

Abstract
Proper regulation of DNA replication ensures the faithful transmission of genetic material essential for optimal cellular and organismal physiology. Central to this regulation is the activity of a set of enzymes that induce or reverse posttranslational modifications of various proteins critical for the initiation, progression, and termination of DNA replication. This is particularly important when DNA replication proceeds in cancer cells with elevated rates of genomic instability and increased proliferative capacities. Here, we describe how DNA replication in mammalian cells is regulated via the activity of the ubiquitin-proteasome system as well as the consequence of derailed ubiquitylation signaling involved in this important cellular activity.
Keywords: DNA replication, Ubiquitin, Ubiquitylation E3 ligases, DNA re-replication, Cancer
19.1 Introduction
Eukaryotic DNA replication is a highly regulated process that ensures the faithful transmission of genetic material to daughter cells (Machida et al. 2005). The process is coupled both with cell cycle progression and with the ability of cells to cope with various intrinsic and extrinsic insults that constantly threaten the integrity of the genome. Various repair mechanisms have evolved to cope with these insults, permitting the repair of various lesions prior to the completion of DNA replication (Hustedt and Durocher 2016; Ganai and Johansson 2016; Berti and Vindigni 2016). Critical to this regulation is the ability of cells to sense environmental perturbations, to enforce appropriate checkpoints, and to activate a number of cellular processes conductive of DNA repair prior to the initiation or resumption of DNA replication. Posttranslational modifications of certain proteins play fundamental role in the timely execution of most, if not all, of these cellular activities.
Similar to many physiological processes in the cell, proper regulation of DNA replication is governed through the balanced production and termination of key cellular proteins controlling the various steps involved. DNA replication depends on key enzymatic activities that are controlled through the action of cellular proteins and cofactors that are actively synthesized, modified, or destroyed to achieve optimal activity. ATP-dependent protein polyubiquitylation plays an important role in almost all physiological processes and in many diseases including cancer owing to its role in the global regulation of protein turnover (Schwartz and Ciechanover 2009; Hershko 2005; Amir et al. 2001; Glickman and Ciechanover 2002; Kornitzer and Ciechanover 2000; Ciechanover and Schwartz 2002). The highly coordinated and reversible process ensures timely downregulation of proteins via the activity of the 26S proteasome, where the polyubiquitylated proteins, roughly 80% of all intracellular proteins, are digested into small peptides, and ubiquitin molecules are recycled (Skaar et al. 2014).
Proteasomal degradation through the ubiquitin-proteasome system (UPS) is triggered following the covalent attachment of multiple ubiquitin molecules linked together through lysine 48 (Lys-48) to substrate proteins (Teixeira and Reed 2013; Groll and Huber 2003). Other forms of polyubiquitylation (e.g., linkage through Lys-63 of ubiquitin) do not result in proteasomal degradation but regulate other functions, such as protein trafficking, protein-protein interaction, and kinase activation (Yang et al. 2010; Behrends and Harper 2011). Polyubiquitylation is comprised of three distinct and consecutive enzymatic steps (Fig. 19.1): ubiquitin activation by an E1 ubiquitin-activating enzyme, the transfer of the activated ubiquitin to an E2 ubiquitin-conjugating enzyme (UBC), and the transfer of ubiquitin to the substrate through the activity of an E3 ubiquitin ligase (Kornitzer and Ciechanover 2000; Groll and Huber 2003; Glickman and Ciechanover 2002). This latter activity by E3 ubiquitin ligases is particularly important as it confers specificity for the substrate to be targeted for ubiquitylation (Fig. 19.1).
Fig. 19.1.
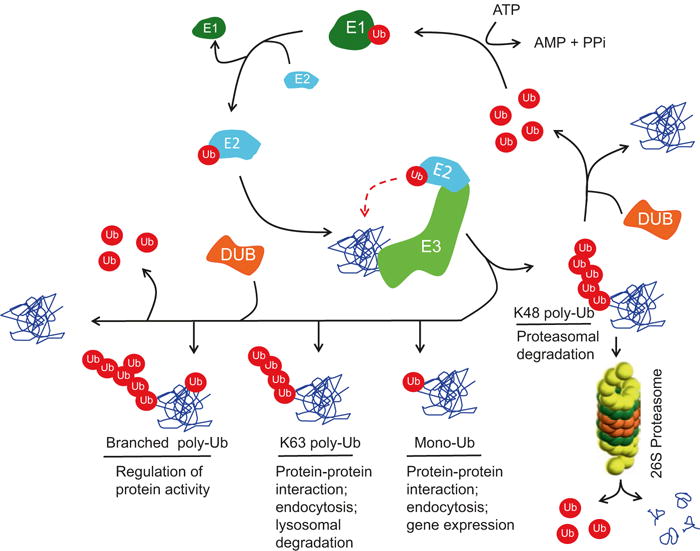
Schematic of the various steps involved in the ubiquitylation of protein substrates via the UPS. Ubiquitin (Ub) is conjugated to various ubiquitylation substrates through various linkages to form monoubiquitylated (mono-Ub), lys-48-linked polyubiquitylated (K48 poly-Ub), lys-63-linked polyubiquitylated, and branched polyubiquitylated (branched poly-ub) species. Other forms of ubiquitin linkages are not shown. The process begins with ubiquitin first activated and bound through a thioester bond by the ubiquitin-activating enzyme (E1). Activated ubiquitin is subsequently transferred to one of several ubiquitin-conjugating enzymes (E2) through another thioester bond. An E3 ubiquitin ligase (E3) then promotes the transfer of ubiquitin to the target substrate through interaction with the E2-charged ubiquitin, whereby a covalent isopeptide bond is formed between the C-terminus of ubiquitin and a specific lysine residue on the substrate. Elongation of the ubiquitin chain is effected when the C-terminus of another ubiquitin moiety is linked to one of seven lysine residues (e.g., K48) or the fist methionine residue (M1) on the first ubiquitin. Polyubiquitylation through K48 or the less common K29 linkages targets the ubiquitylated substrate for proteasomal degradation via the 26S proteasome. Other polyubiquitylation linkages (e.g., K63) serve non-proteolytic functions. Ubiquitin and polyubiquitin chains can be removed from the substrates through the activity of one of many highly specific cysteine proteases, called deubiquitylating enzymes (DUBs), which can cleave both the isopeptide bond between the ubiquitin and ε-amino group of the lysine on the substrate or on ubiquitin (in a polyubiquitin chain) or the peptide bond between ubiquitin and the N-terminal methionine of ubiquitin
While polyubiquitylation is an efficient mechanism by which cellular proteins are eliminated or modified, the process is reversible, and the removal of ubiquitin chains from substrate proteins is carried by a class of highly specific cysteine proteases, collectively termed “deubiquitinases” or “DUBs” (Fig. 19.1). DUBs hydrolyze the isopeptide bonds between the ε-amino group of lysine side chains of the target substrate and the C-terminal group of ubiquitin or the peptide bond between the α-amino group of the target protein and the C-terminal of ubiquitin (Wilkinson 1997). DUBs play a pivotal role as regulators of the turnover rate, activation, recycling, and localization of many proteins and thus are essential for regulating several signaling pathways and for cellular homeostasis (Komander et al. 2009; Reyes-Turcu et al. 2009). The role of the UPS in controlling DNA replication following replication stress and in response to DNA damage is described in excellent recent reviews (Garcia-Rodriguez et al. 2016; Renaudin et al. 2016; Sommers et al. 2015). In this chapter, we focus on protein ubiquitylation leading to proteasomal degradation or modification of function of key proteins to control DNA replication, with an emphasis on key E3 ubiquitin ligases and DUBs.
19.2 Structure and Function of Cullin-RING Ubiquitin Ligases Controlling DNA Replication
The Cullin-RING (Really Interesting New Gene) E3 ubiquitin Ligases (CRLs) represent the largest family of E3 ligases and are responsible for the ubiquitylation of approximately 20% of total cellular proteins degraded through the proteasome (Soucy et al. 2009). The other major E3 ligases belong to the HECT (Homologous to the E6-AP Carboxyl Terminus) domain containing E3 ubiquitin ligases (Li et al. 2008; Deshaies and Joazeiro 2009; Skaar et al. 2014). CRLs play significant roles in various processes including cell cycle regulation, cell proliferation, and tumorigenesis (Petroski and Deshaies 2005; Hotton and Callis 2008; Bosu and Kipreos 2008). Family members include eight cullin proteins (cullins 1, 2, 3, 4A, 4B, 5, 7, and 9 (also known as PARC or p53-associated parkin-like cytoplasmic protein)) and a cullin-like protein ANAPC2 or APC2. The general description of the structure and function of CRLs has been described in several excellent reviews (Chen et al. 2015; Lydeard et al. 2013; Sarikas et al. 2011; Duda et al. 2011; Hua and Vierstra 2011; Lipkowitz and Weissman 2011; Deshaies and Joazeiro 2009; Hotton and Callis 2008). The SCF (SKP1-Cullin1-F-Box protein; also known as CRL1, Fig. 19.2) is one of the well-characterized members of CRL ligases best known for its role in the regulation of cell cycle progression, cellular proliferation, apoptosis, and differentiation (Nakayama and Nakayama 2005; Welcker and Clurman 2008; Maser et al. 2007; Huang et al. 2010; Lee and Diehl 2014; Duan et al. 2012). SCF recognizes and promotes the ubiquitylation and degradation of its substrates through association with one of many substrate receptors (69 in mammalian cells), collectively known as F-box proteins, constituting a large family of distinct SCF ligases with varying specificity (Heo et al. 2016; Kipreos and Pagano 2000; Cardozo and Pagano 2004; Skaar et al. 2013; Wang et al. 2014). These substrate receptors utilize their F-box motifs to associate with the SKP1 (S-phase kinase-associated protein 1) bridge subunit (Wang et al. 2014). The SKP1 subunit of the SCF ligase bridges the F-box proteins (along with their cognate substrates) to the N-terminal domain of the cullin 1 subunit. The C-terminal domain of cullin 1 interacts with a small RING domain protein (RBX1 or RBX2), which is essential for the recruitment of E2 UBCs necessary for the polyubiquitylation of substrates distend for proteolytic degradation (Fig. 19.2).
Fig. 19.2.
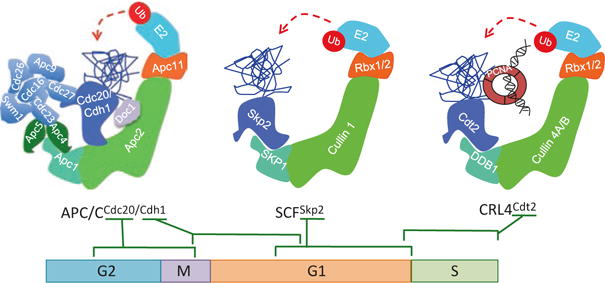
Molecular architecture of the three multi-subunit E3 ubiquitin ligases controlling DNA rereplication and their activity during the cell cycle. The schematic illustrates the general architecture of the APC/C (CDC20 or CDH1), cullin 1 (SCF), and cullin 4 (CRL4) E3 ubiquitin ligases. Ubiquitin molecules (red circles) are transferred to the substrate (blue ribbons) through the activity of one of the three E3 ubiquitin ligase complexes (made of a cullin or cullin-like subunit (light green), a bridge protein(s) (cyan), substrate adaptors (royal blue), and small RING protein (orange)) and E2 conjugating enzymes (baby blue). Multiple proteins bridge the substrate receptors CDC20 or CDH1 to the APC2 scaffold subunit of the APC/C ligase. SKP1 and DDB1 (damage-specific DNA-binding protein 1) bridge the substrate receptors SKP2 and CDT2 to the cullin 1 or cullin 4 (A or B) of the SCFSKP2 or CRL4CDT2 E3 ligases, respectively. CRL4CDT2 recognizes its ubiquitylation substrates when bound to chromatin-bound trimeric proliferating cell nuclear antigen (PCNA), encircling DNA (black helix). G1 first gap phase of the cell cycle, S DNA synthesis phase, G2 second gap phase of the cell cycle, M mitosis
Three subfamilies (FBXW, FBXL, and FBXO) characterize the F-box proteins, depending on whether they contain WD40 repeats (FBXW or FBW series), leucine-rich repeats (FBXL or FBL series), or variable other or “catch-all” domains (FBXO or F-box only series) (Skaar et al. 2014). Through their ability to assemble distinct SCF ligases, many of these F-box proteins are involved in the regulation of DNA replication and cell cycle progression, and their expression or activity is altered in human malignancies (Heo et al. 2016). For example, the SCFSKP2 ubiquitin ligase (Fig. 19.2) composed of the core SCF ligase associating with the substrate receptor and SKP2 (S-phase kinase-associated protein 2; also known as FBL1) is an essential driver of cell cycle progression, primarily through impacting DNA replication either directly or indirectly. SCFSKP2 directly controls DNA replication through its ability to promote the timely ubiquitin-dependent degradation of several components of the pre-replication complexes (pre-RCs) from mid-G1-phase (Fig. 19.3). This ensures that licensing of replication origins occurs only from late M till mid-G1-phase of the cell cycle and is prevented from occurring again until cells complete the genome duplication and following the segregation of daughter chromosomes in late M (Fig. 19.4; see below). SCFSKP2 also activates DNA replication through activating cyclin-dependent kinases (CDKs) primarily through the ubiquitylation and degradation of negative CDK regulators (e.g., the CDK inhibitors p21CIP1, p27KIP1, and p57KIP2). Later in the cell cycle, SCFSKP2 is also responsible for the targeted proteolysis of positive regulators of CDKs (e.g., cyclin D1 and cyclin A) to promote the handover of CDK activity from one CDK to the next (Fig. 19.4). The latter activity ensures the availability of CDK molecules for assembling distinct cyclin-CDK complexes for catalyzing various specific activities necessary for the irreversible progression of the cell cycle. For example, the ubiquitin-dependent proteolysis of cyclin A via the SCFSKP2 ligase ensures the availability of CDK1 molecules to assemble cyclin B-CDK1 complexes essential for G2 progression. Likewise, the degradation of cyclin E, mediated through the activity of the SCF ligase with the substrate receptor FBXW7 (SCFFBXW7) following cyclin E phosphorylation by CDK2, ensures the availability of CDK2 molecules to assemble cyclin A-CDK2 complexes necessary for S-phase progression (Clurman et al. 1996; Koepp et al. 2001).
Fig. 19.3.
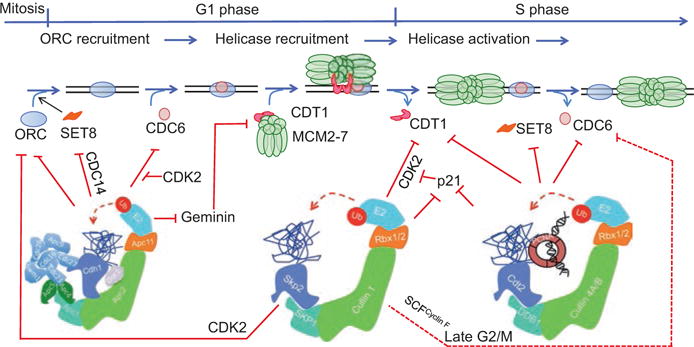
Control of MCM2-7 loading via the UPS. The APC/CCdh1 ligase promotes MCM2-7 loading in late M and in G1 by degrading Geminin. However, APC/CCdh1 also appears to limit the abundance of certain licensing factors like Drosophila ORC1 and mammalian CDC6 in G1, narrowing to late M-phase the window in the cell cycle when there is enough ORC, CDC6, and active CDT1 available to load MCM2-7 on origins. The E3 ubiquitin ligases, SCFSKP2 and CRL4CDT2 limit the abundance of key proteins involved in MCM2-7 loading (pre-RC assembly) in the S-, G2-, and early M-phases of the cell cycle. The SCF ubiquitin ligase also utilizes the substrate recognition subunit cyclin F (SCFCyclin F; red dashed lines) to suppress origin relicensing by promoting the ubiquitylation and degradation of CDC6 in late G2- and early M-phase of the cell cycle
Fig. 19.4.
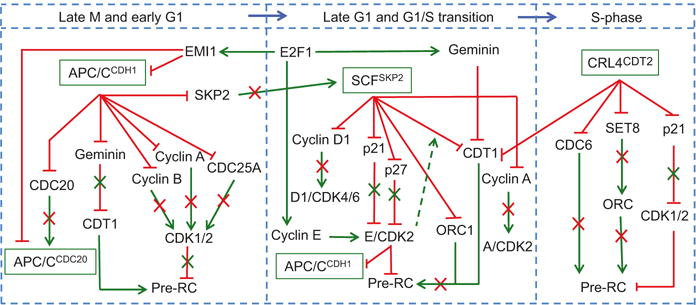
UPS control of DNA replication via direct and indirect mechanisms. MCM2-7 is loaded (Pre-RCs established) in late M (and perhaps early G1)-phase of the cell cycle and is prevented from being loaded again from the G1/S transition until the latest part of the next mitosis. This regulation is dependent primarily on the activity of CDK, which is maintained at low levels through late mitosis and G1 through the activity of APC/CCDH1 ubiquitin ligase. CDK activity peaks again in late G1 and at the G1/S transition through the activity of the SCFSKP2 ligase, which promotes the proteolysis of negative regulators of CDK2. CDK2 activity remains high in S-phase through the targeted proteolysis of p21 via CRL4CDT2. Such elevated levels of CDK suppress pre-RC in late G1 and in S and prevent aberrant origin relicensing and DNA rereplication. All three E3 ligases also directly control the abundance of pre-RC proteins such that replication occurs only once in the cell cycle. The circuit receives transcriptional input from the E2F1 transcription factor, which in addition to promoting S-phase entry into the cycle through upregulation of cyclin E and the consequent elevation of cyclin E/CDK2 activity provides a negative feedback control to shut down the activity of APC/C ligase (via EMI1 upregulation) and inactivation of CDT1 (via Geminin upregulation). This ensures that cells proceed in S-phase without aberrant licensing. Green arrows, positive regulation. Red lines, negative regulation
The other major E3 ubiquitin ligase regulating DNA replication is the APC/C (anaphase-promoting complex/cyclosome) ubiquitin ligase (Fig. 19.2). This ubiquitin ligase utilizes the APC2 cullin-like scaffold to assemble the largest multi-subunit CRL ligase in mammalian cells (van Leuken et al. 2008). APC/C associates with one of two substrate adaptor proteins, CDH1 and CDC20, for recognizing and promoting the polyubiquitylation (both K48- and K11-linked ubiquitin conjugation) of a large number of key drivers of cell cycle progression (Visintin et al. 1997; Zachariae and Nasmyth 1999; Pines 2006). Unlike the SCF-FBX ligases, which recognize the vast majority of target substrates through phosphorylation-dependent sequence motifs or “phospho-degrons” within these substrates (Skaar et al. 2013), APC/C ligases recognize proteins containing D-boxes and KEN-boxes, such as cyclin A and cyclin B (Pfleger and Kirschner 2000; Pfleger et al. 2001). The assembly of APC/CCDC20 is driven by CDK-mediated phosphorylation of CDC20 and is active primarily in mitosis (Rahal and Amon 2008); this is critical for the initial targeted proteolysis of mitotic cyclins and for exit from mitosis. Assembly of APC/CCDH1 on the other hand is stimulated through dephosphorylation of the CDH1 subunit by the CDC14A phosphatase (Cdc14 in budding yeast) and is active in late mitosis and though G1-phase of the cell cycle (Jaspersen et al. 1999; Robbins and Cross 2010; Sullivan and Morgan 2007). The oscillating activities of APC/C ligases during the cell cycle, as described in more detail below, are critical for not only controlling the timing of DNA replication but also for driving cell cycle progression and for guarding against genomic instability and cancer development (van Leuken et al. 2008; Nakayama and Nakayama 2006).
19.3 Regulation of Origin Licensing via the UPS
The control of eukaryotic DNA replication begins at the end of mitosis and through the G1-phase of the cell cycle, whereby the sequential binding of origin recognition complex proteins (ORCs) followed by CDC6 and CDT1 loads the replicative helicase MCM2-7 onto replication origins: the establishment of pre-RCs (Fig. 19.3). Origin licensing is inhibited by CDK activity, which suppresses pre-RC formation either by promoting the phosphorylation-dependent nuclear exclusion of certain replication licensing factors (e.g., mammalian CDC6) or through the targeted proteolysis of a number pre-RC components though the UPS (Zhu et al. 2005). The degradation of SKP2 by the APC/CCDH1 ligase is particularly important for the stabilization of key replication factors in late M- and G1-phases of eukaryotic cell cycles (Fig. 19.4). For example, human ORC1, the largest subunit of ORC, is stable in G1 because SKP2 is degraded by APC/CCDH1 in G1 but is specifically degraded in S-phase cells via the activity of the SCFSKP2 ubiquitin ligase (Mendez et al. 2002; Tatsumi et al. 2003). Drosophila ORC1 is paradoxically ubiquitylated and degraded via the APC/CFZr/CDH1 E3 ligase as cells exit mitosis and during G1 phase of the cell cycle, and this requires the non-conserved N-terminal domain of ORC1 (Araki et al. 2003, 2005; Narbonne-Reveau et al. 2008). Thus, ORC would have had to finish its licensing activity (MCM2-7 loading) before cells exit mitosis. The Drosophila ORC1 protein reappears in late G1 following induction of ORC1 RNA by E2F1 after the latter is activated by cyclin-CDK.
CDC6, another protein important for MCM2-7 loading, is also regulated extensively by the UPS. Yeast Cdc6 is degraded via an SCFCdc4-dependent proteolytic pathway, while the phosphorylation of mammalian CDC6 by increasing CDK activity in late G1 and in S-phase triggers its relocalization to the cytoplasm, thereby preventing origin licensing from occurring until CDK activity drops again in late mitosis (Aparicio et al. 1997; Tanaka et al. 1997; Fujita et al. 1999; Jiang et al. 1999; Petersen et al. 1999; Saha et al. 1998; Alexandrow and Hamlin 2004). Ubiquitin-mediated proteolysis of mammalian CDC6 in G1 also controls its abundance in G1 and in quiescent non-cycling cells. This is carried out by the APC/CCDH1 ligase and is mediated through interaction between the destruction box and KEN box motifs of CDC6 and CDH1 (Petersen et al. 2000). This too suggests that as with fly ORC1 above, the degradation of mammalian CDC6 in G1 implies that the licensing activity must be completed before cells exit mitosis. The degradation of CDC6 by APC in G1 poses a special problem for cells entering the cell cycle from G0. It turns out that CDC6 is phosphorylated by cyclin E/CDK2 in cells entering the cell cycle from quiescence, and this inhibits APC/CCDH1-mediated proteolysis of CDC6 (Mailand and Diffley 2005). This protection from ubiquitylation ensures that sufficient origins of replications are licensed before cells enter S-phase (Fig. 19.3). Once cells enter S-phase, CDC6 is targeted for ubiquitylation and degradation again, but this is mediated via the CRL4CDT2 ligase (Clijsters and Wolthuis 2014). Late in the cell cycle (in G2 and early M), CDC6 is targeted for proteolysis via the SCFCyclin F ligase (Walter et al. 2016).
The third protein important for loading MCM2-7 is CDT1. CDK kinase activity promotes the ubiquitylation and degradation of CDT1 via the SCFSKP2 ligase at the G1/S transition of the cell cycle, and this, along with the targeted proteolysis of CDT1 in S-phase via the CRL4CDT2 ligase (see below), ensures that CDT1 is not available for relicensing origins of replication in late G1 or in S-phase. Metazoans employ a second mechanism by which CDT1 is inactivated in S-phase through interaction with and inhibition by Geminin (Wohlschlegel et al. 2000; Tada et al. 2001). Geminin, however, is degraded in G1 through the activity of the APC/C E3 ligase (McGarry and Kirschner 1998), which is associated with low CDK activity. Origin licensing in cycling cells therefore can only proceed with low CDK activity (Figs. 19.3 and 19.4).
The mechanisms that maintain low CDK activity during late mitosis and in G1 are themselves under the control of the UPS. For example, the APC/C ligase is responsible for the decrease of mitotic CDK activity by promoting the ubiquitylation and degradation of the mitotic cyclins A and B (den Elzen and Pines 2001). This is first mediated through APC/CCDC20, which promotes the degradation of cyclin A and cyclin B in prometaphase and metaphase, respectively. As cells proceed in metaphase, the degradation of cyclin B is mediated by APC/CCDH1. APC/CCDH1 additionally suppresses CDK2 activity in late M and in early G1 by promoting the degradation of the dual-specificity CDC25A phosphatase, which catalyzes the removal of inhibitory phosphorylations on CDK2 (Donzelli et al. 2002). Low CDK activity in late M and early G1 is also aided through the accumulation of elevated levels of the CDK inhibitors (CKI) p21 and p27, which can bind to and inhibit CDK2 in early G1 (Abbas and Dutta 2009). The protein levels of these two CDK inhibitors in G1 are under the control of the SCFSKP2 ligase, which is active only in late G1.
19.4 Feedback Control of Origin Licensing Through the UPS
The ubiquitylation reactions involved in regulating origin licensing function in “self-regulating” networks with multiple feedbacks, whereby elevated CDK activity in mitosis turns on specific ubiquitylation reactions that feedback to decrease CDK activity during late mitosis and early G1 (Fig. 19.4). In both yeast and man, elevated G2 and mitotic CDK activity renders the APC/CCDH1 inactive due to phosphorylation-dependent conformational changes in CDH1 precluding assembly of the active ligase. However, exit from mitosis requires inactivation of mitotic CDK, and this requires (a) targeted proteolysis of mitotic cyclins via the APC/CCDH1 (or its homologue in yeast APC/CHct1) and (b) the stabilization of the CDK inhibitor p21 (or its homologue in yeast, Sic1) through degradation of CDC20. APC/CCDC20 targets p21 for proteasomal activity during mitosis (Shirayama et al. 1999; Amador et al. 2007). APC/CCDC20 ligase also promotes (through an unknown mechanism) the release of the yeast Cdc14 phosphatase from the nucleolus or the human CDC14A from centrosomes (Mocciaro et al. 2010; Kaiser et al. 2002; Chen et al. 2016; Shirayama et al. 1999; Bembenek and Yu 2001). In yeast, Cdc14 dephosphorylates and activates the Cdh1/Hct1 subunit, which competes with and targets Cdc20 for ubiquitylation and proteolysis, thereby assembling active APC/CCdh1/Hct1 and inactivating the APC/CCdc20 ligase (Jaspersen et al. 1999; Robbins and Cross 2010; Sullivan and Morgan 2007). Thus, the simultaneous activation of APC/CCDH1 and inactivation of APC/CCDC20 trigger CDK inactivation, culminating in exit from mitosis. As cells progress through early G1-phase of the cell cycle with low CDK activity, APC/CCDH1 promotes the degradation of SKP2 (Bashir et al. 2004; Wei et al. 2004), and this prevents the premature formation of the SCFSKP2 complex and consequent destabilization of its ubiquitylation substrates p21 and p27, thus maintaining low CDK activity (Fig. 19.4).
At the G1/S transition, cyclin E-CDK2 phosphorylates CDH1 leading to APC/CCDH1 inactivation (Cappell et al. 2016). In addition CDH1 binds to and is inhibited by the F-box protein and early mitotic inhibitor 1 (EMI1), marking a “point of no return” driving cells entry into S-phase (Cappell et al. 2016). As cells proceed through S-phase, the activity of both APC/CCDH1 and APC/CCDC20 is kept low through interaction between EMI1 with CDH1 or CDC20 (Cappell et al. 2016; Reimann et al. 2001). Inhibition of APC/CCDC20 by EMI1 is essential for the stabilization of mitotic cyclins A and B, thereby ensuring the completion of DNA synthesis, prevention of DNA rereplication (see below), and progression through G2-phase (Di Fiore and Pines 2007).
As CDK1 activity builds up, the APC/CCDC20 activity is increased in prometaphase through the coordinated sequential phosphorylation of the APC3 and APC1 subunits of the APC/C ligase by CDK1 and the docking of CDC20 onto the APC/C ligase (Fujimitsu et al. 2016). Yeast APC/CCdc20 is similarly activated by mitotic Clb-CDK activity, and this is critical for metaphase-anaphase transition (Rahal and Amon 2008). Elevated CDK1 activity in G2 and in early mitosis is enhanced by the targeted ubiquitylation and degradation of the CDK1 tyrosine kinase and inhibitor Wee1 via the activity of an SCF ligase, which utilizes the β-transducin repeat-containing protein 1 (βTRCP1) as substrate recognition subunits (SCFβTRCP1) (Watanabe et al. 2004). Interestingly, this same E3 ligase also promotes the ubiquitylation and degradation of EMI1 (Guardavaccaro et al. 2003) and thus contributing to the rising activity of APC/CCDC20 necessary for mitotic progression. This, along with the targeted ubiquitylation and degradation of EMI1 via the SCFβTRCP ligase, ensures optimal activity of APC/CCDC20 ligase activity to drive mitotic progression. Thus, fluctuating CDK activity throughout the cell cycle, itself regulated through ubiquitin-mediated proteolysis, ensures that the ubiquitylation machinery required for the irreversible progression through the cell cycle is temporally coordinated with successive stabilization and destabilization of key drivers of cell cycle progression.
19.5 Inhibition of Origin Relicensing and DNA Rereplication Through the UPS
An extensive body of literature demonstrates a critical role for the UPS in restricting origin licensing to late M- and early G1-phase of the cell cycle and thus preventing aberrant relicensing and refiring of replication origins or rereplication (Fig. 19.3). As CDK activity builds up in G1 cells, the phosphorylation of the tumor suppressor protein pRb, first by cyclin D1/CDK4/6 and subsequently by cyclin E/CDK2, results in its dissociation from the E2F1 transcription factor, which transactivates Geminin and dozen other genes essential for S-phase progression (Wong et al. 2011). Increased Geminin transcription by E2F1 and increased Geminin protein stability through cyclin E/CDK2-dependent suppression of APC/CCDH1 prevent origin relicensing by directly binding CDT1, which sterically hinders its ability to recruit MCM2-7 complexes to origins of replication (Fig. 19.4). Second, and as cells proceed through S-phase, E2F1-dependent transcription of cyclin A coupled with enhanced protein stability of cyclin A due to inhibition of APC/CCDC20 by EMI1 causes cyclin A-CDK2 activity to increase dramatically. Cyclin A/CDK2 phosphorylates CDT1 at a conserved N-terminal threonine residue (Thr-29) creating a phospho-degron that is specifically recognized by SKP2, which is itself stabilized due to inhibition of the APC/CCDH1 activity in S-phase. The newly assembled SCFSKP2 promotes phospho-CDT1 ubiquitylation and degradation (Li et al. 2003 Li et al. 2004; Takeda et al. 2005). Thus, increased EMI1 levels at the G1/S transition and throughout S-phase with the consequent inhibition of APC/C ubiquitylation activity are critical for suppressing origin relicensing in mammalian cells, both by inhibiting CDT1 activity by Geminin and by promoting its proteolysis by the CDK-dependent and SCFSKP2-mediated activity. Interestingly, alleviating Geminin-mediated suppression of CDT1, or inhibiting cyclin A-dependent proteolytic degradation of CDT1 via the SCFSKP2 ligase alone, is sufficient to promote origin relicensing and rereplication in certain cancer cell types. This, however, is insufficient to induce rereplication in some other cancer cell types or in non-cancer cells (Zhu and Depamphilis 2009; Benamar et al. 2016; Machida and Dutta 2007). Inhibition of EMI1 (e.g., by short-interfering RNAs (siRNA)) on the other hand is sufficient to trigger robust rereplication both in cancer and non-cancer cells (Machida and Dutta 2007; Benamar et al. 2016). Thus, APC/C activity in S-phase inhibits origin licensing through the timely inhibition and degradation of CDT1 (Sivaprasad et al. 2007).
Whereas the SCFSKP2 ligase promotes the degradation of cyclin A/CDK2-phosphorylated soluble CDT1 in S-phase, the ubiquitylation and degradation of chromatin-bound CDT1 in S-phase occur through a phosphorylation-independent mechanism that requires the activity of the CRL4CDT2 ubiquitin ligase (Nishitani et al. 2006; Arias and Walter 2006; Senga et al. 2006; Jin et al. 2006). The overall composition and architecture of CRL4 ligases are very similar to CRL1 ligases (Fig. 19.2) (Angers et al. 2006; Higa and Zhang 2007). The core complex is composed of one of two E3 ubiquitin ligases (cullin 4A or cullin 4B), DDB1 (DNA damage-specific protein-1), which is a bridge protein analogous to the SKP1 subunit in CRL1, and functions to bridge one of many substrate receptors (also known as DCAFs; DDB1 and cullin 4 associated factors) to the cullin subunit, and through that to RBX1 or RBX2, required for the recruitment of E2 UBCs. DCAFs include at least 49 family members of WD motif-rich proteins that function to recruit substrates to the CRL4 ligase similar to the function of the F-box proteins in the CRL1 ligases (Angers et al. 2006; He et al. 2006; Higa et al. 2006; Jin et al. 2006). CRL4 is emerging as a master regulator of genome stability, and recent findings suggest that it orchestrates a variety of physiological processes, particularly those related to chromatin regulation and genomic stability (Jackson and Xiong 2009). The substrate adaptor CDT2 assembles with CRL4 to form a rather unique E3 ubiquitin ligase that does not recognize CDT1 (or several other ubiquitylation substrates) directly but specifically recognizes the substrate when it interacts with proliferating cell nuclear antigen (PCNA) (Arias and Walter 2006; Senga et al. 2006). PCNA, the processivity factor for DNA polymerase δ, serves this role as an accessory factor for recognition by CDT2 only when PCNA is bound to chromatin, a condition that is established only in S-phase and following certain types of DNA damage (Havens and Walter 2011; Abbas and Dutta 2011; Abbas et al. 2013). Thus DNA damage is another stimulus that uses this pathway to degrade CDT1 and other replication factors (Higa et al. 2003).
The interaction between CDT1 and PCNA occurs through a specialized PCNA-interacting protein (PIP) box motif. This PIP box, commonly referred to as the “PIP degron,” is a modified version of the PIP box motif utilized by many PCNA-interacting proteins; in that it contains, in addition to the canonical sequence (Q-X-X-(I/L/M)-X-X-(F/Y)-(F/Y)), conserved threonine and aspartic acid residues at positions 5 and 6, respectively, as well as a basic amino acid residue C-terminal of the PIP box (at position +4) and a second basic amino acid residue at position +3 or +5 (or both) (Abbas et al. 2010; Havens and Walter 2011; Michishita et al. 2011; Havens and Walter 2009). The importance of CRL4CDT2-mediated ubiquitylation and degradation of CDT1 is manifested by the fact that cells from various organisms that are deficient in cullin 4, DDB1, or CDT2 exhibit rereplication and genomic instability reminiscent to that seen in cells overexpressing CDT1 (Jin et al. 2006; Lovejoy et al. 2006; Vaziri et al. 2003; Tatsumi et al. 2006; Zhong et al. 2003; Kim et al. 2008; Sansam et al. 2006), and this is associated with double-strand DNA breaks, rereplication, and activation of the ATM and ATR-dependent checkpoints (Zhu et al. 2004; Zhu and Dutta 2006), which can be partially suppressed by co-depletion of CDT1 (Lovejoy et al. 2006). The ubiquitylation-dependent proteolysis of CDT1 via CRL4CDT2 is a feature of all eukaryotes except for budding yeast, where the CDT1-MCM complexes are exported to the cytoplasm (Tanaka and Diffley 2002; Devault et al. 2002). This is likely due to the fact that although budding yeast contains orthologs for cullin 4 and DDB1, they lack an identifiable ortholog for CDT2 (Zaidi et al. 2008).
Synchronization experiments in human cancer cells have shown that the levels of CDT1 protein begin to degrease as cells enter S-phase but re-accumulate late in S-phase and reach significantly higher levels in G2 (Abbas et al. 2010). In late S and in G2 cells, CDT1 is protected from CRL4CDT2-mediated ubiquitylation and degradation through two phosphorylation-dependent and distinct mechanisms. The first one employs the phosphorylation of CDT1 by the stress-activated mitogen-activated protein kinases (MAPK) p38 and JNK precluding recognition by CRL4CDT2 (Chandrasekaran et al. 2011). The second mechanism is dependent on CDK1-dependent phosphorylation of CDT1 preventing its recruitment to chromatin (Rizzardi et al. 2015). Just as the ubiquitylation and degradation of CDT1 in S-phase are critical for preventing rereplication and cell cycle progression, its re-accumulation in late S and in G2 is equally important for cell cycle progression, although its role in G2 is not fully understood (Rizzardi et al. 2015). Although the steady state level and protein half-life of CDT1 are clearly increased in late S and in G2 cells, a recent report suggests that its abundance particularly in G2-phase is also under the control of another E3 ubiquitin ligase, the SCFFBXO31 ligase (Johansson et al. 2014). In this study, the authors demonstrated that depletion of cancer cells of the substrate adaptor and putative tumor suppressor FBXO31 protein by siRNA induces low levels of DNA rereplication (7.6% vs. 4.6% in control cells), which is insufficient to inhibit growth (Johansson et al. 2014). It remains unclear however, how the stabilized CDT1 in these G2 cells gain access to chromatin to trigger rereplication in the presence of elevated CDK1 activity. An interesting possibility is that the loss of SCFFBXO31 activity in tumors lacking FBXO31 or with FBXO31 inactivating mutations may result in a minute amount of rereplication that do not interfere with proliferation but are sufficient to induce gene amplification and/or genome instability exacerbating the tumorigenic phenotype (Green et al. 2010).
In addition to CDT1, the CRL4CDT2 ligase promotes the ubiquitylation of several other proteins whose proteolysis in S-phase is critical for preventing origin relicensing and DNA rereplication. These include the CDK inhibitor p21, the histone H4 methyltransferase SET8 and CDC6 (Abbas et al. 2008, 2010; Nishitani et al. 2008; Kim et al. 2008; Tardat et al. 2010; Centore et al. 2010; Oda et al. 2010; Jorgensen et al. 2011; Clijsters and Wolthuis 2014). Ubiquitin-dependent degradation of p21 in S-phase via the CRL4CDT2 ligase is critical for ensuring elevated CDK activity necessary for S-phase progression and for promoting DNA replication by freeing PCNA from inhibitory p21 (Abbas and Dutta 2009). Recent evidence also demonstrates that increased p21 stability downstream of CRL4CDT2 inhibition stimulates rereplication in cancer cells, presumably due to suppression of CDK activity, a condition compatible with origin licensing (Kim et al. 2008; Benamar et al. 2016). Because these studies utilized the overexpression of a mutant p21 protein, which fails to interact with PCNA – a PIP degron mutant of p21 – these rereplicating cells are able to replicate DNA free from p21-mediated suppression of PCNA. Although ectopic expression of PIP degron mutant p21 induces only minor rereplication, the p21 protein is required for rereplication induced by CDT2 depletion (Benamar et al. 2016). Unlike the case for p21, the expression of PIP degron mutant of SET8, but not wild type SET8, induces robust rereplication, and this required the catalytic activity of this enigmatic methyltransferase (Abbas et al. 2010; Tardat et al. 2010). SET8 is also required for DNA rereplication in cells depleted of CDT2 (Benamar et al. 2016). SET8, also known as PR-SET7, is an enzyme that deposits a single methyl group on lysine 20 of nucleosomal histone 4 (H4K20me1) (Nishioka et al. 2002; Xiao et al. 2005). H4K20 can also be di- and tri-methylated (H4K20me2/3), but this is carried out by the SUV4-20H1 and SUV4-20H2 histone methyltransferases, which utilize H4K20me1 as substrate (Schotta et al. 2008). How SET8 promotes rereplication following CRL4CDT2 inactivation is not entirely clear, but likely dependent on its ability to mono-methylate H4K20 at replication origins (Tardat et al. 2010). In fact, tethering SET8 to a specific genomic locus permitted loading of pre-RC proteins on chromatin and induced rereplication from that site. Interestingly, the rereplication phenotype associated with ectopic expression of PIP degron mutant SET8 was correlated with increased di- and tri-methylation of H4K20 with a concurrent reduction of H4K20me1 and required SUV4-20H1 and SUV4-20H2 (Abbas et al. 2010; Beck et al. 2012). Consistent with this, it was shown that the ability of SET8 to nucleate origins of replication is mediated through SUV4-20H1- and SUV4-20H2-dependent recruitment of ORC1 and the ORC-associated protein (ORCA) proteins to chromatin and that both of these proteins are capable of binding H4K20me2/3 in vitro (Beck et al. 2012). Intriguingly, CRL4CDT2-mediated proteolytic degradation of SET8 is critical for S→G2 cell cycle progression and for proper chromatin condensation, and inactivation of this pathway in cancer cells results not only in the induction of rereplication, but also in the repression of histone gene transcription and the consequent chromatin decondensation as well as repression of E2F1-driven gene expression (Abbas et al. 2010). These latter toxicities are likely mediated through enrichment of the repressive chromatin marks (H4K20me2/3) at the promoters of these genes (Abbas et al. 2010). Thus, cell viability is critically dependent on CRL4CDT2-dependent downregulation of SET8 in S-phase for the maintenance of a stable epigenetic state. In M-phase SET8 protein is phosphorylated at Ser-29 by cyclin B/CDK1 from prometaphase to early anaphase, preceding the accumulation of H4K20me1 (Wu et al. 2010). This phosphorylation is reversed in late M-phase by the CDC14 phosphatase, which is activated by APC/CCDC20. SET8 dephosphorylation renders the protein subject to proteolytic degradation via APC/CCDH1, which facilitates mitotic progression (Wu et al. 2010).
As discussed above, CDC6 is another pre-RC component, which is targeted for ubiquitylation and degradation in S-phase via the CRL4CDT2 ligase, and this is dependent on a conserved PIP degron contained near its N-terminus (Clijsters and Wolthuis 2014). However, although CDC6 depletion prevented rereplication induced by CDT2 depletion, it is not clear whether failure to degrade CDC6 via this ligase is sufficient to induce rereplication. As mentioned above, CDC6 is also ubiquitylated and degraded via the SCFCyclin F ligase in G2- and in early M-phase, and this is also critical for preventing DNA rereplication as the depletion of cyclin F or the expression of a stable mutant form of CDC6 promotes rereplication and genome stability in cells lacking Geminin (Walter et al. 2016). Finally, in Drosophila melanogaster, CRL4CDT2 also promotes the ubiquitylation and degradation of the transcription factor E2f1, and this is dependent on E2f1 interaction with PCNA through a PIP degron, which is not conserved in human E2F1 protein (Shibutani et al. 2008). From these studies, it is becoming abundantly clear that the CRL4CDT2 ligase is a major inhibitor of origin licensing in S- and G2-phase of the cell cycle and does so through the degradation of key positive regulators of origin activity via their specialized interaction with chromatin-bound PCNA.
19.6 Targeting the Ubiquitylation Machinery Controlling DNA Replication Licensing for Therapeutic Gain
Deregulated origin licensing can have serious consequences. On one hand, failure of the ordered assembly of pre-RCs on sufficient origins of replications in late M and in G1 inhibits cell proliferation. This is demonstrated by the fact that targeted knockout or knockdown of many of the pre-RC components in various eukaryotes interferes with viability, and thus some of these components, e.g., the MCM2-7 helicases, are considered promising chemotherapeutic targets in cancer (Lei 2005; Simon and Schwacha 2014). On the other hand, excessive origin licensing can result in DNA rereplication, which is deleterious to cells owing to the accumulation of replication intermediates and collapsed replication forks (Abbas et al. 2013). Substantial evidence shows that induction of rereplication in cancer cells through genetic manipulation (e.g., by depletion of Geminin, EMI1, or CDT2) results in growth inhibition through the accumulation of DNA damage, cell cycle checkpoint activation, and the induction of growth arrest and/or cell death (Zhu et al. 2005, 2004; Abbas and Dutta 2011; Abbas et al. 2013;Zhu and Dutta 2006). The first indication showing that pharmacological induction of rereplication is associated with inhibitory antiproliferation activity came from the accidental discovery that a small molecule, called MLN4924, designed to inhibit protein neddylation, inhibits the proliferation of human cancer cell lines and is associated with DNA rereplication (Soucy et al. 2009; Lin et al. 2010; Wei et al. 2012; Jazaeri et al. 2013). Neddylation of cullins is a posttranslational modification necessary for the activity of cullin-RING ligases. A small protein called NEDD8 is covalently attached to target protein substrate by an enzyme cascade system similar to ubiquitylation (Merlet et al. 2009). MLN4924 (also known as pevonedistat), currently in multiple clinical trials for hematologic (NCT00722488, NCT00911066) and solid (NCT01011530) malignancies, inhibits the NEDD8-activating enzyme (NAE), which catalyzes the first step in this enzymatic cascade, and its ability to suppress cullin neddylation and induction of rereplication suggested that the anticancer activity of this investigational drug is mediated through the suppression of CRL4CDT2 and the stabilization of its ubiquitylation substrate CDT1 (Soucy et al. 2009). Subsequent studies, however, demonstrated that MLN4924, in addition to inhibiting all CRLs, inhibits a number of signal transduction pathways critical for cell proliferation, including the NFκB, AKT, and mTOR signal transduction pathways (Soucy et al. 2009; Lin et al. 2010; Milhollen et al. 2010, 2011; Gu et al. 2014; Godbersen et al. 2014; Li et al. 2014a, b). We have recently shown that the stabilization of the CRL4CDT2 substrates SET8 and p21 is critical for the induction of DNA rereplication and senescence in melanoma cells treated with MLN4924 (Benamar et al. 2016). In particular, melanoma cells with hypomorphic expression of either of these two proteins are resistant to MLN4924-induced rereplication and senescence in culture, and, more importantly, tumor xenografts of these melanoma lines are refractory to inhibition by MLN4924 in nude mice. Interestingly, MLN4924 induces rereplication only in cancer cells but not in nonmalignant primary cells, suggesting that normal cells and tissues may be protected from the toxicity induced by this agent (Benamar et al. 2016). Thus, MLN4924 represents the first compound with anticancer activity, which selectively inhibits cancer cell proliferation through the induction of rereplication (Nawrocki et al. 2012; Zhao and Sun 2013; Tanaka et al. 2013; Zhao et al. 2014; Jiang and Jia 2015; Benamar et al. 2016).
Although specific inhibitors for CRL4CDT2 ligases are yet to be developed, specific inhibitors of the other major E3 ligase controlling DNA replication, SCFSKP2, already exist and exhibit potent anticancer activity in nude mice (Chan et al. 2013). However, because SKP2 promotes the degradation of many other proteins in addition to the ones that impact DNA replication, it is unclear whether such antitumor activity is dependent on dysregulated DNA replication. An image-based high- throughput screening for compounds with the ability to induce rereplication selectively in cancer cells has identified other small molecules that selectively inhibit cancer cell proliferation (Zhu et al. 2011). However, whether these compounds exhibit antitumor activity or induce rereplication via targeting components or the UPS remains to be determined.
19.7 Regulation of DNA Synthesis via the UPS
Whereas the role of the UPS for controlling origin licensing and entry of cells into S-phase is well established, its role in regulating ongoing DNA replication and in coordinating the associated chromatin dynamics in unperturbed cells is just beginning to be appreciated. The latter is accomplished through the activity of histone proteins, histone chaperones, nucleosome-remodeling complexes, histone and DNA methylation-binding proteins, and chromatin-modifying enzymes. Together, these factors facilitate nucleosomal disassembly ahead of incoming replication forks and promote their reassembly following their passage. Many of these proteins and protein complexes are regulated through the UPS, and several excellent recent reviews describe these activities in details (Garcia-Rodriguez et al. 2016; Talbert and Henikoff 2017; Henikoff 2016; Almouzni and Cedar 2016). In this section, we focus on the role of the UPS in regulating replication initiation, progression, and termination.
A high-resolution proteome-wide mass spectrometry-based identification of ubiquitylated peptides in unperturbed cells identified many components of the replication machinery as substrates for ubiquitin modification, although in some cases this is not associated with proteasomal degradation (Wagner et al. 2011). These include components of various complexes regulating DNA replication, such as the MCM2-7 helicase, GINS, and replication factor C (RFC) clamp loader complexes. In addition, almost all DNA polymerases and their associated factors had detectable ubiquitylated peptides identified in this screen. Although the individual components coordinating these ubiquitylation events and their functional significance remain to be defined, this study suggests that ubiquitylation plays an important role in regulating DNA replication and its fidelity. Not all of these ubiquitylation events are associated with proteolysis of the DNA replication factors. For example, the two subunits of the mammalian DNA polymerase δ, p66 and p12, undergo non-proteolytic ubiquitylation during S-phase that is thought to regulate protein-protein interactions within the polymerase complex or with other replication factors (Liu and Warbrick 2006). Following DNA damage, the p12 subunit is degraded through a PCNA-dependent ubiquitylation via the CRL4CDT2 ligase, and this is critical for inhibiting fork progression (Terai et al. 2013). Similarly, the S. pombe Pol2, the catalytic subunit of polymerase ε, which is responsible for leading strand synthesis, undergoes ubiquitin-dependent proteolysis, and this is mediated via the SCFPof3 ligase (Roseaulin et al. 2013). Because the catalytic subunit of polymerase δ (responsible for lagging strand synthesis) remains stable, despite undergoing ubiquitylation, the results suggest that DNA synthesis of the leading strand requires a “fresh” supply of DNA polymerase, whereas the synthesis of the discontinuous lagging strand does not (Roseaulin et al. 2013). This example highlights the complexity in the mechanisms regulating the abundance and activity of replication factors through the UPS in response to various stimuli. Minichromosome maintenance protein 10 (MCM10) is another important DNA replication protein, which undergoes non-proteolytic ubiquitylation (Das-Bradoo et al. 2006). MCM10 is dispensable for the assembly of the MCM2-7 replicative helicase but facilitates strand separation during the early stages of replication initiation (Kanke et al. 2012; van Deursen et al. 2012; Thu and Bielinsky 2013). In fission yeast, Mcm10/Cdc23 was shown to function after pre-RC assembly and facilitates Cdc45 chromatin binding (Gregan et al. 2003). The budding yeast Mcm10 protein was also suggested to play a role in replication elongation through interacting with and stabilization of the catalytic subunit of DNA polymerase α (POL1) (Ricke and Bielinsky 2004). Similarly, mammalian MCM10 interacts with and stabilizes the catalytic subunit of DNA polymerase α (p180) and is essential for efficient DNA synthesis (Chattopadhyay and Bielinsky 2007; Zhu et al. 2007). In G1- and in S-phase, MCM10 is monoubiquitylated at two residues, and this promotes its interaction with PCNA, which may be important for the release of polymerase α and the recruitment of polymerase δ necessary for the extension of Okazaki fragments (Das-Bradoo et al. 2006; Thu and Bielinsky 2013). The identity of the E3 ligase responsible for MCM10 monoubiquitylation is currently unknown. MCM10 is also subject to ubiquitylation-dependent proteolysis via the CRL4VBRBP E3 ligase, both in unperturbed cells and following exposure to UV, although the biological significance of this regulation remains elusive (Kaur et al. 2012; Romani et al. 2015).
Perhaps the most prominent example where the regulation of DNA replication involves coordination between proteolytic and non-proteolytic ubiquitylation machineries concerns the enigmatic PCNA protein (Fig. 19.5). PCNA is a homotrimeric ring-shaped protein complex that functions as a platform for coordinating the activity of many proteins involved in DNA replication, repair, and chromatin-related transactions (Ulrich and Takahashi 2013; Choe and Moldovan 2017). PCNA has long been shown to undergo DNA damage-induced monoubiquitylation at a conserved lysine residue (Lys-164), and this is critical to recruit translesion Y-family DNA polymerases for bypassing replication-stalling DNA lesions by translesion DNA synthesis (TLS) (Yang et al. 2013). This recruitment is dependent on the enhanced interaction of TLS polymerases with the modified PCNA through their ubiquitin-binding domains (Bienko et al. 2005; Plosky et al. 2006). PCNA monoubiquitylation at Lys-164 is catalyzed by the Rad18 E3 ligase and mediated by the E2 Rad6. The monoubiquitin moiety can be converted to Lys-63-linked polyubiquitylation by a heterodimeric E2, Ubc13-Mms2, to initiate an error-free pathway of repair, template switching, which uses the newly replicated sister chromatid as a template for replication (Hedglin and Benkovic 2015; Branzei 2011). Rad18, however, is not the only E3 ligase that can monoubiquitylate PCNA at Lys-64 as this modification can still be detected in the absence of functional Rad18 (Simpson et al. 2006). Consistent with this, two other E3 ligases, RNF8 and CRL4CDT2, were shown to promote PCNA monoubiquitylation (Zhang et al. 2008; Terai et al. 2010). Intriguingly, PCNA undergoes monoubiquitylation in normal replicating cells and without exposure to external stresses (Frampton et al. 2006; Leach and Michael 2005; Terai et al. 2010). Although the role of this ubiquitylation in unperturbed DNA replication is not clear, PCNA monoubiquitylation appears to be important for efficient DNA replication and for promoting TLS that may be necessary to cope with replication-associated stress (Leach and Michael 2005; Terai et al. 2010).
Fig. 19.5.
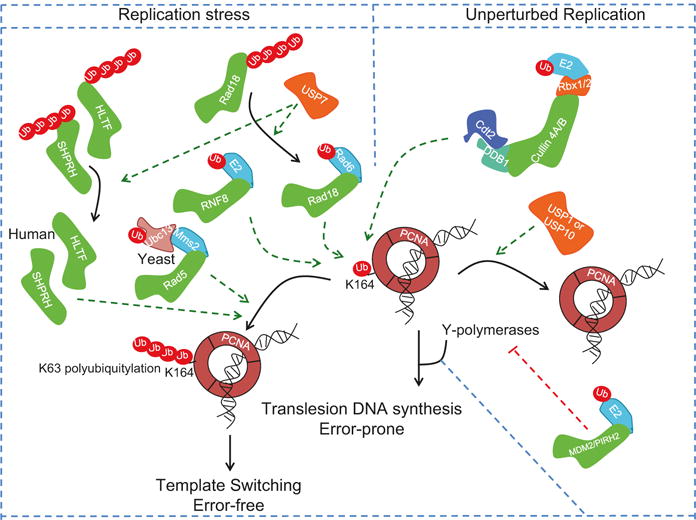
Control of PCNA ubiquitylation and its role in normal DNA replication and following replication stress. The trimeric PCNA is subject to monoubiquitylation via the activity of multiple E3 ligases both in unperturbed cells (via CRL4-CDT2 E3 ligase) and following replication stresses via Rad18/Rad5 E2/E3 or RNF8 E3 ligase. PCNA monoubiquitylation is essential for the recruitment of Y-family DNA polymerases, such as DNA polymerase eta, and this is critical for translesion DNA synthesis (TLS). This is reversed by the action of the deubiquitylating enzymes USP1 and USP7 (Orange). The monoubiquitin on PCNA at Lys-164 (K164) can be extended by the Rad5 E3 ligase and heterodimeric Ubc13/Mms2 in yeast or through the activity of the HLTF and SHPRH E3 ligases in mammals to form K63-linked polyubiquitin chain, and this promotes template switching and minimizes mutagenesis due to excessive activity of TLS polymerases. Other E3 ligases, MDM2 and PRH2, limit the activity of TLS polymerases to limit error-prone TLS either via the targeted proteolysis of TLS polymerase eta (MDM2) or ubiquitin-dependent suppression of its activity (PRH2)
In mammalian cells, two E3 ubiquitin ligases, SNF2 histone linker plant homeodomain RING helicase (SHPRH) and helicase-like transcription factor (HLTF), promote the Lys-63-linked polyubiquitylation at Lys-164 to suppress PCNA-dependent TLS and mutagenesis (Unk et al. 2008; Motegi et al. 2008). This latter activity is aided by the activity of the DUB USP7, which deubiquitylates and stabilizes HLTF and Rad18, promoting template switching through interaction between the unubiquitylated Rad18 and HLTF (Qing et al. 2011; Zeman et al. 2014). In response to replication stress, USP7 also deubiquitylates and stabilizes both Rad18 (so that it can monoubiquitylate PCNA) and the TLS polymerase, polymerase eta (POL-η), and this facilitates the bypass of lesions through the error-prone TLS pathway (Qian et al. 2015; Zlatanou et al. 2016). Another E3 ubiquitin ligase, the TNF receptor-associated factor (TRAF)-interacting protein (TRIP), and its homologue in Drosophila, “no pole” or NOPO, promotes the Lys-63-polyubiquitylation of POL-η, and this stimulates TLS by promoting the localization of POL-η to nuclear foci (Wallace et al. 2014). Intriguingly, Rad18 itself undergoes monoubiquitylation, and this prevents its interaction with SHPRH or HLTF and at the same time suppresses its ability to promote PCNA monoubiquitylation and TLS (Lin et al. 2011; Moldovan and D’Andrea 2011; Zeman et al. 2014). Kashiwaba et al. have additionally shown that USP7 can deubiquitylate PCNA, but this is not coupled to DNA replication and likely plays a role in DNA repair (Kashiwaba et al. 2015).
Just as it is important to initiate TLS in the face of replication-associated fork stalling, it is critical that the TLS activity is restricted to prevent increased mutations caused by low-fidelity polymerases. Two potential mechanisms ensure that TLS activity is restrained in cells (Fig. 19.5). The first involves the ubiquitin-dependent inhibition of TLS polymerases via both proteolytic and non-proteolytic ubiquitylation of TLS polymerases. For example, POL-η undergoes ubiquitylation-dependent proteolysis via the E3 ligase activity of MDM2 (Jung et al. 2012). On the other hand, the E3 ligase PIRH2 monoubiquitylates POL-η to suppress its interaction with monoubiquitylated PCNA (Jung et al. 2010, 2011). In both cases, this results in suppression of POL-η-dependent TLS.
Homologues of the TLS polymerases in yeast also undergo ubiquitin-dependent proteolysis. For example, both Rad30 (the S. cerevisiae homologue of POL-η) and Rev1 undergo cell cycle-dependent proteolysis to limit mutagenic activity, and in the case of Rad30, this is mediated via the SCF ubiquitin ligase with the F-box protein Ufo1 (Waters and Walker 2006; Skoneczna et al. 2007; Plachta et al. 2015). The second, and perhaps more important, mechanism restraining TLS activity is mediated through deubiquitylation of monoubiquitylated PCNA via the USP1 isopeptidase (Huang et al. 2006). An interesting study has recently shown that the deubiquitylation of mammalian PCNA, at least in the context of DNA damage induced by ultraviolet irradiation (UV), is aided by the deubiquitylating activity of USP10. In response to UV irradiation, USP10 is recruited to PCNA following its ISGylation by the ubiquitin-like modifier ISG15 (Park et al. 2014). Following deubiquitylation by USP10 and the release of POL-η, PCNA is de-ISGylated by UBP43, which renders PCNA available for reloading the replicative DNA polymerases and the resumption of DNA replication. Surprisingly, inactivation of Ubp10, the deubiquitinase for PCNA monoubiquitylation in yeast, does not appear to cause increased mutagenesis, arguing that other mechanisms contribute to inhibiting TLS-induced mutagenesis in yeast (Gallego-Sanchez et al. 2012).
In addition to PCNA monoubiquitylation at Lys-164, large-scale studies have identified additional lysine residues on PCNA that are ubiquitylated, but the E2/E3 enzymes carrying these ubiquitylation events and their biological functions are yet to be determined (McIntyre and Woodgate 2015). PCNA is also subject to extensive regulation through the ubiquitin-related SUMOylation pathway, and this is critical to suppress spontaneous and DNA damage-induced homologous recombination and help unload PCNA during DNA replication (Zilio et al. 2017; Garcia-Rodriguez et al. 2016). In this case, ubiquitylation and SUMOylation of the same protein and at the same residue appear to be tightly coordinating for ensuring optimal activity for this important multifunctional protein.
Termination of DNA replication is also regulated via the UPS (Fig. 19.6). Replication termination is initiated upon the convergence of replications forks, and this is triggered by the disassembly of the replicative helicase complex CMG (CDC45-MCM-GINS) DNA. The mechanism underlying replisome disassembly is not entirely clear, but two recent studies in yeast and in Xenopus laevis egg extracts provided the first insights into how replication termination is achieved (Maric et al. 2014; Moreno et al. 2014). Both studies have shown that the disassembly of the CMG complex is dependent on Lys-48-linked polyubiquitylation of the MCM7 subunit of the MCM2-7 complex, and this triggers its recognition by the ubiquitin-dependent segregase Cdc48 (also known as p97), a AAA+ adenosine triphosphatase (ATPase) that forms a hexameric ring, which undergoes conformational changes upon ATP hydrolysis to drive protein unfolding (Maric et al. 2014; Moreno et al. 2014; Barthelme et al. 2014). This results in the translocation of the MCM7 subunit through the p97 hexameric ring and the opening of the MCM2-7 ring allowing DNA exit and replication termination (Bell 2014; Lengronne and Pasero 2014). Mcm7 polyubiquitylation in S. cerevisiae is catalyzed by the activity of the SCF E3 ligase and the F-box protein Dia2, which has been previously shown to localize to the replisome (Maric et al. 2014; Moreno et al. 2014; Morohashi et al. 2009). Although SCFDia2 inactivation prevents CMG disassembly and results in its retention on chromatin causing replication defects consistent with inhibition of replication termination, blocking Mcm7 proteolysis does not do so, arguing that Mcm7 polyubiquitylation is most important for its extraction from the Mcm2-7 complex and unloading of the Mcm2-7 ring (Maric et al. 2014; Moreno et al. 2014). The role of MCM7 polyubiquitylation in replisome disassembly and replication termination is conserved in higher eukaryotes, but this was recently shown to be mediated through the activity of the CRL2Lrr1 ubiquitin ligase (Dewar et al. 2017).
Fig. 19.6.
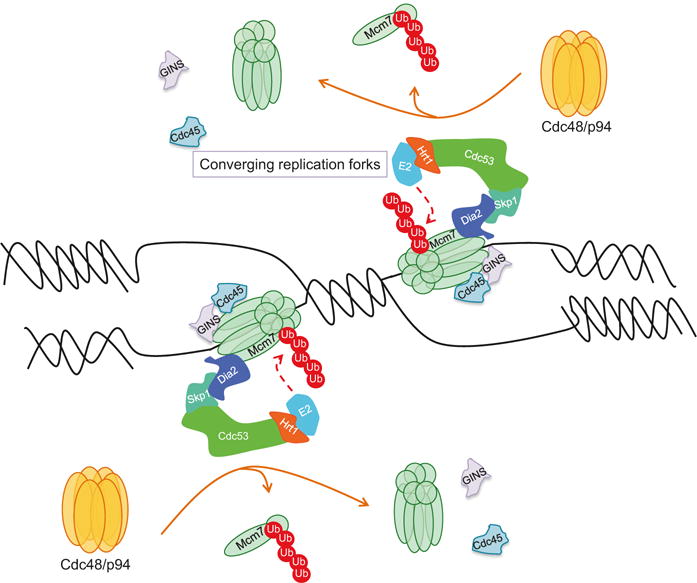
Control of replication termination in S. cerevisiae via the activity of the SCFDia2 E3 ubiquitin ligase. A model depicting the K-48-polyubiquitylation of Mcm7 by the SCFDia2 E3 ubiquitin ligase in S. cerevisiae (CRL2Lrr1 E3 ubiquitin ligase in metazoans) at the sites of converging replication forks, followed by its extraction from the MCM2-7 helicase by the activity of the p97 chaperon, leading to the replisome disassembly and replication termination. GINS and CDC25, components of the CMG helicase complex, are also shown
19.8 Summary and Concluding Remarks
The last few decades have witnessed an extensive understanding of the role of the UPS in regulating almost all aspect of cell physiology, and in many cases, deregulation of key regulators of this ubiquitous regulatory system contributes to disease development. This regulation is even more important for cellular processes that are dependent on the efficient temporal and spatial interplay between many factors and is subject to endogenous and external perturbations, as is the case for the regulation of eukaryotic DNA replication. While many factors influencing DNA replication undergo extensive regulation through the UPS leading to proteasomal degradation, non-proteolytic ubiquitin signaling coordinating protein-protein interactions is being subjected to increasing scrutiny. Various components of the UPS operate with extensive crosstalk and feedback mechanisms as well as interactions with other posttranslational modifications, and together, they provide a complex network of protein-protein communications to control the specificity and robustness of DNA replication and cell cycle control. When faced with stresses that impact the replication machinery and threaten the fidelity of DNA replication, for example, when replication forks encounter replication-stalling lesions, cells must be able to quickly respond and adopt the appropriate measures to cope with these stresses. The dynamic and reversible activities associated with the UPS allow the cell to adjust to these insults. Even in the absence of these stresses, the non-proteolytic as well as proteolytic mechanisms ensure that DNA replication proceed unperturbed with exquisite accuracy. The diversity in the specificity of the various E2-E3 pairs in conjugating various polymers of ubiquitin chains on target substrates adds another layer of complexity that will require more effort to fully understand and appreciate. Key regulators, such as the APC/C, CRL4CDT2, and SCF ubiquitin ligases, and the USP7 and USP1 deubiquitinases, dominate the scene and regulate the steady state levels or activities of many proteins controlling DNA replication and cell cycle progression. They are also important for regulating appropriate cellular responses to perturbations by activating cellular checkpoints. It is therefore understandable that significant research is targeting some of these key regulators for therapeutic gain, but this will require a greater understanding of the molecular and biochemical details and structural information of these regulators. Proteomewide studies suggest that we are yet to understand the functional significance of new ubiquitin modifications of many replication-associated factors. The development of novel techniques for genome editing and for gain- or loss-of-function screens as well as the development of new protocols for enriching for and identifying various ubiquitin modifications will no doubt be great assets for making these discoveries.
Acknowledgments
Work in the author’s laboratories is supported by NIH grants, R00 CA140774 to TA and R01 CA60499 and GM84465 to AD.
Contributor Information
Tarek Abbas, Department of Radiation Oncology, University of Virginia, Charlottesville, VA, USA; Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, VA, USA.
Anindya Dutta, Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, VA, USA.
References
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414. doi: 10.1038/nrc2657. https://doi.org/10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle. 2011;10(2):241–249. doi: 10.4161/cc.10.2.14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22(18):2496–2506. doi: 10.1101/gad.1676108. https://doi.org/10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40(1):9–21. doi: 10.1016/j.molcel.2010.09.014. https://doi.org/10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Keaton MA, Dutta A. Genomic instability in cancer. Cold Spring Harb Perspect Biol. 2013;5(3):a012914. doi: 10.1101/cshperspect.a012914. https://doi.org/10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrow MG, Hamlin JL. Cdc6 chromatin affinity is unaffected by serine-54 phosphorylation, S-phase progression, and overexpression of cyclin a. Mol Cell Biol. 2004;24(4):1614–1627. doi: 10.1128/MCB.24.4.1614-1627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almouzni G, Cedar H. Maintenance of epigenetic information. Cold Spring Harb Perspect Biol. 2016;8(5) doi: 10.1101/cshperspect.a019372. https://doi.org/10.1101/cshperspect.a019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador V, Ge S, Santamaria PG, Guardavaccaro D, Pagano M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol Cell. 2007;27(3):462–473. doi: 10.1016/j.molcel.2007.06.013. https://doi.org/10.1016/j.molcel.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R, Ciechanover A, Cohen S. The ubiquitin-proteasome system: the relationship between protein degradation and human diseases. Harefuah. 2001;140(12):1172–1176. 1229. [PubMed] [Google Scholar]
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443(7111):590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91(1):59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Araki M, Wharton RP, Tang Z, Yu H, Asano M. Degradation of origin recognition complex large subunit by the anaphase-promoting complex in drosophila. EMBO J. 2003;22(22):6115–6126. doi: 10.1093/emboj/cdg573. https://doi.org/10.1093/emboj/cdg573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M, Yu H, Asano M. A novel motif governs APC-dependent degradation of drosophila ORC1 in vivo. Genes Dev. 2005;19(20):2458–2465. doi: 10.1101/gad.1361905. https://doi.org/10.1101/gad.1361905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat Cell Biol. 2006;8(1):84–90. doi: 10.1038/ncb1346. https://doi.org/10.1038/ncb1346. [DOI] [PubMed] [Google Scholar]
- Barthelme D, Chen JZ, Grabenstatter J, Baker TA, Sauer RT. Architecture and assembly of the archaeal Cdc48*20S proteasome. Proc Natl Acad Sci U S A. 2014;111(17):E1687–E1694. doi: 10.1073/pnas.1404823111. https://doi.org/10.1073/pnas.1404823111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428(6979):190–193. doi: 10.1038/nature02330. https://doi.org/10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Beck DB, Burton A, Oda H, Ziegler-Birling C, Torres-Padilla ME, Reinberg D. The role of PR-Set7 in replication licensing depends on Suv4–20h. Genes Dev. 2012;26(23):2580–2589. doi: 10.1101/gad.195636.112. https://doi.org/10.1101/gad.195636.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18(5):520–528. doi: 10.1038/nsmb.2066. https://doi.org/10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- Bell SP. DNA replication. Terminating the replisome. Science. 2014;346(6208):418–419. doi: 10.1126/science.1261245. https://doi.org/10.1126/science.1261245. [DOI] [PubMed] [Google Scholar]
- Bembenek J, Yu H. Regulation of the anaphase-promoting complex by the dual specificity phosphatase human Cdc14a. J Biol Chem. 2001;276(51):48237–48242. doi: 10.1074/jbc.M108126200. https://doi.org/10.1074/jbc.M108126200. [DOI] [PubMed] [Google Scholar]
- Benamar M, Guessous F, Du K, Corbett P, Obeid J, Gioeli D, Slingluff CL, Jr, Abbas T. Inactivation of the CRL4-CDT2-SET8/p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine. 2016;10:85–100. doi: 10.1016/j.ebiom.2016.06.023. https://doi.org/10.1016/j.ebiom.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol. 2016;23(2):103–109. doi: 10.1038/nsmb.3163. https://doi.org/10.1038/nsmb.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310(5755):1821–1824. doi: 10.1126/science.1120615. https://doi.org/10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- Bosu DR, Kipreos ET. Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 2008;3:7. doi: 10.1186/1747-1028-3-7. https://doi.org/10.1186/1747-1028-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D. Ubiquitin family modifications and template switching. FEBS Lett. 2011;585(18):2810–2817. doi: 10.1016/j.febslet.2011.04.053. https://doi.org/10.1016/j.febslet.2011.04.053. [DOI] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, Meyer T. Irreversible APC(Cdh1) inactivation underlies the point of no return for cell-cycle entry. Cell. 2016;166(1):167–180. doi: 10.1016/j.cell.2016.05.077. https://doi.org/10.1016/j.cell.2016.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5(9):739–751. doi: 10.1038/nrm1471. https://doi.org/10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40(1):22–33. doi: 10.1016/j.molcel.2010.09.015. https://doi.org/10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, Logothetis CJ, Hung MC, Zhang S, Lin HK. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154(3):556–568. doi: 10.1016/j.cell.2013.06.048. https://doi.org/10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran S, Tan TX, Hall JR, Cook JG. Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor. Mol Cell Biol. 2011;31(22):4405–4416. doi: 10.1128/MCB.06163-11. https://doi.org/10.1128/MCB.06163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Bielinsky AK. Human Mcm10 regulates the catalytic subunit of DNA polymerase-alpha and prevents DNA damage during replication. Mol Biol Cell. 2007;18(10):4085–4095. doi: 10.1091/mbc.E06-12-1148. https://doi.org/10.1091/mbc.E06-12-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Sui J, Zhang F, Zhang C. Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. J Cancer. 2015;6(3):233–242. doi: 10.7150/jca.11076. https://doi.org/10.7150/jca.11076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NP, Uddin B, Voit R, Schiebel E. Human phosphatase CDC14A is recruited to the cell leading edge to regulate cell migration and adhesion. Proc Natl Acad Sci USA. 2016;113(4):990–995. doi: 10.1073/pnas.1515605113. https://doi.org/10.1073/pnas.1515605113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe KN, Moldovan GL. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol Cell. 2017;65(3):380–392. doi: 10.1016/j.molcel.2016.12.020. https://doi.org/10.1016/j.molcel.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Schwartz AL. Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology. 2002;35(1):3–6. doi: 10.1053/jhep.2002.30316. [DOI] [PubMed] [Google Scholar]
- Clijsters L, Wolthuis R. PIP-box-mediated degradation prohibits re-accumulation of Cdc6 during S phase. J Cell Sci. 2014;127(Pt 6):1336–1345. doi: 10.1242/jcs.145862. https://doi.org/10.1242/jcs.145862. [DOI] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10(16):1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- Das-Bradoo S, Ricke RM, Bielinsky AK. Interaction between PCNA and diubiquitinated Mcm10 is essential for cell growth in budding yeast. Mol Cell Biol. 2006;26(13):4806–4817. doi: 10.1128/MCB.02062-05. https://doi.org/10.1128/MCB.02062-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Elzen N, Pines J. Cyclin a is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol. 2001;153(1):121–136. doi: 10.1083/jcb.153.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. https://doi.org/10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Devault A, Vallen EA, Yuan T, Green S, Bensimon A, Schwob E. Identification of Tah11/Sid2 as the ortholog of the replication licensing factor Cdt1 in Saccharomyces cerevisiae. Curr Biol. 2002;12(8):689–694. doi: 10.1016/s0960-9822(02)00768-6. [DOI] [PubMed] [Google Scholar]
- Dewar JM, Low E, Mann M, Raschle M, Walter JC. CRL2Lrr1 promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017;31(3):275–290. doi: 10.1101/gad.291799.116. https://doi.org/10.1101/gad.291799.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore B, Pines J. Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J Cell Biol. 2007;177(3):425–437. doi: 10.1083/jcb.200611166. https://doi.org/10.1083/jcb.200611166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF. Dual mode of degradation of Cdc25 a phosphatase. EMBO J. 2002;21(18):4875–4884. doi: 10.1093/emboj/cdf491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, Chapuy B, Shipp M, Chiarle R, Pagano M. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481(7379):90–93. doi: 10.1038/nature10688. https://doi.org/10.1038/nature10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, Schulman BA. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol. 2011;21(2):257–264. doi: 10.1016/j.sbi.2011.01.003. https://doi.org/10.1016/j.sbi.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton J, Irmisch A, Green CM, Neiss A, Trickey M, Ulrich HD, Furuya K, Watts FZ, Carr AM, Lehmann AR. Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol Biol Cell. 2006;17(7):2976–2985. doi: 10.1091/mbc.E05-11-1008. https://doi.org/10.1091/mbc.E05-11-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimitsu K, Grimaldi M, Yamano H. Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science. 2016;352(6289):1121–1124. doi: 10.1126/science.aad3925. https://doi.org/10.1126/science.aad3925. [DOI] [PubMed] [Google Scholar]
- Fujita M, Yamada C, Goto H, Yokoyama N, Kuzushima K, Inagaki M, Tsurumi T. Cell cycle regulation of human CDC6 protein. Intracellular localization, interaction with the human mcm complex, and CDC2 kinase-mediated hyperphosphorylation. J Biol Chem. 1999;274(36):25927–25932. doi: 10.1074/jbc.274.36.25927. [DOI] [PubMed] [Google Scholar]
- Gallego-Sanchez A, Andres S, Conde F, San-Segundo PA, Bueno A. Reversal of PCNA ubiquitylation by Ubp10 in Saccharomyces cerevisiae. PLoS Genet. 2012;8(7):e1002826. doi: 10.1371/journal.pgen.1002826. https://doi.org/10.1371/journal.pgen.1002826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganai RA, Johansson E. DNA replication-a matter of fidelity. Mol Cell. 2016;62(5):745–755. doi: 10.1016/j.molcel.2016.05.003. https://doi.org/10.1016/j.molcel.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez N, Wong RP, Ulrich HD. Functions of ubiquitin and SUMO in DNA replication and replication stress. Front Genet. 2016;7:87. doi: 10.3389/fgene.2016.00087. https://doi.org/10.3389/fgene.2016.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi: 10.1152/physrev.00027.2001. https://doi.org/10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, Danilov AV. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20(6):1576–1589. doi: 10.1158/1078-0432.CCR-13-0987. https://doi.org/10.1158/1078-0432.CCR-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green BM, Finn KJ, Li JJ. Loss of DNA replication control is a potent inducer of gene amplification. Science. 2010;329(5994):943–946. doi: 10.1126/science.1190966. https://doi.org/10.1126/science.1190966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregan J, Lindner K, Brimage L, Franklin R, Namdar M, Hart EA, Aves SJ, Kearsey SE. Fission yeast Cdc23/Mcm10 functions after pre-replicative complex formation to promote Cdc45 chromatin binding. Mol Biol Cell. 2003;14(9):3876–3887. doi: 10.1091/mbc.E03-02-0090. https://doi.org/10.1091/mbc.E03-02-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol. 2003;35(5):606–616. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Gu Y, Kaufman JL, Bernal L, Torre C, Matulis SM, Harvey RD, Chen J, Sun SY, Boise LH, Lonial S. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood. 2014;123(21):3269–3276. doi: 10.1182/blood-2013-08-521914. https://doi.org/10.1182/blood-2013-08-521914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4(6):799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- Havens CG, Walter JC. Docking of a specialized PIP box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol Cell. 2009;35(1):93–104. doi: 10.1016/j.molcel.2009.05.012. https://doi.org/10.1016/j.molcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25(15):1568–1582. doi: 10.1101/gad.2068611. https://doi.org/10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006;20(21):2949–2954. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M, Benkovic SJ. Regulation of Rad6/Rad18 activity during DNA damage tolerance. Annu Rev Biophys. 2015;44:207–228. doi: 10.1146/annurev-biophys-060414-033841. https://doi.org/10.1146/annurev-biophys-060414-033841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S. Mechanisms of nucleosome dynamics in vivo. Cold Spring Harb Perspect Med. 2016;6(9) doi: 10.1101/cshperspect.a026666. https://doi.org/10.1101/cshperspect.a026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J, Eki R, Abbas T. Deregulation of F-box proteins and its consequence on cancer development, progression and metastasis. Semin Cancer Biol. 2016;36:33–51. doi: 10.1016/j.semcancer.2015.09.015. https://doi.org/10.1016/j.semcancer.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 2005;12(9):1191–1197. doi: 10.1038/sj.cdd.4401702. https://doi.org/10.1038/sj.cdd.4401702. [DOI] [PubMed] [Google Scholar]
- Higa LA, Zhang H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007;2:5. doi: 10.1186/1747-1028-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003;5(11):1008–1015. doi: 10.1038/ncb1061. https://doi.org/10.1038/ncb1061. [DOI] [PubMed] [Google Scholar]
- Higa LA, Banks D, Wu M, Kobayashi R, Sun H, Zhang H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle. 2006;5(15):1675–1680. doi: 10.4161/cc.5.15.3149. [DOI] [PubMed] [Google Scholar]
- Hotton SK, Callis J. Regulation of cullin RING ligases. Annu Rev Plant Biol. 2008;59:467–489. doi: 10.1146/annurev.arplant.58.032806.104011. https://doi.org/10.1146/annurev.arplant.58.032806.104011. [DOI] [PubMed] [Google Scholar]
- Hua Z, Vierstra RD. The cullin-RING ubiquitin-protein ligases. Annu Rev Plant Biol. 2011;62:299–334. doi: 10.1146/annurev-arplant-042809-112256. https://doi.org/10.1146/annurev-arplant-042809-112256. [DOI] [PubMed] [Google Scholar]
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D’Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8(4):339–347. doi: 10.1038/ncb1378. https://doi.org/10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- Huang H-L, Zheng W-L, Zhao R, Zhang B, Ma W-L. FBXO31 is down-regulated and may function as a tumor suppressor in hepatocellular carcinoma. Oncol Rep. 2010;24:715–720. doi: 10.3892/or_00000912. [DOI] [PubMed] [Google Scholar]
- Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol. 2016;19(1):1–9. doi: 10.1038/ncb3452. https://doi.org/10.1038/ncb3452. [DOI] [PubMed] [Google Scholar]
- Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34(11):562–570. doi: 10.1016/j.tibs.2009.07.002. https://doi.org/10.1016/j.tibs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9(5):227–236. doi: 10.1016/s0960-9822(99)80111-0. [DOI] [PubMed] [Google Scholar]
- Jazaeri AA, Shibata E, Park J, Bryant JL, Conaway MR, Modesitt SC, Smith PG, Milhollen MA, Berger AJ, Dutta A. Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor MLN4924. Mol Cancer Ther. 2013;12(10):1958–1967. doi: 10.1158/1535-7163.MCT-12-1028. https://doi.org/10.1158/1535-7163.MCT-12-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Jia L. Neddylation pathway as a novel anti-cancer target: mechanistic investigation and therapeutic implication. Anti Cancer Agents Med Chem. 2015;15(9):1127–1133. doi: 10.2174/1871520615666150305111257. [DOI] [PubMed] [Google Scholar]
- Jiang W, Wells NJ, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci U S A. 1999;96(11):6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23(5):709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Johansson P, Jeffery J, Al-Ejeh F, Schulz RB, Callen DF, Kumar R, Khanna KK. SCF-FBXO31 E3 ligase targets DNA replication factor Cdt1 for proteolysis in the G2 phase of cell cycle to prevent re-replication. J Biol Chem. 2014;289:18514–18525. doi: 10.1074/jbc.M114.559930. https://doi.org/10.1074/jbc.M114.559930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MS, Kousholt AN, Syljuasen RG, Trelle MB, Jensen ON, Helin K, Sorensen CS. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J Cell Biol. 2011;192(1):43–54. doi: 10.1083/jcb.201009076. https://doi.org/10.1083/jcb.201009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YS, Liu G, Chen X. Pirh2 E3 ubiquitin ligase targets DNA polymerase eta for 20S proteasomal degradation. Mol Cell Biol. 2010;30(4):1041–1048. doi: 10.1128/MCB.01198-09. https://doi.org/10.1128/MCB.01198-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YS, Hakem A, Hakem R, Chen X. Pirh2 E3 ubiquitin ligase monoubiquitinates DNA polymerase eta to suppress translesion DNA synthesis. Mol Cell Biol. 2011;31(19):3997–4006. doi: 10.1128/MCB.05808-11. https://doi.org/10.1128/MCB.05808-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YS, Qian Y, Chen X. DNA polymerase eta is targeted by Mdm2 for polyubiquitination and proteasomal degradation in response to ultraviolet irradiation. DNA Repair (Amst) 2012;11(2):177–184. doi: 10.1016/j.dnarep.2011.10.017. https://doi.org/10.1016/j.dnarep.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser BK, Zimmerman ZA, Charbonneau H, Jackson PK. Disruption of centrosome structure, chromosome segregation, and cytokinesis by misexpression of human Cdc14A phosphatase. Mol Biol Cell. 2002;13(7):2289–2300. doi: 10.1091/mbc.01-11-0535. https://doi.org/10.1091/mbc.01-11-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanke M, Kodama Y, Takahashi TS, Nakagawa T, Masukata H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 2012;31(9):2182–2194. doi: 10.1038/emboj.2012.68. https://doi.org/10.1038/emboj.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaba S, Kanao R, Masuda Y, Kusumoto-Matsuo R, Hanaoka F, Masutani C. USP7 is a suppressor of PCNA ubiquitination and oxidative-stress-induced mutagenesis in human cells. Cell Rep. 2015;13(10):2072–2080. doi: 10.1016/j.celrep.2015.11.014. https://doi.org/10.1016/j.celrep.2015.11.014. [DOI] [PubMed] [Google Scholar]
- Kaur M, Khan MM, Kar A, Sharma A, Saxena S. CRL4-DDB1-VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res. 2012;40(15):7332–7346. doi: 10.1093/nar/gks366. https://doi.org/10.1093/nar/gks366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22(18):2507–2519. doi: 10.1101/gad.1703708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1(5) doi: 10.1186/gb-2000-1-5-reviews3002. REVIEWS3002.: https://doi.org/10.1186/gb-2000-1-5-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294(5540):173–177. doi: 10.1126/science.1065203. https://doi.org/10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–563. doi: 10.1038/nrm2731. doi:nrm2731 [pii] 1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Kornitzer D, Ciechanover A. Modes of regulation of ubiquitin-mediated protein degradation. J Cell Physiol. 2000;182(1):1–11. doi: 10.1002/(SICI)1097-4652(200001)182:1<1::AID-JCP1>3.0.CO;2-V. https://doi.org/10.1002/(SICI)1097-4652(200001)182:1<1∷AID-JCP1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Leach CA, Michael WM. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol. 2005;171(6):947–954. doi: 10.1083/jcb.200508100. https://doi.org/10.1083/jcb.200508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Diehl JA. SCFs in the new millennium. Oncogene. 2014;33(16):2011–2018. doi: 10.1038/onc.2013.144. https://doi.org/10.1038/onc.2013.144. [DOI] [PubMed] [Google Scholar]
- Lei M. The MCM complex: its role in DNA replication and implications for cancer therapy. Curr Cancer Drug Targets. 2005;5(5):365–380. doi: 10.2174/1568009054629654. [DOI] [PubMed] [Google Scholar]
- Lengronne A, Pasero P. Closing the MCM cycle at replication termination sites. EMBO Rep. 2014;15(12):1226–1227. doi: 10.15252/embr.201439774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhao Q, Liao R, Sun P, Wu X. The SiCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J Biol Chem. 2003;278(33):30854–30858. doi: 10.1074/jbc.C300251200. https://doi.org/10.1074/jbc.C300251200. [DOI] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One. 2008;3(1):e1487. doi: 10.1371/journal.pone.0001487. https://doi.org/10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tan M, Jia L, Wei D, Zhao Y, Chen G, Xu J, Zhao L, Thomas D, Beer DG, Sun Y. Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. J Clin Invest. 2014a;124(2):835–846. doi: 10.1172/JCI70297. https://doi.org/10.1172/JCI70297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang M, Yu G, Chen P, Li H, Wei D, Zhu J, Xie L, Jia H, Shi J, Li C, Yao W, Wang Y, Gao Q, Jeong LS, Lee HW, Yu J, Hu F, Mei J, Wang P, Chu Y, Qi H, Yang M, Dong Z, Sun Y, Hoffman RM, Jia L. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014b;106(6):dju083. doi: 10.1093/jnci/dju083. https://doi.org/10.1093/jnci/dju083. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70(24):10310–10320. doi: 10.1158/0008-5472.CAN-10-2062. https://doi.org/10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Zeman MK, Chen JY, Yee MC, Cimprich KA. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell. 2011;42(2):237–249. doi: 10.1016/j.molcel.2011.02.026. https://doi.org/10.1016/j.molcel.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11(9):629–643. doi: 10.1038/nrc3120. https://doi.org/10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Warbrick E. The p66 and p12 subunits of DNA polymerase delta are modified by ubiquitin and ubiquitin-like proteins. Biochem Biophys Res Commun. 2006;349(1):360–366. doi: 10.1016/j.bbrc.2006.08.049. https://doi.org/10.1016/j.bbrc.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Liu E, Li X, Yan F, Zhao Q, Wu X. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J Biol Chem. 2004;279(17):17283–17288. doi: 10.1074/jbc.C300549200. https://doi.org/10.1074/jbc.C300549200. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA, Lock K, Yenamandra A, Cortez D. DDB1 maintains genome integrity through regulation of Cdt1. Mol Cell Biol. 2006;26(21):7977–7990. doi: 10.1128/MCB.00819-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14(12):1050–1061. doi: 10.1038/embor.2013.173. https://doi.org/10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Dutta A. The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev. 2007;21(2):184–194. doi: 10.1101/gad.1495007. https://doi.org/10.1101/gad.1495007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123(1):13–24. doi: 10.1016/j.cell.2005.09.019. https://doi.org/10.1016/j.cell.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122(6):915–926. doi: 10.1016/j.cell.2005.08.013. https://doi.org/10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Maric M, Maculins T, De Piccoli G, Labib K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science. 2014;346(6208):1253596. doi: 10.1126/science.1253596. https://doi.org/10.1126/science.1253596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, McNamara K, Zaghlul S, Edkins S, Stevens C, Brennan C, Martin ES, Wiedemeyer R, Kabbarah O, Nogueira C, Histen G, Aster J, Mansour M, Duke V, Foroni L, Fielding AK, Goldstone AH, Rowe JM, Wang YA, Look AT, Stratton MR, Chin L, Futreal PA, DePinho RA. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447(7147):966–971. doi: 10.1038/nature05886. https://doi.org/10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998;93(6):1043–1053. doi: 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- McIntyre J, Woodgate R. Regulation of translesion DNA synthesis: posttranslational modification of lysine residues in key proteins. DNA Repair (Amst) 2015;29:166–179. doi: 10.1016/j.dnarep.2015.02.011. https://doi.org/10.1016/j.dnarep.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Zou-Yang XH, Kim SY, Hidaka M, Tansey WP, Stillman B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol Cell. 2002;9(3):481–491. doi: 10.1016/s1097-2765(02)00467-7. [DOI] [PubMed] [Google Scholar]
- Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci. 2009;66(11–12):1924–1938. doi: 10.1007/s00018-009-8712-7. https://doi.org/10.1007/s00018-009-8712-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita M, Morimoto A, Ishii T, Komori H, Shiomi Y, Higuchi Y, Nishitani H. Positively charged residues located downstream of PIP box, together with TD amino acids within PIP box, are important for CRL4(Cdt2) -mediated proteolysis. Genes Cells. 2011;16(1):12–22. doi: 10.1111/j.1365-2443.2010.01464.x. https://doi.org/10.1111/j.1365-2443.2010.01464.x. [DOI] [PubMed] [Google Scholar]
- Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116(9):1515–1523. doi: 10.1182/blood-2010-03-272567. https://doi.org/10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71(8):3042–3051. doi: 10.1158/0008-5472.CAN-10-2122. https://doi.org/10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- Mocciaro A, Berdougo E, Zeng K, Black E, Vagnarelli P, Earnshaw W, Gillespie D, Jallepalli P, Schiebel E. Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain DNA damage checkpoint proficiency but are impaired in DNA repair. J Cell Biol. 2010;189(4):631–639. doi: 10.1083/jcb.200910057. https://doi.org/10.1083/jcb.200910057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, D’Andrea AD. DNA damage discrimination at stalled replication forks by the Rad5 homologs HLTF and SHPRH. Mol Cell. 2011;42(2):141–143. doi: 10.1016/j.molcel.2011.03.018. https://doi.org/10.1016/j.molcel.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Moreno SP, Bailey R, Campion N, Herron S, Gambus A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science. 2014;346(6208):477–481. doi: 10.1126/science.1253585. https://doi.org/10.1126/science.1253585. [DOI] [PubMed] [Google Scholar]
- Morohashi H, Maculins T, Labib K. The amino-terminal TPR domain of Dia2 tethers SCF(Dia2) to the replisome progression complex. Curr Biol. 2009;19(22):1943–1949. doi: 10.1016/j.cub.2009.09.062. https://doi.org/10.1016/j.cub.2009.09.062. [DOI] [PubMed] [Google Scholar]
- Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JH, Myung K. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A. 2008;105(34):12411–12416. doi: 10.1073/pnas.0805685105. https://doi.org/10.1073/pnas.0805685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16(3):323–333. doi: 10.1016/j.semcdb.2005.02.010. https://doi.org/10.1016/j.semcdb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–381. doi: 10.1038/nrc1881. https://doi.org/10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- Narbonne-Reveau K, Senger S, Pal M, Herr A, Richardson HE, Asano M, Deak P, Lilly MA. APC/CFzr/Cdh1 promotes cell cycle progression during the drosophila endocycle. Development. 2008;135(8):1451–1461. doi: 10.1242/dev.016295. https://doi.org/10.1242/dev.016295. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21(10):1563–1573. doi: 10.1517/13543784.2012.707192. https://doi.org/10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, Lis JT, Allis CD, Reinberg D. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell. 2002;9(6):1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, Lygerou Z, Nishimoto T. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006;25(5):1126–1136. doi: 10.1038/sj.emboj.7601002. https://doi.org/10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283(43):29045–29052. doi: 10.1074/jbc.M806045200. https://doi.org/10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010;40(3):364–376. doi: 10.1016/j.molcel.2010.10.011. https://doi.org/10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Yang SW, Yu KR, Ka SH, Lee SW, Seol JH, Jeon YJ, Chung CH. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol Cell. 2014;54(4):626–638. doi: 10.1016/j.molcel.2014.03.031. https://doi.org/10.1016/j.molcel.2014.03.031. [DOI] [PubMed] [Google Scholar]
- Petersen BO, Lukas J, Sorensen CS, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin a/CDK2 regulates its subcellular localization. EMBO J. 1999;18(2):396–410. doi: 10.1093/emboj/18.2.396. https://doi.org/10.1093/emboj/18.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen BO, Wagener C, Marinoni F, Kramer ER, Melixetian M, Lazzerini Denchi E, Gieffers C, Matteucci C, Peters JM, Helin K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000;14(18):2330–2343. doi: 10.1101/gad.832500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. https://doi.org/10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14(6):655–665. [PMC free article] [PubMed] [Google Scholar]
- Pfleger CM, Lee E, Kirschner MW. Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes Dev. 2001;15(18):2396–2407. doi: 10.1101/gad.918201. https://doi.org/10.1101/gad.918201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006;16(1):55–63. doi: 10.1016/j.tcb.2005.11.006. https://doi.org/10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Plachta M, Halas A, McIntyre J, Sledziewska-Gojska E. The steady-state level and stability of TLS polymerase eta are cell cycle dependent in the yeast S. cerevisiae. DNA Repair (Amst) 2015;29:147–153. doi: 10.1016/j.dnarep.2015.02.015. https://doi.org/10.1016/j.dnarep.2015.02.015. [DOI] [PubMed] [Google Scholar]
- Plosky BS, Vidal AE, Fernandez de Henestrosa AR, McLenigan MP, McDonald JP, Mead S, Woodgate R. Controlling the subcellular localization of DNA polymerases iota and eta via interactions with ubiquitin. EMBO J. 2006;25(12):2847–2855. doi: 10.1038/sj.emboj.7601178. https://doi.org/10.1038/sj.emboj.7601178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Pentz K, Zhu Q, Wang Q, He J, Srivastava AK, Wani AA. USP7 modulates UV-induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene. 2015;34(36):4791–4796. doi: 10.1038/onc.2014.394. https://doi.org/10.1038/onc.2014.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing P, Han L, Bin L, Yan L, Ping WX. USP7 regulates the stability and function of HLTF through deubiquitination. J Cell Biochem. 2011;112(12):3856–3862. doi: 10.1002/jcb.23317. https://doi.org/10.1002/jcb.23317. [DOI] [PubMed] [Google Scholar]
- Rahal R, Amon A. Mitotic CDKs control the metaphase-anaphase transition and trigger spindle elongation. Genes Dev. 2008;22(11):1534–1548. doi: 10.1101/gad.1638308. https://doi.org/10.1101/gad.1638308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 2001;105(5):645–655. doi: 10.1016/s0092-8674(01)00361-0. [DOI] [PubMed] [Google Scholar]
- Renaudin X, Koch Lerner L, Menck CF, Rosselli F. The ubiquitin family meets the Fanconi anemia proteins. Mutat Res Rev Mutat Res. 2016;769:36–46. doi: 10.1016/j.mrrev.2016.06.004. https://doi.org/10.1016/j.mrrev.2016.06.004. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. https://doi.org/10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, Bielinsky AK. Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol Cell. 2004;16(2):173–185. doi: 10.1016/j.molcel.2004.09.017. https://doi.org/10.1016/j.molcel.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Rizzardi LF, Coleman KE, Varma D, Matson JP, Oh S, Cook JG. CDK1-dependent inhibition of the E3 ubiquitin ligase CRL4CDT2 ensures robust transition from S phase to mitosis. J Biol Chem. 2015;290(1):556–567. doi: 10.1074/jbc.M114.614701. https://doi.org/10.1074/jbc.M114.614701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JA, Cross FR. Regulated degradation of the APC coactivator Cdc20. Cell Div. 2010;5:23. doi: 10.1186/1747-1028-5-23. https://doi.org/10.1186/1747-1028-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Shaykh Baygloo N, Aghasadeghi MR, Allahbakhshi E. HIV-1 Vpr protein enhances proteasomal degradation of MCM10 DNA replication factor through the Cul4-DDB1[VprBP] E3 ubiquitin ligase to induce G2/M cell cycle arrest. J Biol Chem. 2015;290(28):17380–17389. doi: 10.1074/jbc.M115.641522. https://doi.org/10.1074/jbc.M115.641522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseaulin LC, Noguchi C, Martinez E, Ziegler MA, Toda T, Noguchi E. Coordinated degradation of replisome components ensures genome stability upon replication stress in the absence of the replication fork protection complex. PLoS Genet. 2013;9(1):e1003213. doi: 10.1371/journal.pgen.1003213. https://doi.org/10.1371/journal.pgen.1003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha P, Chen J, Thome KC, Lawlis SJ, Hou ZH, Hendricks M, Parvin JD, Dutta A. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol Cell Biol. 1998;18(5):2758–2767. doi: 10.1128/mcb.18.5.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, Hopkins N, Lees JA. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 2006;20(22):3117–3129. doi: 10.1101/gad.1482106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12(4):220. doi: 10.1186/gb-2011-12-4-220. https://doi.org/10.1186/gb-2011-12-4-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, Espejo A, Bedford MT, Nussenzweig A, Busslinger M, Jenuwein T. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22(15):2048–2061. doi: 10.1101/gad.476008. https://doi.org/10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. https://doi.org/10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem. 2006;281(10):6246–6252. doi: 10.1074/jbc.M512705200. https://doi.org/10.1074/jbc.M512705200. [DOI] [PubMed] [Google Scholar]
- Shibutani ST, de la Cruz AF, Tran V, Turbyfill WJ, 3rd, Reis T, Edgar BA, Duronio RJ. Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Dev Cell. 2008;15(6):890–900. doi: 10.1016/j.devcel.2008.10.003. https://doi.org/10.1016/j.devcel.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Toth A, Galova M, Nasmyth K. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature. 1999;402(6758):203–207. doi: 10.1038/46080. https://doi.org/10.1038/46080. [DOI] [PubMed] [Google Scholar]
- Simon NE, Schwacha A. The Mcm2-7 replicative helicase: a promising chemotherapeutic target. Biomed Res Int. 2014;2014:549719. doi: 10.1155/2014/549719. https://doi.org/10.1155/2014/549719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson LJ, Ross AL, Szuts D, Alviani CA, Oestergaard VH, Patel KJ, Sale JE. RAD18-independent ubiquitination of proliferating-cell nuclear antigen in the avian cell line DT40. EMBO Rep. 2006;7(9):927–932. doi: 10.1038/sj.embor.7400777. https://doi.org/10.1038/sj.embor.7400777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaprasad U, Machida YJ, Dutta A. APC/C–the master controller of origin licensing? Cell Div. 2007;2:8. doi: 10.1186/1747-1028-2-8. https://doi.org/10.1186/1747-1028-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–381. doi: 10.1038/nrm3582. https://doi.org/10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014;13(12):889–903. doi: 10.1038/nrd4432. https://doi.org/10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoneczna A, McIntyre J, Skoneczny M, Policinska Z, Sledziewska-Gojska E. Polymerase eta is a short-lived, proteasomally degraded protein that is temporarily stabilized following UV irradiation in Saccharomyces cerevisiae. J Mol Biol. 2007;366(4):1074–1086. doi: 10.1016/j.jmb.2006.11.093. https://doi.org/10.1016/j.jmb.2006.11.093. [DOI] [PubMed] [Google Scholar]
- Sommers JA, Suhasini AN, Brosh RM., Jr Protein degradation pathways regulate the functions of helicases in the DNA damage response and maintenance of genomic stability. Biomol Ther. 2015;5(2):590–616. doi: 10.3390/biom5020590. https://doi.org/10.3390/biom5020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. https://doi.org/10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8(11):894–903. doi: 10.1038/nrm2276. https://doi.org/10.1038/nrm2276. [DOI] [PubMed] [Google Scholar]
- Tada S, Li A, Maiorano D, Mechali M, Blow JJ. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat Cell Biol. 2001;3(2):107–113. doi: 10.1038/35055000. https://doi.org/10.1038/35055000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Parvin JD, Dutta A. Degradation of Cdt1 during S phase is Skp2-independent and is required for efficient progression of mammalian cells through S phase. J Biol Chem. 2005;280(24):23416–23423. doi: 10.1074/jbc.M501208200. https://doi.org/10.1074/jbc.M501208200. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol. 2017;18(2):115–126. doi: 10.1038/nrm.2016.148. https://doi.org/10.1038/nrm.2016.148. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Diffley JF. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat Cell Biol. 2002;4(3):198–207. doi: 10.1038/ncb757. https://doi.org/10.1038/ncb757. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Knapp D, Nasmyth K. Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell. 1997;90(4):649–660. doi: 10.1016/s0092-8674(00)80526-7. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Nakatani T, Kamitani T. Negative regulation of NEDD8 conjugation pathway by novel molecules and agents for anticancer therapy. Curr Pharm Des. 2013;19(22):4131–4139. doi: 10.2174/1381612811319220017. [DOI] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12(11):1086–1093. doi: 10.1038/ncb2113. doi: ncb2113 [pii]1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- Tatsumi Y, Ohta S, Kimura H, Tsurimoto T, Obuse C. The ORC1 cycle in human cells: I. cell cycle-regulated oscillation of human ORC1. J Biol Chem. 2003;278(42):41528–41534. doi: 10.1074/jbc.M307534200. https://doi.org/10.1074/jbc.M307534200. [DOI] [PubMed] [Google Scholar]
- Tatsumi Y, Sugimoto N, Yugawa T, Narisawa-Saito M, Kiyono T, Fujita M. Deregulation of Cdt1 induces chromosomal damage without rereplication and leads to chromosomal instability. J Cell Sci. 2006;119(Pt 15):3128–3140. doi: 10.1242/jcs.03031. [DOI] [PubMed] [Google Scholar]
- Teixeira LK, Reed SI. Ubiquitin ligases and cell cycle control. Annu Rev Biochem. 2013;82:387–414. doi: 10.1146/annurev-biochem-060410-105307. https://doi.org/10.1146/annurev-biochem-060410-105307. [DOI] [PubMed] [Google Scholar]
- Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol Cell. 2010;37(1):143–149. doi: 10.1016/j.molcel.2009.12.018. https://doi.org/10.1016/j.molcel.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Shibata E, Abbas T, Dutta A. Degradation of p12 subunit by CRL4Cdt2 E3 ligase inhibits fork progression after DNA damage. J Biol Chem. 2013;288(42):30509–30514. doi: 10.1074/jbc.C113.505586. https://doi.org/10.1074/jbc.C113.505586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu YM, Bielinsky AK. Enigmatic roles of Mcm10 in DNA replication. Trends Biochem Sci. 2013;38(4):184–194. doi: 10.1016/j.tibs.2012.12.003. https://doi.org/10.1016/j.tibs.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD, Takahashi T. Readers of PCNA modifications. Chromosoma. 2013;122(4):259–274. doi: 10.1007/s00412-013-0410-4. https://doi.org/10.1007/s00412-013-0410-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Fatyol K, Hurwitz J, Yoon JH, Prakash L, Prakash S, Haracska L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc Natl Acad Sci U S A. 2008;105(10):3768–3773. doi: 10.1073/pnas.0800563105. https://doi.org/10.1073/pnas.0800563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen F, Sengupta S, De Piccoli G, Sanchez-Diaz A, Labib K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 2012;31(9):2195–2206. doi: 10.1038/emboj.2012.69. https://doi.org/10.1038/emboj.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786(1):49–59. doi: 10.1016/j.bbcan.2008.05.002. https://doi.org/10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, Wagle N, Hwang DS, Dutta A. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11(4):997–1008. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278(5337):460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10(10) doi: 10.1074/mcp.M111.013284. M111013284. https://doi.org/10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace HA, Merkle JA, Yu MC, Berg TG, Lee E, Bosco G, Lee LA. TRIP/NOPO E3 ubiquitin ligase promotes ubiquitylation of DNA polymerase eta. Development. 2014;141(6):1332–1341. doi: 10.1242/dev.101196. https://doi.org/10.1242/dev.101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter D, Hoffmann S, Komseli ES, Rappsilber J, Gorgoulis V, Sorensen CS. SCF(cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat Commun. 2016;7:10530. doi: 10.1038/ncomms10530. https://doi.org/10.1038/ncomms10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14(4):233–247. doi: 10.1038/nrc3700. https://doi.org/10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A. 2004;101(13):4419–4424. doi: 10.1073/pnas.0307700101. https://doi.org/10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci U S A. 2006;103(24):8971–8976. doi: 10.1073/pnas.0510167103. https://doi.org/10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428(6979):194–198. doi: 10.1038/nature02381. https://doi.org/10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, Smith PG, Morgan MA, Sun Y. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72(1):282–293. doi: 10.1158/0008-5472.CAN-11-2866. https://doi.org/10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8(2):83–93. doi: 10.1038/nrc2290. https://doi.org/10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11(14):1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science. 2000;290(5500):2309–2312. doi: 10.1126/science.290.5500.2309. https://doi.org/10.1126/science.290.5500.2309. [DOI] [PubMed] [Google Scholar]
- Wong JV, Dong P, Nevins JR, Mathey-Prevot B, You L. Network calisthenics: control of E2F dynamics in cell cycle entry. Cell Cycle. 2011;10(18):3086–3094. doi: 10.4161/cc.10.18.17350. https://doi.org/10.4161/cc.10.18.17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Wang W, Kong X, Congdon LM, Yokomori K, Kirschner MW, Rice JC. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010;24(22):2531–2542. doi: 10.1101/gad.1984210. https://doi.org/10.1101/gad.1984210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, Martin SR, Sarma K, Reinberg D, Gamblin SJ, Wilson JR. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 2005;19(12):1444–1454. doi: 10.1101/gad.1315905. https://doi.org/10.1101/gad.1315905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene. 2010;29(32):4493–4503. doi: 10.1038/onc.2010.190. https://doi.org/10.1038/onc.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Weinacht CP, Zhuang Z. Regulatory role of ubiquitin in eukaryotic DNA translesion synthesis. Biochemistry. 2013;52(19):3217–3228. doi: 10.1021/bi400194r. https://doi.org/10.1021/bi400194r. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Nasmyth K. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 1999;13(16):2039–2058. doi: 10.1101/gad.13.16.2039. [DOI] [PubMed] [Google Scholar]
- Zaidi IW, Rabut G, Poveda A, Scheel H, Malmstrom J, Ulrich H, Hofmann K, Pasero P, Peter M, Luke B. Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep. 2008;9(10):1034–1040. doi: 10.1038/embor.2008.155. https://doi.org/10.1038/embor.2008.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Lin JR, Freire R, Cimprich KA. DNA damage-specific deubiquitination regulates Rad18 functions to suppress mutagenesis. J Cell Biol. 2014;206(2):183–197. doi: 10.1083/jcb.201311063. https://doi.org/10.1083/jcb.201311063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chea J, Meng X, Zhou Y, Lee EY, Lee MY. PCNA is ubiquitinated by RNF8. Cell Cycle. 2008;7(21):3399–3404. doi: 10.4161/cc.7.21.6949. https://doi.org/10.4161/cc.7.21.6949. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sun Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19(18):3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Morgan MA, Sun Y. Targeting Neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014;21(17):2383–2400. doi: 10.1089/ars.2013.5795. https://doi.org/10.1089/ars.2013.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. 2003;423(6942):885–889. doi: 10.1038/nature01747. [DOI] [PubMed] [Google Scholar]
- Zhu W, Depamphilis ML. Selective killing of cancer cells by suppression of geminin activity. Cancer Res. 2009;69(11):4870–4877. doi: 10.1158/0008-5472.CAN-08-4559. https://doi.org/10.1158/0008-5472.CAN-08-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Dutta A. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol. 2006;26(12):4601–4611. doi: 10.1128/MCB.02141-05. https://doi.org/10.1128/MCB.02141-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Chen Y, Dutta A. Rereplication by depletion of geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol Cell Biol. 2004;24(16):7140–7150. doi: 10.1128/MCB.24.16.7140-7150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Abbas T, Dutta A. DNA replication and genomic instability. Adv Exp Med Biol. 2005;570:249–279. doi: 10.1007/1-4020-3764-3_9. https://doi.org/10.1007/1-4020-3764-3_9. [DOI] [PubMed] [Google Scholar]
- Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, Dutta A. Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev. 2007;21(18):2288–2299. doi: 10.1101/gad.1585607. https://doi.org/10.1101/gad.1585607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Lee CY, Johnson RL, Wichterman J, Huang R, DePamphilis ML. An image-based, high-throughput screening assay for molecules that induce excess DNA replication in human cancer cells. Mol Cancer Res. 2011;9(3):294–310. doi: 10.1158/1541-7786.MCR-10-0570. https://doi.org/10.1158/1541-7786.MCR-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilio N, Eifler-Olivi K, Ulrich HD. Functions of SUMO in the maintenance of genome stability. Adv Exp Med Biol. 2017;963:51–87. doi: 10.1007/978-3-319-50044-7_4. https://doi.org/10.1007/978-3-319-50044-7_4. [DOI] [PubMed] [Google Scholar]
- Zlatanou A, Sabbioneda S, Miller ES, Greenwalt A, Aggathanggelou A, Maurice MM, Lehmann AR, Stankovic T, Reverdy C, Colland F, Vaziri C, Stewart GS. USP7 is essential for maintaining Rad18 stability and DNA damage tolerance. Oncogene. 2016;35(8):965–976. doi: 10.1038/onc.2015.149. https://doi.org/10.1038/onc.2015.149. [DOI] [PubMed] [Google Scholar]