

Abstract
Alpha-1-antitrypsin deficiency (AATD) is a hereditary disorder that is characterized by a low serum level of alpha-1-antitrypsin (AAT). The loss of anti-inflammatory and antiproteolytic functions, together with pro-inflammatory effects of polymerized AAT contribute to protein degradation and increased inflammation resulting in an increased risk of developing chronic obstructive pulmonary disease (COPD) and emphysema, especially in smokers. AATD is a rare disease that is significantly underdiagnosed. According to recent data that are based on extrapolations, in many countries only 5–15% of homozygous individuals have been identified. Furthermore, the diagnostic delay typically exceeds 5 years, resulting in an average age at diagnosis of about 45 years. Although the American Thoracic Society/European Respiratory Society recommendations state that all symptomatic adults with persistent airway obstruction should be screened, these recommendations are not being followed. Potential reasons for that include missing knowledge about the disease and the appropriate tests, and the low awareness of physicians with regard to the disorder. Once the decision to initiate testing has been made, a screening test (AAT serum level or other) should be performed. Further diagnostic evaluation is based on the following techniques: polymerase chain reaction (PCR) for frequent and clinically important mutations, isoelectric focusing (IEF) with or without immunoblotting, and sequencing of the gene locus coding for AAT. Various diagnostic algorithms have been published for AATD detection (severe deficiency or carrier status). Modern laboratory approaches like the use of serum separator cards, a lateral flow assay to detect the Z-protein, and a broader availability of next-generation sequencing are recent advances, likely to alter existing algorithms.
Keywords: Alpha-1-antitrypsin deficiency (AATD), awareness, diagnosis, screening, test
Introduction
Alpha-1-antitrypsin deficiency (AATD) is a rare disease that, like other rare diseases, is underdiagnosed [Blanco et al. 2006; Carroll et al. 2011]. Based on comparisons of the estimated frequency of AATD and the number of identified patients it has been concluded that the vast majority (>85%) of potential patients are yet to be identified [Silverman and Sandhaus, 2009]. Current surveys indicate that the diagnosis is often delayed for several years resulting in an average age at diagnosis of about 45 years [Campos et al. 2005; Kohnlein et al. 2010; Stoller et al. 2005].
This is noteworthy since clear recommendations regarding indications to test for AATD have existed for quite some time: The World Health Organisation recommends testing of all chronic obstructive pulmonary disease (COPD) patients [WHO, 1997], and the European Respiratory Society and American Thoracic Society Guidelines recommend the testing of all symptomatic adults with persistent airway obstruction [ATS/ERS, 2003]. The observed discrepancy between the number of expected patients and the number of identified patients leads to the assumption, that these recommendations are not being followed. Potential reasons for that include missing knowledge about the disease and the appropriate tests, and the low awareness of physicians with regard to the disorder. Moreover, the multi-step approach that is required for final diagnosis have been put forward as potential obstacles on the way to early identification of affected individuals [Stoller et al. 2007].
This review focuses on efforts in the past and on potential actions with the goal to overcome the low awareness and to improve the laboratory diagnosis of AATD.
Biological background
AATD was first described in 1963 by Laurell and Eriksson. Analysing the serum electrophoresis of five patients with severe emphysema, they recognized the absence of a specific band [Laurell and Eriksson, 2013]. Shortly after, AATD had also been associated with a specific type of liver cirrhosis [Sharp et al. 1969]. The finding of alpha-1-antitrypsin (AAT) being a protease inhibitor together with the observation of severe emphysema in deficient individuals led to the development of the protease–antiprotease imbalance hypothesis: antiproteases such as AAT protect tissue (mainly lung parenchyma) from proteolytic damage by enzymes such as neutrophil elastase [Carrell and Lomas, 2002; Gadek et al. 1981]. The absence of AAT as the most important antiprotease of the lung would then lead to uninhibited protease activity resulting in lung parenchymal destruction, i.e. emphysema.
The gene coding for AAT is located on chromosome 14q32.1 as part of a gene cluster called serine protease inhibitor (SERPIN). The Protease Inhibitor (Pi) locus itself is called SERPINA1. The 12.2 kb long gene consists of 4 coding exons (II, III, IV, V), 3 noncoding exons (IA, IB, IC) and 6 introns. The region coding for the reactive loop is located in exon V. The clinically most important mutation (Pi*Z) is caused by a single nucleotide polymorphism (SNP) at position 342 (Glu342Lys) [Jeppsson, 1976]. The consecutive conformational change allows the reactive loop of a second AAT molecule to bind at this position leading to AAT polymers formation [Carrell and Lomas, 2002]. In the liver, these polymers form inclusion bodies and are not secreted into the circulation. The other common mutation Pi*S results in a protein that is degraded intracellularly prior to secretion. Compared with the Pi*Z mutation the serum level is only mildly reduced, and the risk for lung disease is lower compared to the Pi*Z mutation [Crystal et al. 1989; Curiel et al. 1989]. Several mutations (resulting in a stop codon) lead to the total absence of AAT (null mutations). Although more than 150 different mutations in SERPINA1 have been described, the overall frequency in the population is very low [Fregonese et al. 2008; Stockley et al. 2007].
Formation of AAT polymers is not restricted to liver cells, but may also occur in the lungs of Pi*ZZ patients [Janciauskiene et al. 2002]. Polymerized AAT exhibits pro-inflammatory effects by stimulating neutrophil adhesion and chemotaxis [Lomas and Carrell, 2002; Mahadeva et al. 2005; Mulgrew et al. 2004]. Moreover, it could be demonstrated that misfolded AAT protein accumulates in the endoplasmic reticulum of neutrophils in Pi*ZZ individuals leading to the expression of pro-apoptotic pathways (including tumour necrosis factor [TNF]-alpha). Infusion of purified AAT in these patients reduces levels of membrane-bound TNF-alpha and apoptosis [Bergin et al. 2014]. This is in parallel with the restitution of the bacterial killing capacity of AAT-treated cells [Hurley et al. 2014]. Thus, pro-inflammatory effects of polymerized AAT, in addition to the loss of antiproteolytic and anti-inflammatory functions, may contribute significantly to the development of lung parenchyma degradation and inflammation in AATD patients (Figure 1).
Figure 1.

Simplified model of AATD pathophysiology. AAT protein (Z-protein >> M-protein) may polymerize, especially under the influence of cigarette smoke. Polymers may accumulate in the liver, leading to cirrhosis. The loss of protective properties together with the gain of pro-inflammatory and immunomodulatory properties leads to the development of emphysema. AAT, alpha-1-antitrypsin; AATD, alpha-1-antitrypsin deficiency; NE, neutrophil elastase. (Modified from Janciauskiene et al. [2002] and Lomas and Parfrey [2004].)
Prevalence
As in other orphan diseases the exact prevalence of AATD is difficult to determine. The main reason for that is that large-scale population-based screening studies are missing. Determining the prevalence of the genetic predisposition in diseased populations (e.g. respiratory diseases) will inevitably overestimate the prevalence in the general population. On the other hand, restricting the target population to healthy individuals (for example, blood donors) may underestimate the prevalence. Although both approaches are potentially flawed, the latter is very likely to be more precise. Table 1 displays published studies with >1000 individuals screened.
Table 1.
Screening studies with a number of screened individuals >1000, in the order of publication date. (Modified from Aboussouan and Stoller [2009].).
| Year | Location | Screened Population | Number screened | ZZ [%] | SZ [%] | MZ [%] | SS [%] | MS [%] | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1975 | England | Population survey | 5.588 | 0.04 | 0.21 | 2.02 | 0.32 | 7.19 | Cook [1975] |
| 1976 | Sweden | Newborns | 200.000 | 0.06 | 0.02 | – | – | – | Sveger [1976] |
| 1976 | Netherlands | Population survey | 1.474 | 0.07 | 0.07 | 2.24 | 0 | 2.84 | Hoffmann and van den Broek [1976] |
| 1976 | California | High school | 1.841 | 0 | 0.27 | 1.85 | 0.05 | 6.9 | Lieberman et al. [1976] |
| 1977 | New York | Newborns | 1.010 | 0 | 0 | 1.19 | 0.89 | 3.07 | Evans et al. [1977] |
| 1977 | Arizona | Population survey | 2.944 | 0.07 | 0.2 | 3.0 | – | 7.1 | Morse et al. [1977] |
| 1978 | Oregon | Newborns | 107.038 | 0.02 | 0.01 | – | – | O’Brien et al. [1978] | |
| 1979 | Sweden | Military recruits | 11.128 | 0.04 | 0.08 | 0.03 | – | – | Sveger and Mazodier, [1979] |
| 1980 | Netherlands | Newborns | 95.083 | 0.03 | – | – | 0.04 | 0 | Dijkman et al. [1980] |
| 1988 | Belgium | Newborns | 10.329 | 0.06 | 0.12 | 0.97 | 0.01 | 0.88 | Kimpen et al. [1988] |
| 1989 | Missouri | Blood donors | 20.000 | 0.04 | 0.01 | 0.01 | – | – | Silverman et al. [1989] |
| 1993 | New York | Newborns | 11.081 | 0.03 | 0.05 | 0.53 | 0.01 | 0.09 | Spence et al. [1993] |
| 2002 | Denmark | Random sample | 9.187 | 0.07 | 0.11 | 4.91 | 0.13 | 5.0 | Dahl et al. [2002] |
| 2011 | Ireland | Blood bank | 1.100 | – | 0.18 | 4.18 | 0.18 | 10.3 | Carroll et al. [2011] |
| 2011 | Turkey | Blood donors | 1.203 | – | – | 0.58 | – | 0.50 | Simsek et al. [2011] |
| 2012 | Switzerland | Population survey | 6.057 | 0.02 | 0.17 | 2.36 | 0.16 | 7.48 | Ferrarotti et al. [2012] |
| 2014 | Poland | Newborns | 4.231 | 0.01 | 0.02 | 2.13 | 0.01 | 2.13 | Chorostowska-Wynimko et al. [2014] |
In Europe, the highest prevalence of the Pi*Z mutation has been recorded in North-Western European countries with a gene frequency between 0.026 and 0.049 [Dahl et al. 2002; Hutchison, 1998; Sveger, 1976]. The prevalence in North America seems to be similar (0.019 and 0.03) [Lieberman et al. 1976; Morse et al. 1977]. In eastern Asian countries, however, the gene frequency of Pi*Z seems to be extremely low (0.006).
Knowledge and awareness
AATD is significantly under-diagnosed
Comparing estimates of the prevalence with the number of known patients in specific countries, it becomes evident that only a minority of expected patients have already been identified. In the US, 70,000–100,000 AAT-deficient individuals are expected, although only 10% have been identified [Stoller and Brantly, 2013]. In Europe, where the estimated number of deficient individuals is 125,000, only approximately 5000 individuals have been included in the international registry [Blanco et al. 2006; Stockley et al. 2013].
Multiple investigations have demonstrated that the average delay between onset of symptoms and time of diagnosis exceeds 5 years [Stoller et al. 1994]. Although a variety of measures had been undertaken during the last 10 years, this scenario has not changed significantly [Campos et al. 2005; Kohnlein et al. 2010; Stoller et al. 2005].
Reasons for under-diagnosis
There are a number of possible reasons explaining under diagnosis: poor awareness of the disease and/or the methods of testing; the perception that existing treatments are lacking efficacy (or are unavailable in specific regions of the world); the testing algorithm for AATD that may be perceived as complicated. While these reasons are plausible, there are only scarce data to support them.
There is little to no evidence at all in the literature that therapeutic nihilism (no efficacious treatment available, therefore no need to test) contributes significantly to under-recognition of AATD. In a survey that assessed reasons for medical doctors for not testing as recommended, only 8% of survey respondents answered ‘There is no treatment available for this disease’ [Greulich et al. 2013]. This is in contrast to personal communication between many AATD specialists and their colleagues not specialized in AATD.
Regarding knowledge about the disease, Taliercio and colleagues assessed knowledge about clinical manifestations and test-related matters about AATD in a 30-item questionnaire emailed to respiratory therapists and interns of internal medicine. The percentage of correct answers was low, regardless of whether the respondents were respiratory therapists (52%) or internal medicine residents (54%) [Taliercio et al. 2010]. When doctors in Germany and Italy were asked to rate their own knowledge about AATD, 16.5% of pulmonologists (20/121), 41.1% of internists (37/90) and 56.0% of general practitioners (84/150) rated their own knowledge as poor (‘little’ or ‘none at all’) [Greulich et al. 2013]. In parallel, the percentage of doctors that reported testing on a regular basis decreases from pulmonologists over internists to general practitioners (Figure 2). In the same survey, only 20.6% of doctors that initiated testing stated that they would follow the ATS/ERS guidelines and test every COPD patient once in his lifetime [Greulich et al. 2013]. Taking into account that respondents to surveys tend to overestimate their performance, these results are somewhat disappointing.
Figure 2.
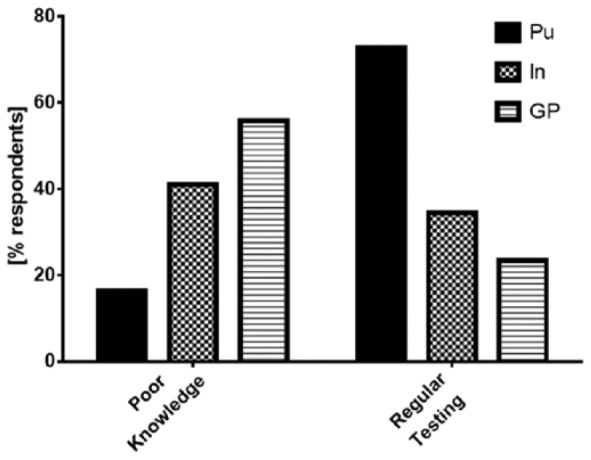
The left-hand side of the figure displays the percentage of medical doctors that rated their own knowledge as either ‘little’ or ‘none at all’. The right-hand side of the figure displays the percentage of medical doctors that stated they would ‘currently test for alpha-1-antitrypsin deficiency’. GP, general practitioner (n = 150); In, internist (n = 90); Pu, pulmonologist (n = 121). (Modified from Greulich et al. [2013].)
Worldwide, there is considerable heterogeneity in the availability of augmentation therapy as a specific treatment for AATD (Table 2). It is obvious that the lack of a specific treatment may discourage physicians to screen for the disease.
Table 2.
The availability of different products licenced for alpha 1-antitrypsin augmentation therapy in certain countries of the world. S/D: solvent/detergent. (Modified from Teschler [2015].).
| Manufacturer | Drug | Purification method | Approved in |
|---|---|---|---|
| Baxter (Deerfield, IL, USA) | Aralast | S/D purification + nanofiltration | USA |
| CSL Behring (King of Prussia, PA, USA) | Zemeira | Pasteurization | USA |
| Grifols (Barcelona, Spain) | Prolastin | Pasteurization | Austria, Belgium, Denmark, Finland, Germany, Greece, Ireland, Italy, The Netherlands, Norway, Poland, Portugal, Spain, Sweden, Switzerland |
| Grifols (Barcelona, Spain) | Prolastin C | Pasteurization | Argentina, Canada, Colombia, USA |
| Grifols (Barcelona, Spain) | Trypsone | S/D purification + nanofiltration | Argentina, Brazil, Chile, Mexico, Spain |
| Kamada (Ness Ziona, Israel) | Glassia | Nanofiltration + S/D purification | USA, Brazil |
| LFB (Courtaboeuf, France) | Alfalastin | Pasteurization | France |
In summary, available data demonstrate the need to continue education of medical staff regarding AATD and support the assumption that inadequate knowledge does contribute significantly to under-recognition of AATD.
Targeted screening
While screening efforts in newborn screening programs (and less so in healthy populations such as blood donors, random population samples and others) are suited to determine the prevalence of deficiency genes in the general population, another approach is usually undertaken to detect potential patients in a time- and money-saving manner: ‘Targeted detection’ concentrates its efforts on populations that carry an increased risk for the mutation and the disease.
As AATD is a genetic disorder with pulmonary and extrapulmonary manifestations, a number of indicator diseases should attract the treating physician’s attention towards AATD. The joint ATS/ERS statement therefore recommends testing for AATD in all COPD patients, all nonresponsive asthmatic adults/adolescents, patients with bronchiectasis of unknown aetiology, all individuals with cryptogenic cirrhosis/liver disease, granulomatosis with polyangiitis, necrotizing panniculitis, and first-degree relatives of patients/carriers with AATD [ATS/ERS, 2003].
A high number of targeted detection programs have been undertaken (Table 3 displays published studies with >500 samples examined). It is impossible to compare the ‘success rate’ of these programs since the programs use different ‘inclusion criteria’; most of the programs do process all of the samples sent to the laboratories, even if information about the indication for testing is completely missing. It may be recognized that those programs, that are associated with a Pi*ZZ detection rate >5%, recommend external measurement of AAT serum level as a first step, thus enriching the test population with individuals having a high a priori probability for deficiency alleles, what may explain the high Pi*ZZ detection rates in Italy and Germany. On the other hand, both labs report that the recommendation is not always being followed.
Table 3.
Published studies with >500 samples examined in the order of their publication date. COPD, chronic obstructive pulmonary disease; PFT, pulmonary function test.
| Year | Location | Screened Population | Number screened | ZZ [%] | SZ [%] | MZ [%] | SS [%] | MS [%] | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1986 | USA | COPD (hospitalized for carotid body surgery) | 965 | 1.87 | 0.31 | 7.67 | 0.31 | 10.1 | Lieberman et al. [1986] |
| 1999 | Italy | Case finding | 1841 | 6.41 | 0.92 | – | – | – | Luisetti et al. [1999] |
| 2002 | Germany | COPD, emphysema, asthma, bronchiectasis | 1060 | 0 | 0.28 | 3.68 | 0.09 | 3.40 | Wencker et al. [2002] |
| 2005 | Spain | COPD | 2137 | 0.37 | 0.14 | – | 0.14 | – | de la Roza et al. [2005] |
| 2007 | Germany | Case finding (education program and free testing) | 2696 | 9.94 | 1.97 | 18.1 | 0 | 3.60 | Bals et al. [2007] |
| 2011 | Spain | Case finding in primary care (education program, free testing, communication with lab and pulmonologist) | 596 | 0.34 | 0.17 | 2.68 | 1.68 | 13.4 | Molina et al. [2011] |
| 2011 | USA | Case finding (electronic record alert to test in fixed airflow obstruction) | 979 | 2.63 | 0 | 2.63 | 0 | 5.26 | Jain et al. [2011] |
| 2012 | Germany | Case finding (free testing) | 11.264 | 6.63 | 1.48 | 17.8 | 0.27 | 4.97 | Greulich et al. [2012] |
| 2012 | USA | Fixed airflow obstruction (initiated by PFT lab technician) | 3.152 | 0.63 | 10.9 | Rahaghi et al. [2012] | |||
| 2013 | USA | Case finding (free testing) | 117.966 | 0.71 | 0.50 | 5.81 | – | – | Stoller and Brantly [2013] |
| 2015 | Ireland | Case finding (free testing) | 13.500 | 1.85 | 1.44 | 13.6 | 0.49 | 9.79 | Fee et al. [2015] |
| 2015 | USA | Case finding (free testing) | 7.530 | 0.65 | 0.66 | 6.92 | 0.37 | 7.82 | Sanders and Kim [2015] |
| 2015 | Eastern Europe | Obstructive Lung Disease (Patient reported) | 11.648 | 2.28 | 0.57 | 10.8 | 0.07 | 5.06 | Greulich et al. [2015] |
Recent advances
Increasing efforts have been made during the past decade to increase awareness and to overcome barriers to AAT testing. One possible way is to offer testing and to distribute test kits free of charge. This is typically done in cooperation with the pharmaceutical industry, namely manufacturers of augmentation therapy. One prominent example is the national detection program, performed by the University of Florida AAT Genetics Laboratory in cooperation with Grifols, USA. Testing 117,966 individuals, 843 Pi*ZZ, 593 Pi*SZ and 6859 Pi*MZ individuals have been detected (detection rate: 7.03%) [Stoller and Brantly, 2013]. In a second, state-wide detection program (State of Florida-funded, Alpha-1 Foundation sponsored), carried out by the same laboratory, 17,567 individuals have been tested [Brantly et al. 2003; Stoller and Brantly, 2013]. A total of 1016 individuals carried Pi*ZZ, Pi*SZ or Pi*MZ (detection rate: 5.78%). The Alpha-1 Coded Testing (ACT) program targets concerns of individuals regarding genetic discrimination: samples are sent on dried blood spot to the University of South Carolina and then submitted for coded testing through the AAT Genetics Laboratory of the University of Florida. Compared with the other two programs, the detection rate of the ACT program is significantly higher (39.13%) [Stoller and Brantly, 2013]. This may well be explained by the higher degree of family testing.
A second possible way to increase awareness is to remind physicians regularly: Rahaghi and colleagues added a written recommendation to test for AATD to pulmonary function test (PFT) reports of patients with fixed airflow obstruction. This intervention doubled the rate of testing for AATD (13% versus 6%), although it did not result in an increased number of homozygous test results [Rahaghi et al. 2009]. Jain and colleagues followed a similar strategy: Using a computer algorithm they inserted a comment into the electronic medical record that recommended AATD testing in those individuals whose PFT results demonstrated fixed airflow obstruction. While this increased the test rate significantly (15.1% versus 4.7%), the rate of detected individuals remained unchanged [Jain et al. 2011]. Both studies have been performed in centres that presumably already exhibited a high level of awareness, which could explain failure to increase AATD detection rate.
Another example for a combination of methods to increase awareness is the IDDEA (Information and Detection of the Deficiency of AAT) program that was initiated and evaluated in Spain in 2008 and 2009 (Molina et al. 2011). The program aimed to increase the detection rate of AATD especially in primary care. Participating primary care physicians were provided with information material about AATD (stressing the importance of early detection), simple sampling methods (DBS), and an Internet-based communication system to facilitate communication between the primary care centre, the laboratory, and a pulmonologist specialized in AATD. A total of 596 patients were enrolled by 90 participating primary care physicians: 3.2% were Pi*Z carriers among which two individuals (0.34%) carried the phenotype PiZZ [Molina et al. 2011]. This example demonstrates that targeted detection can be performed in the primary care setting, but may be associated with a low detection rate.
New ways of explaining the disease and reminding colleagues to initiate AAT testing are needed. Miravitlles and colleagues recommended a number of actions to enhance awareness, including increased efforts regarding medical education, dissemination of knowledge and measures to implement existing recommendations (Table 4) [Miravitlles et al. 2010]. While suggestions like this may very well enhance awareness, their effectiveness (with the exception of ‘physician alert’) has not been studied extensively.
Table 4.
Suggestions that could further increase the number of identified individuals with severe deficiency genotypes. (Modified from Miravitlles et al. [2010].).
| Educational efforts | Dissemination of knowledge | Implementation of recommendations |
|---|---|---|
|
|
|
AAT, alpha 1-antitrypsin; AATD, alpha 1-antitrypsin deficiency; ATS/ERS, American Thoracic Society/European Respiratory Society; COPD, chronic obstructive pulmonary disease.
Newborn screening
Newborn screening for AATD (as part of a public health program) is seen as controversial: after having conducted one of the two existing large-scale programs (>100,000 samples, Oregon, USA) the initiators advised against the continuation of the program because pulmonary or hepatic disease would be rather infrequent (at least in childhood) and specific therapy would not be available [O’Brien et al. 1978]. In contrast, the WHO explicitly recommended neonatal screening programs (of limited time) to determine the prevalence of deficiency alleles in specific areas of the world [WHO, 1997]. Furthermore, a recent statement issued on an international AAT experts workshop (called together by the Alpha-1 Foundation) stated that pilot studies should be conducted to determine whether early detection improves clinical relevant outcomes [Teckman et al. 2014].
Diagnostic algorithm
Initial tests for AATD
There is no single universally accepted laboratory algorithm for AAT diagnosis. However, there is wide agreement that a combination of different laboratory methods delivers the best results [Bals et al. 2007; Ferrarotti et al. 2007; Miravitlles et al. 2010; Snyder et al. 2006]. Most often, quantitative measurements of the AAT serum level are used as the initial screening test [Bornhorst et al. 2013; Steiner et al. 2003]. Available methods for quantitative AAT measurements include radial immunodiffusion, nephelometry, and latex-enhanced immunoturbidimetry. The use of dried blood samples (DBSs) for AATD diagnosis has become widely available and has facilitated testing. AAT levels can be measured from DBS with moderate to good correlation with serum AAT serum levels [Costa et al. 2000].
Many laboratories and screening programs test for the most common S- and Z-mutations (some also for the F-mutation) either concomitantly or in ‘DBS AAT low’ samples [Miravitlles et al. 2010]. The decision to include targeted polymerase chain reaction (PCR) for the detection of Pi*S and Pi*Z as a concomitant first step or only in low-level samples is dependent on the funding that is available and on the primary goal of the algorithm: two recent publications showed that the negative predictive value of the AAT serum level (cutoff 104 mg/dl, 113 mg/dl) is 99.8% and 100%, respectively [Ferrarotti et al. 2012; Greulich et al. 2015]. In other words: if the serum level exceeds these thresholds, the probability to carry a homozygous mutation for AAT is extremely low. Therefore, if the primary goal of the algorithm is to detect all homozygous individuals this can be done effectively without conducting PCR as a first step.
However, if the testing algorithm aims to detect heterozygous carriers it is very difficult to determine a threshold. Recent laboratory analyses of high numbers of patient samples demonstrated 97.5% percentiles of PiMZ of approximately 150 mg/dl (220 mg/dl for PiMS). Thus, the serum level has only a very limited value if the goal is to exclude heterozygous carriers [Bornhorst et al. 2013; Donato et al. 2012].
The protective threshold
The threshold that is used to exclude severe deficiency is different from the so-called protective threshold. A protective threshold has been hypothesized when investigators noticed that Pi*SZ individuals with a serum level beyond that threshold did not have emphysema [Crystal, 1990]. At that time, the usually used method for AAT serum measurement was radial immunodiffusion that tends to overestimate the serum level. A level of 80 mg/dl was regarded as the protective threshold, while the normal range was approximately 150–350 mg/dl. Later, a highly purified standard was introduced (11 µM). Nowadays, nephelometry or immunoturbidimetry are the methods that are used in many laboratories. Using these methods, accepted normal ranges of serum AAT are approximately 90–200 mg/dl, the protective threshold is at 50 mg/dl [Stoller and Aboussouan, 2005].
The initial observation that this equals the 10% percentile of Pi*SZ patients has recently been substantiated in the large Swiss SAPALDIA (Swiss study on Air Pollution and Lung Disease in adults) cohort. In this cohort, the 10% percentile of Pi*SZ patients is at 49 mg/dl [Ferrarotti et al. 2012]. More recent confirmatory evidence comes from two large retrospective analyses of laboratories that received samples for analysis of AAT status, thus representing a patient population rather than the general population. The first analysis included 21,406 samples from adult patients, including 161 Pi*SZ samples: 11.8% had an AAT serum concentration below 50 mg/dl, supporting this cutoff as a protective threshold. In the second of these analyses, 72,229 samples were evaluated. Here, 15.37% of all PiSZ samples (n = 540) exhibited serum levels <50 mg/dl. Although the 10% percentile was not reported, the data support a protective threshold of approximately 50 mg/dl [Bornhorst et al. 2013].
The concept of a single protective threshold is challenged by the results of the largest randomized controlled trial so far that has been conducted on augmentation therapy in AATD. In the ‘Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency’ (RAPID) trial, 180 patients were treated with AAT (60 mg/kg once weekly) or placebo for 24 months [Chapman et al. 2015]. The primary endpoint (lung density loss at total lung capacity [TLC] and functional residual capacity [FRC]) was negative, which is in line with a Cochrane analysis on augmentation therapy [Gotzsche and Johansen, 2010]. However, there was a statistically significant effect of the treatment arm towards a reduction of the loss of lung density at TLC (p = 0.03). During this trial, serum levels under augmentation therapy have been assessed. Individuals with higher serum levels seemed to benefit more than individuals with lower levels (although above the ‘threshold’), suggesting a direct correlation between achieved serum level and efficacy.
Phenotyping and gene sequencing
If AAT serum level and PCR do not give consistent results or if confirmation on the protein level is desired, isoelectric focusing (IEF) is the method of choice. IEF with or without immunoblotting separates proteins according to their isoelectric points (Figure 3), and enables the user to identify different AAT isoforms [Zerimech et al. 2008]. Although it is a relatively inexpensive test, it requires significant expertise. Traditionally, this method has been regarded as the gold standard in AAT diagnosis, but has recently been replaced in many laboratories by the combination of serum level and PCR. It still represents the gold standard to detect rare variants (except null variants) after having detected low serum levels of AAT [Greene et al. 2013].
Figure 3.
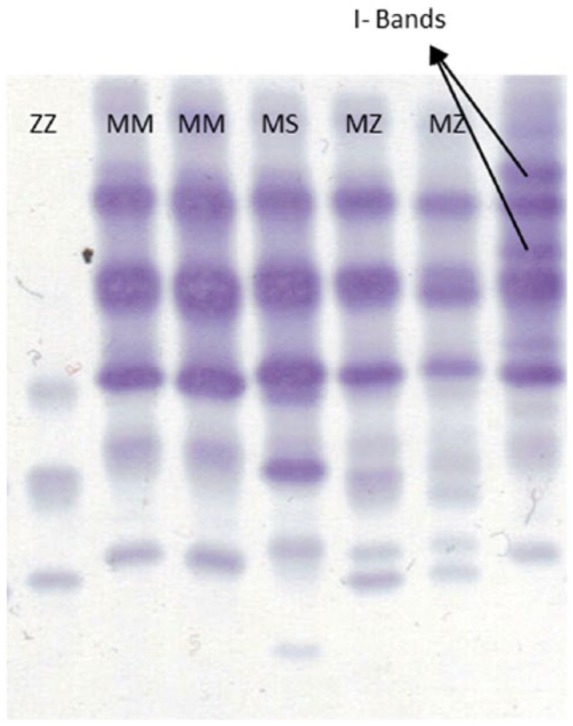
Isoelectric focusing electrophoresis with immunoblotting (Sebia Hydrasys 2). Samples are taken from dried blood spot. Different phenotypes can be distinguished according to their bands (analysis conducted by V. Kotke, University of Marburg, Germany).
Gene sequencing of SERPINA-1 is currently reserved for cases in which the serum level is low and this finding cannot be explained fully by targeted PCR or IEF [Miravitlles et al. 2010]. The sequence is compared with known mutations via Internet-based databases.
Currently published algorithms differ with regard to the indication for IEF and/or sequencing. The use of IEF and/or gene sequencing in all samples below a certain serum level is supported by the fact that PCR for S and Z together with the AAT serum level will still miss those allelic combinations where S or Z are combined with a rare mutation that would lead to intermediate deficiency. It could be argued that this would not be relevant as long as the serum level exceeds the protective threshold; on the other hand, little is known about the functional capacity of many rare alleles or the resulting AAT proteins. Thus, a serum level of 60 mg/dl in a Pi*MZ individual might have different functional properties than a serum level of 60 mg/dl in a Pi*M/rare mutation.
While mutations other than Pi*S or Pi*Z have been traditionally named ‘rare mutations’, the increasing number of novel allelic deficient variants challenges this concept [Ferrarotti et al. 2014; Lara et al. 2014]. It stresses the need for a combination of laboratory methods (including IEF and/or sequencing) with the goal of detecting all clinically relevant mutations.
Recent advances in the laboratory diagnosis of AATD
Very recently, a new test for AATD was introduced, following the principle of a lateral-flow assay [Vogelmeier et al. 2013]. The test works with a Z-protein-specific antibody, thus detecting the clinically most important Z-mutation. The advantage of that test is the availability of the test result within 15 min, reflecting a point of care test. The disadvantage is that all mutations ‘other than Z’ (mostly S, but also a number of rare mutations) are missed. This does not play a role in all combinations of mutations that include one Z-mutation. Combinations of two rare alleles have been described as being extremely rare, but the frequency depends strongly on the country in which the test is used. As the test cannot differentiate between heterozygous and homozygous carriers of the Z-mutation, further confirmatory analyses need to follow.
Advances in technology have made it possible to conduct gene sequencing from DBS-derived DNA and from serum separator cards, enabling the laboratory to conduct a multiple-step analysis (AAT level, PRC, IEF, gene sequencing) with only one patient visit [Sanders and Kim, 2015].
Currently, ‘next-generation sequencing’ is becoming widely available. Therefore, one alternative future algorithm would be to use a screening test with a very high sensitivity to exclude the disease. All test-positive individuals could then directly undergo sequencing of the SERPINA1 gene.
Conclusion
The delay between the onset of symptoms and time of diagnosis of AATD patients exceeds 5 years. This, together with the huge discrepancy between the estimated number of patients and the number of identified patients supports the hypothesis that AATD is still significantly underdiagnosed. Recent data confirm that this may be caused by limited knowledge and awareness of physicians. Furthermore, only a minority of physicians follow existing guidelines. Continued efforts to increase medical education, and the dissemination and implementation of existing recommendations are mandatory.
The diagnostic algorithm is likely to change as novel techniques (such as the lateral-flow assay to detect the Z-protein) and next-generation sequencing will become broadly available. Comparative research of diagnostic algorithms incorporating new technologies is needed, preferably in real-life settings.
Footnotes
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/ or publication of this article: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: TG has received consultancy fees, an unrestricted grant and travel support from Grifols. He has also received consultancy fees and travel support from Chiesi and CSL-Behring. CFV gave presentations at symposia and/or served on scientific advisory boards sponsored by Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Grifols, Janssen, Mundipharma, Novartis and Takeda.
Contributor Information
Timm Greulich, Department of Medicine, Pulmonary and Critical Care Medicine, University Medical Centre Giessen and Marburg, Philipps-University, Baldingerstrasse, 35043 Marburg, Germany.
Claus F. Vogelmeier, Department of Medicine, Pulmonary and Critical Care Medicine, University Medical Centre Giessen and Marburg, Philipps-University, Member of the German Centre for Lung Research (DZL), Marburg, Germany
References
- Aboussouan L., Stoller J. (2009) Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 103: 335–341. [DOI] [PubMed] [Google Scholar]
- ATS/ERS (2003) American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 168: 818–900. [DOI] [PubMed] [Google Scholar]
- Bals R., Koczulla R., Kotke V., Andress J., Blackert K., Vogelmeier C. (2007) Identification of individuals with alpha-1-antitrypsin deficiency by a targeted screening program. Respir Med 101: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Bergin D., Reeves E., Hurley K., Wolfe R., Jameel R., Fitzgerald S., et al. (2014) The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med 6: 217ra1. [DOI] [PubMed] [Google Scholar]
- Blanco I., de Serres F., Fernandez-Bustillo E., Lara B., Miravitlles M. (2006) Estimated numbers and prevalence of PI*S and PI*Z alleles of alpha1-antitrypsin deficiency in European countries. Eur Respir J 27: 77–84. [DOI] [PubMed] [Google Scholar]
- Bornhorst J., Greene D., Ashwood E., Grenache D. (2013) alpha1-Antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest 143: 1000–1008. [DOI] [PubMed] [Google Scholar]
- Brantly M., Mishra V., Viranovskaya N., Zienko L., Corcoran V., et al. (2003) Statewide targeted screening and detection of AAT deficiency. Am J Respir Crit Care Med 167: A222. [Google Scholar]
- Campos M., Wanner A., Zhang G., Sandhaus R. (2005) Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 128: 1179–1186. [DOI] [PubMed] [Google Scholar]
- Carrell R., Lomas D. (2002) Alpha1-antitrypsin deficiency - a model for conformational diseases. N Engl J Med 346: 45–53. [DOI] [PubMed] [Google Scholar]
- Carroll T., O’Connor C., Floyd O., McPartlin J., Kelleher D., O’Brien G., et al. (2011) The prevalence of alpha-1 antitrypsin deficiency in Ireland. Respir Res 12: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman K., Burdon J., Piitulainen E., Sandhaus R., Seersholm N., Stocks J., et al. (2015) Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet, in press. [DOI] [PubMed] [Google Scholar]
- Chorostowska-Wynimko J., Struniawski R., Poplawska B., Borszewska-Kornacka M. (2014) The large scale newborn screening for alpha-1-antitrypsin (A1AT) deficiency alleles in central Poland. Am J Respir Crit Care Med 189: A5778. [PubMed] [Google Scholar]
- Cook P. (1975) The genetics of alpha1-antitrypsin: a family study in England and Scotland. Ann Hum Genet 38: 275–287. [DOI] [PubMed] [Google Scholar]
- Costa X., Jardi R., Rodriguez F., Miravitlles M., Cotrina M., Gonzalez C., et al. (2000) Simple method for alpha1-antitrypsin deficiency screening by use of dried blood spot specimens. Eur Respir J 15: 1111–1115. [DOI] [PubMed] [Google Scholar]
- Crystal R. (1990) Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest 85: 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal R., Brantly M., Hubbard R., Curiel D., States D., Holmes M. (1989) The alpha 1-antitrypsin gene and its mutations. Clinical consequences and strategies for therapy. Chest 95: 196–208. [DOI] [PubMed] [Google Scholar]
- Curiel D., Chytil A., Courtney M., Crystal R. (1989) Serum alpha 1-antitrypsin deficiency associated with the common S-type (Glu264—Val) mutation results from intracellular degradation of alpha 1-antitrypsin prior to secretion. J Biol Chem 264: 10477–10486. [PubMed] [Google Scholar]
- Dahl M., Tybjaerg-Hansen A., Lange P., Vestbo J., Nordestgaard B. (2002) Change in lung function and morbidity from chronic obstructive pulmonary disease in alpha1-antitrypsin MZ heterozygotes: A longitudinal study of the general population. Ann Intern Med 136: 270–279. [DOI] [PubMed] [Google Scholar]
- de la Roza C., Rodriguez-Frias F., Lara B., Vidal R., Jardi R., Miravitlles M. (2005) Results of a case-detection programme for alpha1-antitrypsin deficiency in COPD patients. Eur Respir J 26: 616–622. [DOI] [PubMed] [Google Scholar]
- Dijkman J., Penders T., Kramps J., Sonderkamp H., van den Broek W., ter Haar B. (1980) Epidemiology of alpha 1-antitrypsin deficiency in the Netherlands. Hum Genet 53: 409–413. [DOI] [PubMed] [Google Scholar]
- Donato L., Jenkins S., Smith C., Katzmann J., Snyder M. (2012) Reference and interpretive ranges for alpha(1)-antitrypsin quantitation by phenotype in adult and pediatric populations. Am J Clin Pathol 138: 398–405. [DOI] [PubMed] [Google Scholar]
- Evans H., Bognacki N., Perrott L., Glass L. (1977) Prevalence of of alpha 1-antitrypsin Pi types among newborn infants of different ethnic backgrounds. J Pediatr 90: 621–624. [DOI] [PubMed] [Google Scholar]
- Fee L., Carroll T., O’Connor C., Ryan P., O’Brien P., Pentony E., et al. (2015) The Irish Alpha-1 Antitrypsin Deficiency National Targeted Detection Programme., C46. How do you do it? COPD Diagnosis Assess 191: A4477. [Google Scholar]
- Ferrarotti I., Carroll T., Ottaviani S., Fra A., O’Brien G., Molloy K., et al. (2014) Identification and characterisation of eight novel SERPINA1 Null mutations. Orphanet J Rare Dis 9: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarotti I., Scabini R., Campo I., Ottaviani S., Zorzetto M., Gorrini M., et al. (2007) Laboratory diagnosis of alpha1-antitrypsin deficiency. Transl Res 150: 267–274. [DOI] [PubMed] [Google Scholar]
- Ferrarotti I., Thun G., Zorzetto M., Ottaviani S., Imboden M., Schindler C., et al. (2012) Serum levels and genotype distribution of alpha1-antitrypsin in the general population. Thorax 67: 669–674. [DOI] [PubMed] [Google Scholar]
- Fregonese L., Stolk J., Frants R., Veldhuisen B. (2008) Alpha-1 antitrypsin null mutations and severity of emphysema. Respir Med 102: 876–884. [DOI] [PubMed] [Google Scholar]
- Gadek J., Fells G., Zimmerman R., Rennard S., Crystal R. (1981) Antielastases of the human alveolar structures. Implications for the protease–antiprotease theory of emphysema. J Clin Invest 68: 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzsche P., Johansen H. (2010) Intravenous alpha-1 antitrypsin augmentation therapy for treating patients with alpha-1 antitrypsin deficiency and lung disease. Cochrane Database Syst Rev CD007851. [DOI] [PubMed] [Google Scholar]
- Greene D., Elliott-Jelf M., Straseski J., Grenache D. (2013) Facilitating the laboratory diagnosis of alpha1-antitrypsin deficiency. Am J Clin Pathol 139: 184–191. [DOI] [PubMed] [Google Scholar]
- Greulich T., Averyanov A., Borsa L., Rozborilova E., Vaicius D., Major T., et al. (2015) European screening for alpha-1 antitrypsin deficiency in subjects with lung disease. Clin Respir J, in press. [DOI] [PubMed] [Google Scholar]
- Greulich T., Nell C., Kehr K., Kotke V., Wiedmann S., et al. (2012) Detection of patients with alpha-1 antitrypsin deficiency in Germany - update 2011. In COPD: alpha 1 antitrypsin (American Thoracic Society International Conference Abstracts, vol. 183). American Thoracic Society: abstract A4356. [Google Scholar]
- Greulich T., Ottaviani S., Bals R., Lepper P., Vogelmeier C., Luisetti M., et al. (2013) Alpha1-antitrypsin deficiency - Diagnostic testing and disease awareness in Germany and Italy. Respir Med 107: 1400–1408. [DOI] [PubMed] [Google Scholar]
- Hoffmann J., van den Broek W. (1976) Distribution of alpha-1-antitrypsin phenotypes in two Dutch population groups. Hum Genet 32: 43–48. [DOI] [PubMed] [Google Scholar]
- Hurley K., Lacey N., O’Dwyer C., Bergin D., McElvaney O., O’Brien M., et al. (2014) Alpha-1 antitrypsin augmentation therapy corrects accelerated neutrophil apoptosis in deficient individuals. J Immunol 193: 3978–3991. [DOI] [PubMed] [Google Scholar]
- Hutchison D. (1998) Alpha 1-antitrypsin deficiency in Europe: geographical distribution of Pi types S and Z. Respir Med 92: 367–377. [DOI] [PubMed] [Google Scholar]
- Jain A., McCarthy K., Xu M., Stoller J. (2011) Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 140: 198–204. [DOI] [PubMed] [Google Scholar]
- Janciauskiene S., Dominaitiene R., Sternby N., Piitulainen E., Eriksson S. (2002) Detection of circulating and endothelial cell polymers of Z and wild type alpha 1-antitrypsin by a monoclonal antibody. J Biol Chem 277: 26540–26546. [DOI] [PubMed] [Google Scholar]
- Jeppsson J. (1976) Amino acid substitution Glu leads to Lys alpha1-antitrypsin PiZ. FEBS Lett 65: 195–197. [DOI] [PubMed] [Google Scholar]
- Kimpen J., Bosmans E., Raus J. (1988) Neonatal screening for alpha-1-antitrypsin deficiency. Eur J Pediatr 148: 86–88. [DOI] [PubMed] [Google Scholar]
- Kohnlein T., Janciauskiene S., Welte T. (2010) Diagnostic delay and clinical modifiers in alpha-1 antitrypsin deficiency. Ther Adv Respir Dis 4: 279–287. [DOI] [PubMed] [Google Scholar]
- Lara B., Martinez M., Blanco I., Hernandez-Moro C., Velasco E., Ferrarotti I., et al. (2014) Severe alpha-1 antitrypsin deficiency in composite heterozygotes inheriting a new splicing mutation QOMadrid. Respir Res 15: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell C., Eriksson S. (2013) The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. 1963. COPD 10(Suppl. 1): 3–8. [DOI] [PubMed] [Google Scholar]
- Lieberman J., Gaidulis L., Roberts L. (1976) Racial distribution of alpha1-antitrypsin variants among junior high school students. Am Rev Respir Dis 114: 1194–1198. [DOI] [PubMed] [Google Scholar]
- Lieberman J., Winter B., Sastre A. (1986) Alpha 1-antitrypsin Pi-types in 965 COPD patients. Chest 89: 370–373. [DOI] [PubMed] [Google Scholar]
- Lomas D., Carrell R. (2002) Serpinopathies and the conformational dementias. Nat Rev Genet 3: 759–768. [DOI] [PubMed] [Google Scholar]
- Lomas DA, Parfrey H. (2004) Alpha1-antitrypsin deficiency. 4: Molecular pathophysiology. Thorax 2004; 59: 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisetti M., Massi G., Massobrio M., Guarraci P., Menchicchi F., Beccaria M., et al. (1999) A national program for detection of alpha 1-antitrypsin deficiency in Italy. Gruppo I.D.A. Respir Med 93: 169–172. [DOI] [PubMed] [Google Scholar]
- Mahadeva R., Atkinson C., Li Z., Stewart S., Janciauskiene S., Kelley D., et al. (2005) Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol 166: 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miravitlles M., Herr C., Ferrarotti I., Jardi R., Rodriguez-Frias F., Luisetti M., et al. (2010) Laboratory testing of individuals with severe alpha1-antitrypsin deficiency in three European centres. Eur Respir J 35: 960–968. [DOI] [PubMed] [Google Scholar]
- Molina J., Flor X., Garcia R., Timiraos R., Tirado-Conde G., Miravitlles M. (2011) The IDDEA project: a strategy for the detection of alpha-1 antitrypsin deficiency in COPD patients in the primary care setting. Ther Adv Respir Dis 5: 237–243. [DOI] [PubMed] [Google Scholar]
- Morse J., Lebowitz M., Knudson R., Burrows B. (1977) Relation of protease inhibitor phenotypes to obstructive lung diseases in a community. N Engl J Med 296: 1190–1194. [DOI] [PubMed] [Google Scholar]
- Mulgrew A., Taggart C., Lawless M., Greene C., Brantly M., O’Neill S., et al. (2004) Z alpha1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest 125: 1952–1957. [DOI] [PubMed] [Google Scholar]
- O’Brien M., Buist N., Murphey W. (1978) Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 92: 1006–1010. [DOI] [PubMed] [Google Scholar]
- Rahaghi F., Ortega I., Rahaghi N., Oliveira E., Ramirez J., Smolley L., et al. (2009) Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 6: 26–30. [DOI] [PubMed] [Google Scholar]
- Rahaghi F., Sandhaus R., Strange C., Hogarth D., Eden E., Stocks J., et al. (2012) The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 9: 352–358. [DOI] [PubMed] [Google Scholar]
- Sanders C., Kim J. (2015) Frequencies of alpha-1 antitrypsin deficiency phenotypes detected using a clinical testing strategy that reflexes to next-generation sequencing. In C45. Across the Universe of COPD Epidemiology (American Thoracic Society International Conference Abstracts, vol. 191). American Thoracic Society: abstract A4456. [Google Scholar]
- Sharp H., Bridges R., Krivit W., Freier E. (1969) Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder. J Lab Clin Med 73: 934–939. [PubMed] [Google Scholar]
- Silverman E., Miletich J., Pierce J., Sherman L., Endicott S., Broze G., Jr, et al. (1989) Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis 140: 961–966. [DOI] [PubMed] [Google Scholar]
- Silverman E., Sandhaus R. (2009) Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med 360: 2749–2757. [DOI] [PubMed] [Google Scholar]
- Simsek H., Pinar A., Altinbas A., Alp A., Balaban Y., Buyukasik Y., et al. (2011) Cutoff level to detect heterozygous alpha 1 antitrypsin deficiency in Turkish population. J Clin Lab Anal 25: 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M., Katzmann J., Butz M., Wiley C., Yang P., Dawson D., et al. (2006) Diagnosis of alpha-1-antitrypsin deficiency: An algorithm of quantification, genotyping, and phenotyping. Clin Chem 52: 2236–2242. [DOI] [PubMed] [Google Scholar]
- Spence W., Morris J., Pass K., Murphy P. (1993) Molecular confirmation of alpha 1-antitrypsin genotypes in newborn dried blood specimens. Biochem Med Metab Biol 50: 233–240. [DOI] [PubMed] [Google Scholar]
- Steiner S., Gupta S., Croffie J., Fitzgerald J. (2003) Serum levels of alpha1-antitrypsin predict phenotypic expression of the alpha1-antitrypsin gene. Dig Dis Sci 48: 1793–1796. [DOI] [PubMed] [Google Scholar]
- Stockley R., Dirksen A., Stolk J. (2013) Alpha-1 antitrypsin deficiency: the European experience. COPD 10(Suppl. 1): 50–53. [DOI] [PubMed] [Google Scholar]
- Stockley R., Luisetti M., Miravitlles M., Piitulainen E., Fernandez P. (2007) Ongoing research in Europe: Alpha One International Registry (AIR) objectives and development. Eur Respir J 29: 582–586. [DOI] [PubMed] [Google Scholar]
- Stoller J., Aboussouan L. (2005) Alpha1-antitrypsin deficiency. Lancet 365: 2225–2236. [DOI] [PubMed] [Google Scholar]
- Stoller J., Brantly M. (2013) The challenge of detecting alpha-1 antitrypsin deficiency. COPD 10(Suppl. 1): 26–34. [DOI] [PubMed] [Google Scholar]
- Stoller J., Fromer L., Brantly M., Stocks J., Strange C. (2007) Primary care diagnosis of alpha-1 antitrypsin deficiency: issues and opportunities. Cleve Clin J Med 74: 869–874. [DOI] [PubMed] [Google Scholar]
- Stoller J., Sandhaus R., Turino G., Dickson R., Rodgers K., Strange C. (2005) Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 128: 1989–1994. [DOI] [PubMed] [Google Scholar]
- Stoller J., Smith P., Yang P., Spray J. (1994) Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 61: 461–467. [DOI] [PubMed] [Google Scholar]
- Sveger T. (1976) Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 294: 1316–1321. [DOI] [PubMed] [Google Scholar]
- Sveger T., Mazodier P. (1979) Alpha 1-antitrypsin screening of 18-year-old men. Thorax 34: 397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliercio R., Chatburn R., Stoller J. (2010) Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 55: 322–327. [PubMed] [Google Scholar]
- Teckman J., Pardee E., Howell R., Mannino D., Sharp R., Brantly M., et al. (2014) Appropriateness of newborn screening for alpha1-antitrypsin deficiency. J Pediatr Gastroenterol Nutr 58: 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschler H. (2015) Long-term experience in the treatment of alpha1-antitrypsin deficiency: 25 years of augmentation therapy. Eur Respir Rev 24: 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmeier C., Soriano J., Janciauskiene S., Crystal R., Ferrarotti I., Carroll T. Alpha-1-antitrypsin deficiency: honouring the past and embracing the future. Grifols’ Symposium Report. Eur Respir Rev 2015; 24: 46–51.25726554 [Google Scholar]
- Wencker M., Marx A., Konietzko N., Schaefer B., Campbell E. (2002) Screening for alpha1-Pi deficiency in patients with lung diseases. Eur Respir J 20: 319–324. [DOI] [PubMed] [Google Scholar]
- WHO (1997) Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ 75: 397–415. [PMC free article] [PubMed] [Google Scholar]
- Zerimech F., Hennache G., Bellon F., Barouh G., Jacques L., Porchet N., et al. (2008) Evaluation of a new Sebia isoelectrofocusing kit for alpha 1-antitrypsin phenotyping with the Hydrasys System. Clin Chem Lab Med 46: 260–263. [DOI] [PubMed] [Google Scholar]