media into an autoclaved 1 L glass bottle.
Using a 25 mL serological pipette or 1-mL pipette as appropriate, transfer all remaining components from Table 2 to the autoclaved glass bottle containing the media and swirl gently by hand to mix.
Filter media into a fresh, sterile 1 L, 0.22 µm polyethersulfone (PES) filter flask using the manufacturer's instructions and annotate with "Differentiation Media" and date.
Store media at 4 °C for up to one month, discarding if cloudy, suggesting contamination.
3. Coat Culture Dishes and Plate Feeder Fibroblasts
NOTE: Perform all steps under clean conditions in the biosafety cabinet.
Mix 0.5 mL of collagen stock and 37.5 mL of sterile water in a 50 mL conical tube.
Pipette 4-5 mL of collagen solution into each clean P100 culture dish, ensuring the entire surface is covered.
Cover dishes with lids and incubate overnight at 37 °C and 5% CO2.
In the biosafety cabinet, remove all dish lids. Swirl solution to cover dish, then aspirate.
Sterilize by exposure to ultraviolet (UV) light for 1 h. Place lids back on the dishes, stack and cover dishes with aluminum foil.
Store coated dishes at 4 °C for 3-6 months, re-sterilizing under UV light 15-30 min before cell culture use.
24 h before obtaining a HNE sample, sterilize a pre-coated dish and label as Mouse Embryonic Fibroblast (MEF) and the date.
Add the Expansion Media to cover the plate (approximately 5 mL) with a serologic pipette.
Thaw a vial of 2 x 106 irradiated MEFs in a 37 °C waterbath. As soon as the MEFs are thawed, add half of the vial (approximately 106 cells) to the dish with a 1-mL pipette, swirling by hand to disperse.
Incubate at 37 °C and 5% CO2 overnight in preparation for HNE culture. NOTE: If necessary, steps 3.7-3.10 can be performed as late as 60 min before plating cells, though overnight is ideal.
4. Obtain and Process HNE Sample
Ensure IRB-approved consent/assent is obtained for all subjects.
Have patient blow nose to clear loose mucus.
Visualize inferior turbinate using a rhinoscope. Ensure no lesions or polyps are present that may preclude sample collection.
- Collect nasal cells from the first nostril by curettage or brushing.
- Curettage: Bend curette at its midpoint to create a slight, ~135° angle with the apex away from the curette cup. Insert the curette into the nostril and advance the curette cup under the inferior turbinate. Gently press the curette cup against the inferior turbinate and scrape the turbinate by pulling the curette forward, repeating 3-4 times total.
- Brush: Pass the cytology brush below the inferior turbinate, then rotate and move the brush in-and-out slightly 4-5 times, without exiting the nostril.
Place curette/brush in a 15 mL conical filled with Antibiotic Media. Repeat steps 4.3-4.4 for the other nostril, placing the curette/brush into the same conical tube.
Store conical on ice until ready for processing. Ensure that the processing occurs within 4 h of sample acquisition but can occur as late as 24 h after acquisition if necessary.
5. Process and Expand HNE Cells in Dishes
NOTE: Perform all steps under clean conditions in the biosafety cabinet.
Using a 1-mL pipette and media from the sample conical, wash all cells off of curettes/cytology brushes into the same conical tube. Once clean, discard curettes / brushes.
Centrifuge conical at 360 x g and 4 °C for 5 min.
Aspirate supernatant, taking care not to disturb the cell pellet.
Re-suspend the cell pellet in 3 mL of cell detachment solution, pipetting with a 1 mL tip to homogenize the mixture.
Centrifuge conical at 360 x g and 4 °C for 5 min.
Repeat steps 5.3-5.5 once.
Aspirate remaining supernatant, taking care not to disturb the cell pellet.
Re-suspend the pellet in 1 mL of Antibiotic Media and count cells with a hemocytometer.
Using a 1-mL pipette, plate cells dropwise onto the coated, fibroblast-seeded dish prepared in step 3.10. Add Antibiotic Media to fully cover the dish and disperse the cells across the surface. Label dish with contents and date and incubate at 37 °C and 5% CO2.
After 48 h, ensure that the cells are attached to the culture dish. Aspirate the media and replace with enough fresh Antibiotic Media to cover the plate.
Change media 3x weekly, aspirating old media with a vacuum line and Pasteur pipette, and replacing with enough fresh media to cover the plate. After 5 days on Antibiotic Media, change the media to Expansion Media, continuing to replace three times weekly. NOTE: Contamination risk is high in the first week of culture. Keep fresh samples in a separate incubator to minimize the risk of cross-contamination.
Check the cell growth several times weekly. Once cells are approximately 70-80% confluent, proceed to passage cells.
6. Passage HNE Cells
- Prepare 0.1% Trypsin solution in ethylenediaminetetraacetic acid (EDTA). NOTE: Use caution handling trypsin, which is a skin, respiratory, and eye irritant.
- Weigh 15 mg of trypsin from the porcine pancreas in a conical tube.
- Weigh 56 mg of EDTA in a separate conical tube.
- In the biosafety cabinet, transfer trypsin and EDTA into an autoclaved 250 mL glass bottle. Use a small aliquot of sterile phosphate-buffered saline (PBS) to rinse the trypsin/EDTA tubes to ensure complete transfer.
- Add sterile PBS to bring the solution total to 150 mL. Close lid tightly.
- Incubate trypsin solution in a 37 °C waterbath until fully dissolved. Check every 15 min and remove promptly once dissolved. NOTE: Do not leave unchecked for prolonged periods of time (e.g., overnight), as trypsin in solution will denature if heated for too long.
- Clean bottle with 2-3 sprays of 70% ethanol and return to the biosafety cabinet.
- Filter 0.1% Trypsin-EDTA solution into a fresh, sterile 250 mL, 0.22 µm PES filter flask using the manufacturer's instructions and annotate with contents and date.
- Store trypsin-EDTA solution at 4 °C for up to one month, discarding if cloudy.
Bring Differentiation Media, 0.1% Trypsin-EDTA solution, and sterile PBS to room temperature over 30 min. Clean with 70% ethanol and transfer to a clean biosafety cabinet.
In the biosafety cabinet, use a vacuum line and Pasteur pipette to aspirate and discard media from tissue culture dish. Wash the dish by adding, swirling, and aspirating 5 mL of PBS using a clean serological pipette.
Using a 5-mL serological pipette, add 5 mL of 0.1% Trypsin-EDTA solution to the tissue culture dish and return to the incubator at 37 °C and 5% CO2 for 5 min.
Visualize cells on an inverted microscope at 4-10X. Check that the cells detach from the dish, become round, and float. Gently tap the side of the culture dish to dislodge cells, and/or re-incubate for an additional 5 min until most cells are detached. NOTE: Take caution to quickly remove cells once detached from the culture dish; prolonged exposure to 0.1% Trypsin-EDTA will impair cell viability.
Using a 10 mL serological pipette, add 5 mL of Expansion Media to the dish and collect all liquid contents (10 mL total) into a labeled, sterile 50 mL conical tube.
- If cells remain adherent to the culture dish, repeat steps 6.4 through 6.6 up to two times, collecting contents in the same conical tube.
- If cells remain adherent to the culture dish after three rounds of Trypsin solution exposure, use a sterile cell scraper gently remove the remainder. After scraping the dish, wash the scraper and dish with 5 mL of Expansion Media and collect in the same labeled conical tube. NOTE: If passaging to spheroids, modify this procedure to perform a differential trypsinization. After adding trypsin solution (step 6.4.), check the dish frequently until fibroblasts have detached (typically 1-2 min), leaving only polygonal epithelial cells attached to the dish. Collect and set aside this supernatant, which can be passaged forward for expansion. Repeat steps 6.4-6.6 using the same trypsin solution, and collect only the remaining epithelial cells for spheroid culture.
Centrifuge conical at 360 x g for 5 min and 4 °C. Aspirate media from conical.
Resuspend cell pellet in 1 mL of Expansion Media and count cells using a hemacytometer.
Once quantified, cells can be divided if necessary and either passaged forward onto a new culture dish (including preparation of fibroblast co-culture, step 3, and plating cells for expansion, steps 5.9-5.12) or cultured as spheroids (step 7). NOTE: If passaging to a new culture dish for expansion, a seeding density of 0.5-1 × 106 cells in a P100 dish is recommended.
7. Seeding Cells for HNE Spheroid Cultures
NOTE: Carry out all procedures in this step, other than centrifugation, in a clean biosafety cabinet.
Thaw the basement membrane matrix on ice per manufacturer's instructions (100 µL per spheroid well). NOTE: Consider making sterile 500 µL aliquots of the basement membrane matrix on receipt; this amount will seed a single 4-well plate.
Separate an appropriate number of differentially trypsinized, passaged cells (from step 6.9) for a final concentration of 500,000 cells per mL of the matrix. For a 4-well plate, use 200,000 cells. Collect in a 1.5 mL tube.
Centrifuge cell and media mixture at 360 x g for 5 min at 4 °C. Completely, but carefully aspirate and discard media using a 1 mL pipette, taking care not to disturb the cell pellet.
Using a 1 mL pipette tip, add 100 µL per planned well of the basement membrane matrix to the 1.5 mL tube containing the cells. Keep the tube on ice.
Pipette up and down to carefully and thoroughly resuspend cells in the basement membrane matrix. Move quickly to avoid solidification of the matrix. Avoid completely emptying the pipette tip to ensure air bubbles are not introduced into the matrix/cell mix.
- Seed 100 µL matrix aliquots into each well of a 4-well IVF culture plate.
- Using a clean pair of scissors, trim off the distal 3-4 mm of a 200 µL pipette tip.
- Using the trimmed tip, carefully pipette 100 µL of the cell/matrix mixture into the center of each well. Pipette slowly, with the goal of making a single matrix "drop" in the center of each well. The drop should remain spherical, and should not touch the sides of the well. When moving the plate, do so carefully as to avoid disturbing these drops.
- Incubate the basement membrane matrix drops at 37 °C and 5% CO2 for 30 min, until solid.
- Carefully pipette 500 µL of Differentiation Media into each well, taking care not to disturb the matrix drop. Ensure that the media just cover the matrix drop and add more if necessary.
8. Differentiate and Mature HNE Spheroids
Using a 1 mL pipette tip, gently aspirate all media from well; avoid aspirating the matrix, which will destroy spheroids in that portion of the matrix, though any spheroids left behind may remain viable.
Using a fresh 1 mL pipette tip, gently add 500 µL of differentiation media to the well around the matrix. Do not touch the matrix with the pipette and avoid disturbing the matrix drop.
Return spheroids to the incubator at 37 oC and 5% CO2 for ongoing growth. Repeat steps 8.1 through 8.3 three times weekly.
Evaluate spheroid morphology daily under an inverted microscope at 20X. Spheroids should form within 3-5 days of plating, and reach maturity within approximately 10 days.
9. Pretreat, Stimulate, and Image HNE Spheroids for Analysis
Pretreat spheroids as desired prior to imaging as previously described30. NOTE: For VX809 pretreatment, add 3 µM of VX809 to the media for 48-72 h prior to imaging. Mix 1 µL of 10 mM VX809 stock (see Materials Table) in 3.3 mL of media for this concentration.
Change each well of spheroids to 1 mL of fresh Differentiation Media, including pretreatment (steps 8.1-8.3) before imaging spheroids.
Prepare fresh stimulation drugs solutions (Forskolin, 3-isobutyl-1-methylxanthine [IBMX], VX770, and CFTR Inhibitor 172 [Inh172]) from stocks (see Materials Table). For forskolin/IBMX, add 1 µL of each stock to 98 µL of sterile water in a single labeled 1.5 mL tube. For VX770 and Inh172, add 1 µL of each stock to 99 µL of sterile water in separate labeled 1.5 mL tubes. Make a total volume allowing for 50 µL of solution for each well tested. Prepare solutions under sterile conditions in the biosafety cabinet.
Transfer spheroid plate to an incubated chamber set to 37 °C and 5% CO2, mounted on an inverted microscope equipped with electronic image capture software and a mechanical stage for XY adjustment. Turn on the microscope and camera, as well as the imaging computer.
- Capture baseline images of spheroids in one well.
- Open the image capture software (Slidebook 5.5 for directions below). Once initialized, open a new file by clicking "File", then "New". Name the file accordingly and click "Save".
- Carefully remove the culture plate lid and center one matrix drop over the objective.
- Starting at the top of the matrix drop, move in a grid to find spheroids at 20x magnification with the microscope eyepiece. Focus up and down to capture the sphere at its widest point. Annotate the location of each spheroid on a map of the matrix drop to re-identify after imaging.
- In the software, click "Image-Capture". For Exposure, select "Manual" and enter 50 ms. Ensure other settings are appropriate (Bin Factor: 1x1, W: 512, H: 512, X and Y Offset: 0). Enter an appropriate name for each image in the "Name" box (e.g., Spheroid 1 Baseline). Click "Start" to view a live image, and "Save" to capture a baseline image.
- Repeat steps 9.5.1-9.5.4 until goal number is reached, or entire matrix drop is imaged. NOTE: Group differences can be detected with as few as 3-5 spheroids per condition, however, it has become our practice to image 10 or more spheroids per condition to increase power. Variability of the assay is demonstrated in Figure 2 of the "Representative Results" section, with samples of 8-10 spheroids per subject as an example.
- Stimulate spheroids.
- Using a 200 µL pipette, add 50 µL of the appropriate stimulation drug into the media within the well, taking care not to disturb the matrix. NOTE: After this 10-fold dilution (50 µL into 500 µL of media), the final drug concentration will be: forskolin 10 µM, IBMX 100 µM, VX770 1 µM, Inh172 10 µM.
- For cAMP only, add only forskolin/IBMX.
- For cAMP + VX770, add forskolin/IBMX first, then VX770 after 2 min.
- For inhibited conditions, add Inh172 first, then forskolin/IBMX after 2 min.
Immediately after stimulation, monitor spheroid swelling by timelapse imaging for 1 h. In the software, click "Image-Capture". Click "Timelapse", and set the interval to 30 s, with a duration of 60 min. Enter an appropriate name and click "Save" to start timelapse. NOTE: Standard timelapse is to capture images of a single spheroid every 30 s to ensure a high-quality video. Alternatively, perform lower-magnification capture of the entire well (e.g., 4X), and fewer images captured if desired. If an automated stage is used, perform timelapse of all spheroids using predefined coordinates; otherwise, use timelapse of a subset of spheroids to provide a qualitative review of swelling and to ensure no systematic issues arise during imaging (e.g., matrix detachment from the well).
- Immediately upon completion of the 1 h timelapse image, capture post-stimulation images of all spheroids (per step 9.5) using the map created in step 9.5.3.
- Refer to pre-stimulation images to ensure the same plane of focus is used for follow-up images (the focal quality of surrounding cells, spheroids, or debris greatly facilitate this process).
- Save files with a paired annotation from step 9.5.4 (e.g., Spheroid 1 Post-Stimulation). NOTE:Complete this step immediately after the timelapse, as spheroids may continue to swell.
Replace media in the imaged well with fresh Differentiation Media.
Repeat steps 9.5 through 9.9 for each well/condition, adjusting stimulation drugs as indicated for experimental conditions.
Once all imaging is completed, return spheroids to the incubator at 37 °C and 5% CO2. NOTE: Depending on the sterility of the incubated microscope chamber, post-stimulation contamination of spheroids can be quite common. Either keep imaged spheroids in a separate incubator, to minimize cross-contamination risk, or discard spheroids immediately after imaging.
10. Analyze HNE Spheroid Images
Export all captured images to a format compatible with image analysis software. In the software, click "View", hover over "Export", and select "Default Views of All Images as TIFF...". Save to the hard drive or a flash drive for analysis. NOTE:A commercial analysis software (Table of Materials) and .tiff files perform well and are described here, though analysis can also be performed using other platforms. When naming files, staff analyzing images should be blinded to the experimental conditions, but aware of which images are paired (pre- and post-stimulation) to ensure equivalent analysis.
- Using analysis software, delineate the luminal area of each spheroid image and export into a data analysis software.
- Open analysis software. Click "File", then "Open" and navigate to the file folder containing spheroid images. Select all images for analysis and click "Open".
- Click "Measure" and select "Region Measurements". Within the Region Measurements dialog box, select the droptab "Configuration" and ensure "Image Name" and "Area" boxes are checked.
- Click " Log" and select "Open Data Log". Click "OK" on the Data Log dialogue box and name file accordingly (e.g., Condition X, Date). This will transfer all measurements to a spreadsheet.
- Select " Trace Region" tool button and carefully trace the lumen of the spheroid in the first image for analysis. In the Region Measurement dialogue box, click " Log Data" to log the measurement into Excel.
- Repeat steps 10.2.2-10.2.4 until all spheroids are analyzed.
Calculate the percent change (100 * [Post-stim - Baseline] ÷ Baseline) in the luminal area from baseline for each individual spheroid image pair and compile data for analysis.
Representative Results
HNEs should attach to the culture dish and form small islands of cells within 72 h of seeding; examples of good and poor island formation at one week are shown in Figure 1A and 1B, respectively. These islands should expand to cover the dish over the course of 15-30 days. Small or suboptimal samples may take longer, and often will not yield useful spheroids. Contamination with infectious agents is evidenced by deep yellow/cloudy media, failure of the cells to attach to the culture dish, and/or direct visualization of fungi/bacteria. Any contaminated cultures should be immediately discarded to avoid cross-contamination.
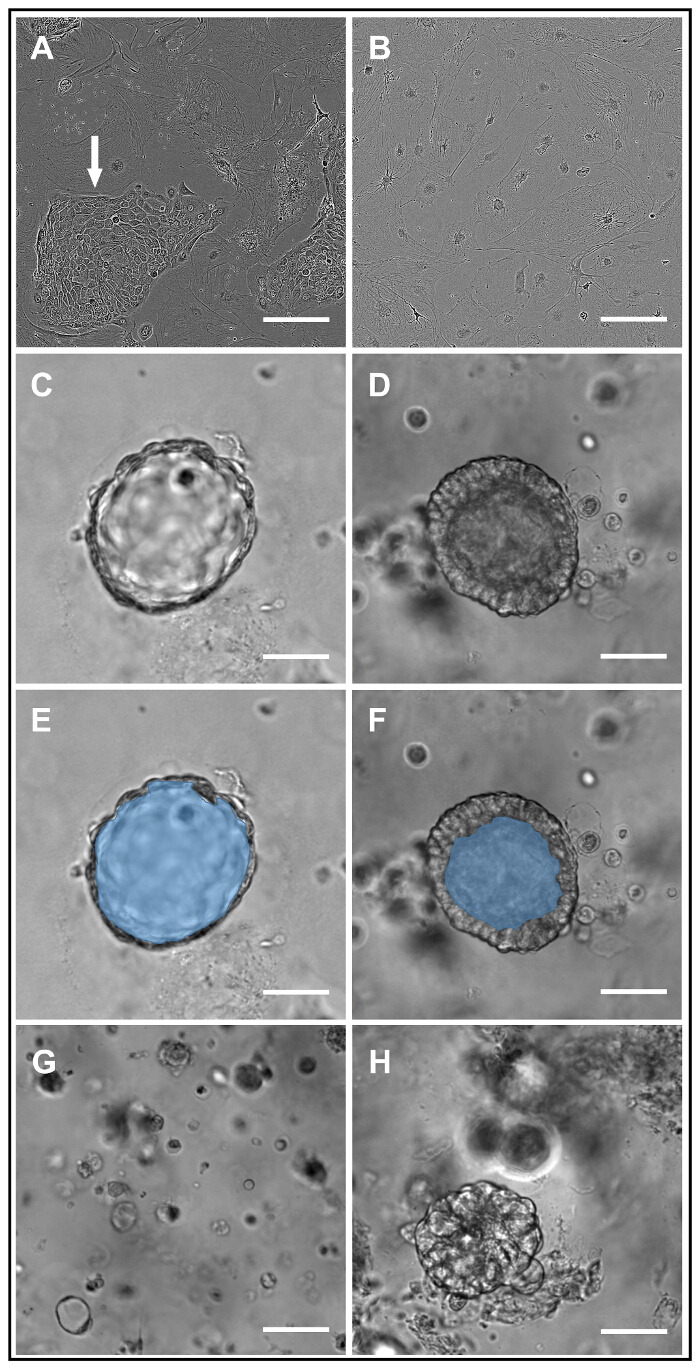
Within the first 3-4 days of culture in the basement membrane matrix, small cystic structures should begin to form in the matrix. These will mature over approximately 10 days into intact spheroids demonstrating a thin wall and a luminal surface. If plated at the described density, successful cultures will contain 50-100 spheroids per matrix drop. The lumens may be relatively clear (Figure 1C) or filled with cellular debris and mucus (Figure 1D); the former is more common in spheroids with wild-type CFTR (wtCFTR), and the latter in CF spheroids. Masking to delineate the luminal area of the spheroids in Figure 1C and 1D is demonstrated in Figure 1E and 1F, respectively. Examples of poorly formed/unsuccessful spheroid cultures are provided in Figure 1G and 1H.
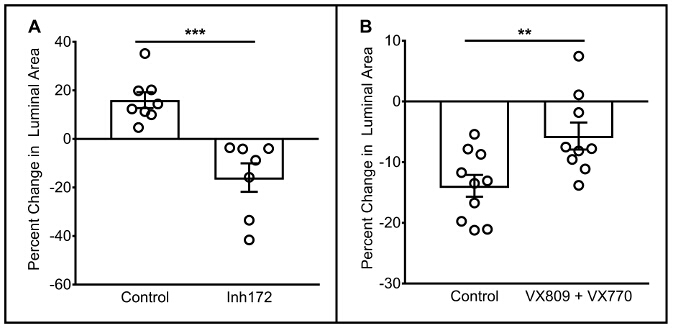
Representative functional data for wild type and F508del CFTR homozygous HNE spheroids is shown in Figure 2A and 2B, respectively; this data is representative of >10 unique HNE samples in wild type and F508del CFTR homozygous subjects30. In short, spheroids with functional CFTR swell, while those with dysfunctional CFTR swell significantly less, or may shrink. Specifically, spheroids from subjects with wtCFTR should swell over an hour when stimulated, and should swell less or shrink if stimulated in the presence of the CFTR inhibitor Inh172. Conversely, spheroids from a subject homozygous for F508del CFTR should either shrink or swell very slightly, with increased swelling (or less shrinking) when pharmacologically corrected with the CFTR modulators VX809 and VX770. Previous analyses of spheroid reliability both within and between subjects of the same genotype demonstrate functional segregation of CFTR genotypes and modest variability in repeated measures30.
| Media Component | Stock Solution | Amount | Storage |
| Expansion Media | |||
| DMEM / F-12 Nutrient Mixture "Base Media" | Use as is | 2x 500 mL containers | Store at 4 ºC up to manufacturer expiration date |
| Fetal Bovine Serum | Use as is | 50 mL | Store at -20 ºC up to manufacturer expiration date |
| Cholera Toxin | 10 mg in 1 mL of sterile water | 1 µL | Store stock at -20 ºC up to six months |
| Epidermal Growth Factor | 500 µg in 1 mL of sterile water | 20 µL | Store stock at -20 ºC up to six months |
| Hydrocortisone | 0.4 mg in 400 µL of sterile water | Entire 400 µL aliquot | Make fresh with each batch. Store powder at room temperature up to manufacturer expiration date |
| Adenine | 24 mg in 1 mL of sterile water | Entire 1 mL aliquot | Make fresh with each batch. Store powder at room temperature up to manufacturer expiration date |
| Y-27632 | 3.2 mg in 1 mL of sterile water | Entire 1 mL aliquot | Make fresh with each batch. Store powder at -20 ºC up to manufacturer expiration date |
| Antibiotic Media | |||
| Amphotericin B | Use as is | 1.2 mL | Store at 4 ºC up to manufacturer expiration date |
| Ceftazidime | 15 mg in 1 mL of sterile water | Entire 1 mL aliquot | Make fresh with each batch. Store powder at -20 ºC up to manufacturer expiration date |
| Tobramycin | 15 mg in 1 mL of sterile water | Entire 1 mL aliquot | Make fresh with each batch. Store powder at -20 ºC up to manufacturer expiration date |
| Vancomycin | 15 mg in 1 mL of sterile water | Entire 1 mL aliquot | Make fresh with each batch. Store powder at -20 ºC up to manufacturer expiration date |
Table 1: Components of Expansion and Antibiotic Media.
| Media Component | Stock Solution | Amount | Storage |
| DMEM / F-12 Nutrient Mixture "Base Media" | Use as is | 2x 500 mL containers | Store at 4 ºC up to manufacturer expiration date |
| Ultroser-G | 20 mL of sterile water in a single, 20 mL bottle of lyophilized Ultroser-G | Entire 20 mL aliquot | Make fresh with each batch. Store powder at 4 ºC up to manufacturer expiration date |
| Fetal Clone II | Use as is | 20 mL | Store at -20 ºC up to manufacturer expiration date |
| Pen Strep | Use as is | 10 mL | Store at -20 ºC up to manufacturer expiration date |
| Bovine Brain Extract | Use as is | 2.48 mL | Store at -20 ºC up to manufacturer expiration date |
| Transferrin | Use as is | 250 µL | Store at -20 ºC up to manufacturer expiration date |
| Insulin | Use as is | 250 µL | Store at -20 ºC up to manufacturer expiration date |
| Ethanolamine | Use as is | 15 µL | Store at room temperature up to manufacturer expiration date |
| Epinephrine | 2.75 mg in 1 ml of sterile water | Entire 1 mL aliquot | Make fresh with each batch. Store powder at 4 ºC up to manufacturer expiration date |
| Triiodothyronine | 8.4 mg in 50 µL of DMSO | Entire 50 µL aliquot | Make fresh with each batch. Store powder at -20 ºC up to manufacturer expiration date |
| Hydrocortisone | 7.24 mg in 1 mL of ethanol | 1 µL | Store stock at -20 ºC up to six months |
| Phsophoryletheanolamine | 35.25 mg in 1 mL of sterile water | 1 µL | Store stock at -20 ºC up to six months |
| Retinoic Acid | 3 mg in 1 mL of DMSO | 1 µL | Store stock at -20 ºC up to six months |
Table 2: Components of Differentiation Medium.
Figure 1: HNE Expansion and Structural Characteristics of HNE Spheroids. Brightfield images of HNE expansion cultures taken seven days after plating demonstrate successful (white arrow, panel A) and unsuccessful (panel B) HNE colony formation on a feeder fibroblast background. Successful wtCFTR and F508del homozygous spheroids are shown in panels C and D, respectively. Masking to delineate the luminal area of spheroids from C/D is provided in panels E and F, respectively. Unsuccessful spheroid cultures are characterized by small, disorganized cellular debris (panel G) and/or disorganized clumps of cells (panel H). Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 2: Functional Characteristics of HNE Spheroids. Representative functional responses of wtCFTR spheroids from a single donor, when stimulated with forskolin/IBMX with/without the presence of the CFTR inhibitor Inh172 are shown in panel (A) Each point represents the response in a single spheroid. Representative functional responses of F508del homozygous spheroids from a single donor, when stimulated with forskolin/IBMX with/without the presence of VX809 and VX770 are shown in panel (B). Error bars = SEM. **p <0.01; ***p <0.001. Please click here to view a larger version of this figure.
Discussion
This protocol describes the generation of patient-derived nasal cell spheroid cultures able to produce an individualized, specific model of CFTR function. There are several key steps in the process that should be closely attended to avoid difficulty. First is a good sample acquisition from the patient's nose. A good sample should have >50,000 cells, limited mucus/debris, and be ready for processing within 4 h (though success is also easily achievable with overnight shipping on ice). Practice acquiring samples with curette or brush by study staff is necessary, which is facilitated by focusing early sample acquisition into the hands of one or two providers to maximize their comfort. Second, good clean/sterile technique in all tissue culture steps is essential. The primary mode of failure, in our experience, for all HNE cultures is bacterial or fungal contamination, which can complicate both the primary sample and other growing cultures in the incubator. This risk only increases with cultures of subjects with chronic infection (e.g., CF), therefore success hinges largely on the ability to keep cultures clean and separate. Our practice is to keep one incubator separate only for infection-prone cultures within the first 5-7 days, protecting older, successful cultures from inadvertent contamination. Third, it is important to closely watch cells during the expansion phase and avoid overconfluence. If the cells are allowed to reach full confluence on the plate, there is a risk that the culture will either become senescent or cells will begin to die and detach. Finally, when plating cells as spheroids, it is important to thoroughly disperse the cells through the matrix and to plate an evenly distributed sample. Either over- or under-population of the matrix drops will lead to culture failure, and large clumps of cells will not produce successful spheroids.
While implementing this protocol, investigators may encounter a few common problems. Contamination, as mentioned above, is best avoided by procuring a good initial sample without mucus and clean culture techniques. If cultures are successful through expansion, but no differentiated spheres are formed, several issues may have occurred. If the matrix appears mostly empty, it is most likely that the cells were seeded at too low of a density; increase the seeding density of subsequent attempts by approximately 20%. Conversely, if the matrix appears to be "dirty" with copious cell debris, it is likely that the cells were seeded at too high of a density, and subsequent attempts should use a seeding density reduced by approximately 20%. A common complication of post-seeding feeding, and maintenance is the detachment of the matrix from the culture vessel. This is caused by overly aggressive media exchange and can be avoided by cautiously and manually changing media with a 1-mL pipette, not wall suction. If encountered, spheres can still be stimulated and imaged, though this may be difficult if the matrix "floats" in the well during imaging, necessitating cautious mapping of spheroids within the well.
Investigators may pursue several modifications to the protocol, depending on the needs of their lab. For higher-resolution imaging of spheroids, we have previously substituted the 4-well plates with optical glass options, including 35-mm glass-bottom dishes or chamber slides. This allows for high-resolution, live imaging of spheroids, but may reduce throughput. Alternatively, to increase throughput, smaller aliquots of cells in the matrix can be seeded into other vessels (e.g., 24-well culture plates). In our experience, spheroids grow best with the matrix in a "droplet" form as opposed to forming a sheet on the bottom of the well; as such, we have had the best success with droplets of at least 25 µL. Investigators may wish to alter the density of matrix by diluting with media. This reduces the total amount of matrix necessary, improving the cost of the assay. This may also, however, alter the spheroid composition. In our previous experience, lower concentrations of the basement membrane matrix lead to altered spheroid structure, with either partial or complete formation of spheroids in a "cell-apex-out" morphology. As such, spheroids generated through any protocol alterations in matrix concentration should be cautiously evaluated for morphology before functional testing is attempted. Finally, different stimulating or inhibitory drugs, or different concentrations of these drugs, could be employed. Forskolin, IBMX, and Inh172 were chosen for our studies at these concentrations based on previous experience in ALI cultures31. Using other drugs (e.g., isoproterenol for stimulation, GlyH101 for inhibition) may better apply to an investigator's study. Similarly, using different concentrations of forskolin, IBMX, or Inh172 may alter the dynamic range of the assay, however, we have not systematically tested these options.
Given the number of existing ex vivo patient-derived CFTR assays, the described model is notable for several key reasons. First, it capitalizes on nasal cells as an easily obtained source of respiratory tissue. HNE procurement can be performed safely with minimal training in almost any setting (clinic, OR, research visit) in all age groups25. This facilitates robust sample procurement, and repeat sampling if necessary due to growth difficulty or contamination. Compared to intestinal organoids, HNE spheroids are less well characterized and appear to have a smaller dynamic range in the present form, however, use of respiratory instead of GI tissue may be a key benefit, as several CF disease drivers (e.g. mucociliary clearance, epithelial sodium channel expression) are not equivalent in the gut. As opposed to planar, ALI cultures of HNE cells, spheroids are grown faster and may be more representative of in vivo conditions, measuring a physiologic process (fluid homeostasis) that may be more relevant than electrophysiology alone32. By improving the time from sample procurement to testing, contamination potential is reduced, therefore increasing the likelihood of culture success. Finally, the 3-dimensional nature of the model may be amenable to the novel and/or complementary lines of research in airway development or morphology that are not feasible in ALI cultures (e.g., luminal mucus tracking, rapid studies of differentiation, etc.).
There are several key limitations to HNE spheroids as an assay of CFTR function. First, this assay remains relatively low-throughput. Using the current methods, image acquisition takes over an hour for each culture condition, and analysis takes an additional 20-30 min per condition. This is compounded by the degree of overlap between certain conditions (such as demonstrated in Figure 2A), which necessitates a larger number of measurements (this overlap in itself may also represent a limitation of the sensitivity of this assay). As such, a single experiment with 4-6 conditions requires almost two days for completion. Throughput can be improved by employing methods of automated image capture and analysis; such adaptations are currently in progress. Secondly, this model requires a large amount of matrix, which can become expensive. As mentioned above, dilution of the matrix may help overcome this barrier, but must be taken with caution, as alterations in the growth matrix will likely result in alterations in spheroid morphology. Third, this model relies on spheroid measurements in an XY plane only, and disregards swelling in the Z plane, which may introduce bias. While previous reproducibility analyses have been reassuring, this shortcoming could be overcome by use of automated imaging with Z-plane scanning to calculate volume30. Finally, and most importantly, extensive work to tie spheroid responses to the individual subject's in vivo drug response is yet to be completed. In absence of this correlation, the predictive value of the model - while promising - is unclear.
Generation and analysis of HNE spheroids allow for ex vivo analysis of individual CFTR activity and modulation, which may ultimately be useful as a pre-clinical model of drug response. Moreover, given the extensive variability in CF disease severity, such an individualized model can provide insights into a subject's unique airway microenvironment. In this way, such a model may be useful for studies of broader CFTR biology and aid in understanding disease heterogeneity. Future use of this model, as well as other similar models of individualized CFTR function, holds promise to better understand CFTR biology in the lab, and to drive personalized and precision medicine in the clinic.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by Cystic Fibrosis Foundation Therapeutics, grant number CLANCY14XX0, and through Cystic Fibrosis Foundation, grant number CLANCY15R0. The authors wish to thank Kristina Ray for her assistance in patient recruitment and regulatory oversight. The authors also wish to thank the HNE working group, supported by the Cystic Fibrosis Foundation, who assisted in generation of HNE culture capabilities: Preston Bratcher, Calvin Cotton, Martina Gentzsch, Elizabeth Joseloff, Michael Myerburg, Dave Nichols, Scott Randell, Steve Rowe, G. Marty Solomon, and Katherine Tuggle.
References
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- Rommens JM, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Anderson MP, et al. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell. 1991;67(4):775–784. doi: 10.1016/0092-8674(91)90072-7. [DOI] [PubMed] [Google Scholar]
- Boucher RC. Human airway ion transport. Part one. Am J Respir Crit Care Med. 1994;150(1):271–281. doi: 10.1164/ajrccm.150.1.8025763. [DOI] [PubMed] [Google Scholar]
- Ehre C, Ridley C, Thornton DJ. Cystic fibrosis: an inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol. 2014;52:136–145. doi: 10.1016/j.biocel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck K, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–680. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Moss RB, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–533. doi: 10.1016/S2213-2600(15)00201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright CE, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015. [DOI] [PubMed]
- Brewington JJ, McPhail GL, Clancy JP. Lumacaftor alone and combined with ivacaftor: preclinical and clinical trial experience of F508del CFTR correction. Expert Rev Respir Med. 2016;10(1):5–17. doi: 10.1586/17476348.2016.1122527. [DOI] [PubMed] [Google Scholar]
- CFTR2 Database. 2017. www.cftr2.org.
- Mou H, Brazauskas K, Rajagopal J. Personalized medicine for cystic fibrosis: establishing human model systems. Pediatr Pulmonol. 2015;50 Suppl 40:S14–S23. doi: 10.1002/ppul.23233. [DOI] [PubMed] [Google Scholar]
- Randell SH, Fulcher ML, O'Neal W, Olsen JC. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol. 2011;742:285–310. doi: 10.1007/978-1-61779-120-8_18. [DOI] [PubMed] [Google Scholar]
- Whitcutt MJ, Adler KB, Wu R. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In Vitro Cell Dev Biol. 1988;24(5):420–428. doi: 10.1007/BF02628493. [DOI] [PubMed] [Google Scholar]
- Worthington EN, Tarran R. Methods for ASL measurements and mucus transport rates in cell cultures. Methods Mol Biol. 2011;742:77–92. doi: 10.1007/978-1-61779-120-8_5. [DOI] [PubMed] [Google Scholar]
- Van Goor F, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug MJ, Tummler B. Intestinal current measurements to diagnose cystic fibrosis. J Cyst Fibros. 2004;3 Suppl 2:157–158. doi: 10.1016/j.jcf.2004.05.033. [DOI] [PubMed] [Google Scholar]
- van Barneveld A, Stanke F, Ballmann M, Naim HY, Tummler B. Ex vivo biochemical analysis of CFTR in human rectal biopsies. Biochim Biophys Acta. 2006;1762(4):393–397. doi: 10.1016/j.bbadis.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Dekkers JF, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- Dekkers JF, et al. Optimal correction of distinct CFTR folding mutants in rectal cystic fibrosis organoids. Eur Respir J. 2016;48(2):451–458. doi: 10.1183/13993003.01192-2015. [DOI] [PubMed] [Google Scholar]
- Dekkers R, et al. A bioassay using intestinal organoids to measure CFTR modulators in human plasma. J Cyst Fibros. 2015;14(2):178–181. doi: 10.1016/j.jcf.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Zomer-van Ommen DD, et al. Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. J Cyst Fibros. 2015. [DOI] [PubMed]
- Bridges MA, Walker DC, Davidson AG. Cystic fibrosis and control nasal epithelial cells harvested by a brushing procedure. In Vitro Cell Dev Biol. 1991;27a(9):684–686. doi: 10.1007/BF02633211. [DOI] [PubMed] [Google Scholar]
- Di Lullo AM, et al. An "ex vivo model" contributing to the diagnosis and evaluation of new drugs in cystic fibrosis. Acta Otorhinolaryngol Ital. 2017;37(3):207–213. doi: 10.14639/0392-100X-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranke IM, et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci Rep. 2017;7(1):7375. doi: 10.1038/s41598-017-07504-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalia JL, Davies RJ. Human nasal and bronchial epithelial cells in culture: an overview of their characteristics and function. Allergy Proc. 1991;12(2):71–79. doi: 10.2500/108854191779011783. [DOI] [PubMed] [Google Scholar]
- Devalia JL, Sapsford RJ, Wells CW, Richman P, Davies RJ. Culture and comparison of human bronchial and nasal epithelial cells in vitro. Respir Med. 1990;84(4):303–312. doi: 10.1016/s0954-6111(08)80058-3. [DOI] [PubMed] [Google Scholar]
- Thavagnanam S, et al. Nasal epithelial cells can act as a physiological surrogate for paediatric asthma studies. PLoS One. 2014;9(1):e85802. doi: 10.1371/journal.pone.0085802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen M, Van der Schueren B, Van den Berghe H, Cassiman JJ. The preservation and regeneration of cilia on human nasal epithelial cells cultured in vitro. Arch Otorhinolaryngol. 1989;246(5):308–314. doi: 10.1007/BF00463582. [DOI] [PubMed] [Google Scholar]
- Brewington JJ, et al. Detection of CFTR function and modulation in primary human nasal cell spheroids. J Cyst Fibros. 2017. [DOI] [PMC free article] [PubMed]
- Sun H, et al. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS One. 2014;9(9):e106842. doi: 10.1371/journal.pone.0106842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med. 2007;261(1):5–16. doi: 10.1111/j.1365-2796.2006.01744.x. [DOI] [PubMed] [Google Scholar]