

Abstract
Cardio-oncology is a new and rapidly expanding field that merges cancer and cardiovascular disease. Cardiovascular disease is an omnipresent side effect of cancer therapy; in fact, it is the second leading cause of death in cancer survivors after recurrent cancer. It has been well documented that many cancer chemotherapeutic agents cause cardiovascular toxicity. Nonetheless, the underlying cause of cancer therapy-induced cardiovascular toxicity is largely unknown. In this review, we discuss the potential role of damage-associated molecular patterns (DAMPs) as an underlying contributor to cancer therapy-induced cardiovascular toxicity. With an increasing number of cancer patients, as well as extended life expectancy, understanding the mechanisms underlying cancer therapy-induced cardiovascular disease is of the utmost importance to ensure that cancer is the only disease burden that cancer survivors have to endure.
Keywords: cancer therapy, cardio-oncology, cardiotoxicity, cardiovascular disease, damage-associated molecular patterns, inflammation
Introduction
Cancer therapy has advanced dramatically in such a way that not only do cancer patients have increased life expectancy, they also have increased survival rates.1 Currently, there are 15.5 million patients living beyond a cancer diagnosis and by 2026, that number is estimated to increase to 20.3 million.1 However, stemming from both short- and long-term cancer treatment, which is often given in multi-drug combinations and escalating doses, new problems are emerging.2 One such problem is cardiovascular complications, which is the second leading cause of death in cancer survivors, only after cancer recurrence, cancer progression, and secondary malignancies.3 In fact, cardiovascular complications have become so prominent that a new area of research is emerging within both cardiology and oncology to prevent and treat cancer therapy-induced cardiotoxicity; this field is referred to as cardio-oncology.
Interestingly, several studies have demonstrated a poor correlation between traditional cardiovascular risk factors such as obesity, dyslipidemia, insulin resistance, and tobacco use, with the onset of cardiovascular disease in cancer survivors,4,5 suggesting an alternative mechanism for systemic cardiovascular toxicity. One such mechanism could be inappropriate immune system activation, which has an active role in both cancer and cardiovascular disease.6–9 It has been well established that the immune system not only responds to foreign substances (i.e. pathogens), it also responds to endogenously derived molecules that are expressed as a result of tissue damage or stressed cells, known as damage-associated molecular patterns (DAMPs).10,11 DAMPs are present in both cancer and cardiovascular disease; however, the roles that DAMPs play are not congruent. In cardiovascular disease literature, high and chronic levels of DAMPs have been shown to be inflammogenic and are associated with disease pathogenesis.12,13 In cancer, the presence of DAMPs has been shown to be a sign of both treatment efficacy and the development of resistance.14,15
Mechanisms of action for therapies such as chemotherapy and radiation have been proposed and reported in the literature;16–21 however, due to the underlying goal of cancer therapy to reduce tumor size, we hypothesize that the unintentional release of DAMPs after any cancer therapeutic contributes to the development of cardiovascular disease, including hypertension. This review describes the increased prevalence of cancer therapy-induced cardiotoxicity after cancer therapy and the possible role that DAMPs play in the disease process.
Cancer therapies cause cell death
Cancer is a complex disease due to the ability of cancer cells to adapt to changing environmental conditions guaranteeing growth and survival. Cancer is the result of one or more DNA mutations, genetically predisposed or environmentally acquired, that allow cells to escape the normal mechanisms constraining cell division and growth. Regardless of mutation or cancer type, cancer cells exhibit four main characteristics, (1) uncontrolled proliferation, (2) de-differentiation and loss of function, (3) invasiveness, and (4) metastasis. Cancer therapies are designed to target these abnormal characteristics.
Cancer therapy type and regimen are often determined by factors specific to the patient and tumor characteristics, with the intention to cure, prolong survival, or relieve symptoms. The classic dogma of cancer therapy uses a continuous dosing strategy to ‘kill as many cancer cells as possible’. In this type of treatment strategy, there is a delicate balance amid median effective dose (ED50), lethal dose (LD50), toxicity, and drug resistance.22 Currently, there are research groups and clinical trials studying differing dosing schedules aimed at thwarting tumor evolution.22 Using effective treatment modality, dose, and frequency are several ways clinicians combat this multifaceted disease.22
The main treatment categories for cancer include surgery, radiation, chemotherapy, hormone therapy, immunotherapy, and targeted therapy, which can be mixed and added together for augmented outcomes. Excluding surgery, which entails the resection of a malignant tumor, all other forms of cancer treatment aim to destroy the pro-proliferative and de-differentiated cancer cells within the body.
Radiation is a form of cancer therapy used mainly against solid tumors. Radiation utilizes high-energy particles or waves to kill cancer cells by inducing irreparable DNA breaks within cells. This method targets rapidly dividing cells; however, its effects may not take place until days or weeks after initiation and cell death could continue months after treatment is completed. Chemotherapy is another form of cancer therapy, which utilizes cytotoxic drugs to treat disseminated cancer; solid tumors are not as sensitive to this treatment option. Chemotherapy targets rapidly growing and dividing cells, and slows tumor proliferation by inhibiting specific aspects of cellular replication, ultimately leading to cell death. As the name implies, targeted therapies are more specific when compared with chemotherapy, and are directed against specific molecules and pathways that drive tumor growth.23 For example, these types of therapies are normally small molecule inhibitors or monoclonal antibodies that block or turn off chemical signals, modify certain proteins, trigger the immune response, or carry toxic substances to cancer cells, thus resulting in cancer cell death. Targeted therapies often lead to tolerance due to overexpression of the targeted protein or through acquired mutation making the drug useless.24 Immunotherapy reinforces or sensitizes the immune system as a defense against tumors. The main categories of immunotherapy include monoclonal antibody therapy (also considered a targeted therapy), immune checkpoint inhibitors, cancer vaccines, and other nonspecific immunotherapies.25
While there is no doubt cancer therapies are effective at targeting and eliminating cancerous cells within the body and increasing patient survival,1 these treatments also induce unpleasant and unintended side effects. These side effects not only include those that affect quality of life (e.g. drowsiness, restlessness, anxiety, loss of libido, etc.), they also promote other life-threatening complications, including cardiovascular disease.3,26 Nonetheless, the mechanisms underlying cancer therapy-induced cardiotoxicity remain relatively unexplored.
Clinical definitions and guidelines for cardiotoxicity after cancer treatment
The National Cancer Institute (NCI) defines cardiotoxicity as ‘toxicity that affects the heart’.27 Aside from this broad definition, there are no universal guidelines for identifying and measuring cancer therapy-induced cardiotoxicity.28 As a result, multiple interpretations of the NCI definition exist. For instance, the NCI Common Terminology Criteria for Adverse Events (CTCAE)29 is considered the standard for clinical trial reporting of adverse events,30 and it includes left ventricular dysfunction (LVD) and heart failure (HF) within its terminology, but does not include hypertension. However, other entities vary in their cardiotoxicity definition. For instance, the Food and Drug Administration (FDA) defines anthracycline cardiotoxicity as >20% decrease in left ventricular ejection fraction (LVEF) when baseline LVEF is normal, or >10% decrease when baseline LVEF is not normal.28,31 Multiple other studies, including the Herceptin Adjuvant (HERA) trial and the Breast Cancer International Research Group (BCIRG), use different measurements to define cardiotoxicity, although LVD is frequently cited.32–34
Plana et al.35 offer a more structured classification system for cancer therapy-induced cardiotoxicity, grouping symptoms into two groups, type 1 or type 2. The type 1 category of cardiotoxicity uses doxorubicin as the representative agent and encompasses cancer therapies with dose-dependent side effects and induces irreversible myocardial damage. Type 2 uses trastuzumab as the typical agent: this classification describes cancer therapy side effects that are not dose related and are reversible. Symptoms in each category can then be further classified as symptomatic or asymptomatic. The various definitions and classifications of cardiotoxicity, paired with the lack of knowledge of cancer therapy-specific pathophysiological mechanisms have made it difficult for clinicians to detect and therefore prevent and manage cancer therapy-induced cardiotoxicity.35,36
Regardless of a lack of universal classification guidelines, there are some established prevention measures in place to combat cancer therapy-induced cardiotoxicity. For example, the American College of Cardiology and American Heart Association have together established procedures for chemotherapeutic agents and radiation therapy that have well described cardiovascular side effects, in order to reduce/prevent them.37–40 To combat these pathologies, the first line of defense is traditional cardioprotective therapies (e.g. iron chelators, angiotensin-converting enzyme inhibitors (ACE-I), beta-blockers, and statins).41 In fact, dexrazoxane, an iron chelator, has been found to be cardioprotective in women diagnosed with advanced breast cancer and receiving anthracycline-based chemotherapy.39,42 Statin therapy has been noted to decrease heart failure incidence in breast cancer patients;43 however, given the potential side effect of rhabdomyolysis, statins should be used with caution.43,44 While the use of these cardioprotective agents seems to reduce cardiotoxicity,41 the need for universal guidelines for cardiotoxicity identification, as well as novel prevention and treatment options, is emphasized by the high rates of cardiovascular morbidity and mortality following cancer therapy.2
Evidence for cardiovascular complications after cancer treatment
In the United States, an estimated one in three adults has been diagnosed with hypertension, 15 million people live with cardiovascular disease (e.g. heart failure, arrhythmias, cardiac dysfunction, etc.),45 and approximately 14 million individuals have a prior history of cancer.9 As the US population ages and cancer prognosis improves, so will the number of patients living with both cardiovascular disease and cancer. Not only is cardiovascular disease a leading cause of death among cancer survivors, there is an elevated risk of developing cardiovascular pathology which approaches 40% of the cancer population.36 The elevated risk of developing cardiovascular disease could be confounded by pre-existing genetic, lifestyle, age, endocrine, and environmental factors unique to each cancer patient (Figure 1). However, these factors, as well as traditional cardiovascular disease risk factors (obesity, dyslipidemia, insulin resistance, and tobacco use),4,5 do not fully account for the increased incidence of cancer therapy-induced cardiovascular toxicity.18
Figure 1.
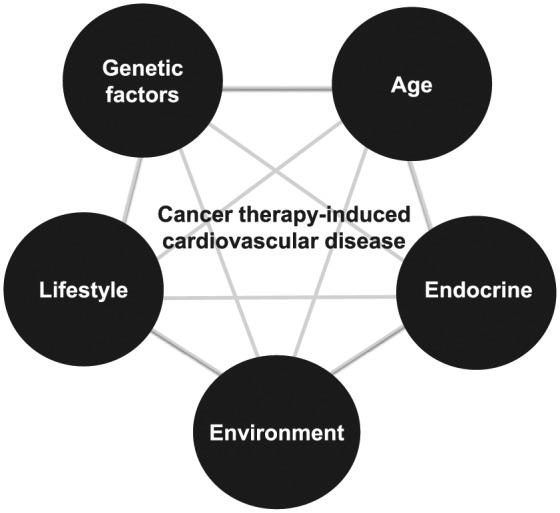
Common risk factors that contribute to both cancer and cardiovascular disease. Adapted from Irvine Page’s Mosaic Theory of Hypertension.46
The main cancer therapies reported to induce cardiovascular dysfunction and disease are radiation, vascular endothelial growth factor (VEGF) inhibitors, which encompass tyrosine kinase inhibitors (TKIs) sorafenib and sunitinib, as well as monoclonal antibodies bevacizumab and ramucirumab, human epidermal growth factor receptor type 2 (HER2) monoclonal antibody trastuzumab, and chemotherapeutic agents such as anthracyclines, platinum-based antineoplastic drugs, microtubule inhibitors, and antimetabolites. Cardiotoxicity induced by these agents could be the result of either on-target effects (e.g. tyrosine kinases located on noncancerous vascular cells) or off-target effects (e.g. implicating pathways of vascular cells differently from cancerous cells) of cancer therapies.23 The time-dependent effects of cancer therapy-induced cardiotoxicity vary according to the specific therapy and the patients’ baseline measurements. Table 1 lists the major forms of cancer therapy and summarizes their reported cardiovascular side effects, as well as their proposed mechanisms.
Table 1.
Common cardiovascular side effects of cancer therapy and potential mechanisms leading to dysfunction.
| Class | Drug | Drug target | Cardiovascular side effects | Potential Mechanism |
|---|---|---|---|---|
| Radiation | NA | Rapidly growing cells | Pericarditis, hypertension, cardiomyopathy, myocardial fibrosis, coronary artery disease, pericardial effusion, valvular disease, arrhythmias,20 autonomic dysfunction47 | Fibrosis, endothelial cell damage,20 oxidative stress48 |
| Chemotherapy | ||||
| Alkylating agents | Carboplatin, cisplatin, cyclophosphamide, oxaliplatin, ifosfamide | Rapidly growing cells; intrastrand cross-linking of DNA | Hypertension, myocardial ischemia,49 thromboembolism, cerebrovascular disease,18 myocardial infarction, cardiomyopathy,50 pericarditis16 | Oxidative stress,51,52 endothelial dysfunction, platelet activation,18 upregulation of adhesion molecule-1, increased plasminogen activator53 |
| Antimetabolites | 5-Fluorouracil, methotrexate, capecitabine, gemcitabine, cytarabine | Rapidly growing cells; blocking DNA/RNA synthesis | Cardiac ischemia54,55 angina, ECG changes, thrombosis | Oxidative stress, myocardial cell damage and necrosis, endothelial cell damage56 |
| Anthracyclines | Daunorubicin, doxorubicin, epirubicin, idarubicin, mitoxantrone |
Rapidly growing cells; enzymes involved in DNA replication | Cardiomyopathy, arrhythmia, acute myocarditis,57 heart failure,58 LVD59
cardiac ischemia60,61 |
Oxidative stress,62,63 DNA damage,64 topoisomerase 2-alpha,65 alterations in ATP production in myocytes, myocyte damage and apoptosis18 |
| Topoisomerase inhibitors | Topotecan, irinotecan, etoposide, teniposide |
Rapidly growing cells; topoisomerase inhibition | Bradycardia,66 cardiac toxicity,67 cardiac ischemia16 | Topoisomerase 2-a68 |
| Mitotic inhibitors | Paclitaxel, docetaxel, estramustine, ixabepilone, vinblastine |
Rapidly growing cells; microtubule assembly, inhibition of spindle formation | Myocardial ischemia,69 coronary spasm,49 arrhythmias, bradycardia, hypertension70 | Modulation of calcium handling,71 cellular hypoxia18 |
| Targeted therapy | ||||
| Tyrosine kinase inhibitors | Imatinib, dasatinib, nilotinib, bosutinib, ponatinib, sorafenib, sunitinib, lapatinib |
ABL1 kinase KIT, PDGFRα, VEGF72 |
Cardiomyopathy,73,74 heart failure,75–77 hypertension, LVD,16 thromboembolism, cardiac contractile dysfunction78 | Inhibiting cell survival, angiogenesis, and cell growth,49 inhibition of NO and prostacyclin, increased production of endothelin-1,79 oxidative stress, endothelial cell apoptosis,80 disruption of mitochondrial metabolism and impairment of myocardial function.81 |
| Immunotherapy | ||||
| Interferons, interleukins | Type I-III IFNs | Immune system activation | Hypertension, arrhythmias, heart failure, LVD,16 myocardial infarction82 | PRR activation,83 interferon-induced coronary spasm84 |
| Monoclonal antibodies | ||||
| Trastuzumab, lapatinib, pertuzumab |
HER2 | Systolic cardiac dysfunction,49,85 cardiomyopathy,85 reduced LVEF, heart failure, hypertension86 | Cardiac myocyte dysfunction,87 increased sympathetic output,86 impaired cardiac repair and myocyte homeostasis88 | |
| Bevacizumab, ramucirumab | VEGF | Cardiac ischemia,89 heart failure,75 systolic dysfunction, left ventricular dysfunction, hypertension,82 thromboembolism16 | Cardiac myocyte dysfunction,90 inhibiting cell survival, angiogenesis, and cell growth,49 inhibition of NO and prostacyclin, increased production of endothelin-1,79 oxidative stress, endothelial cell apoptosis80 |
ABL1, Abelson tyrosine-protein kinase 1; ATP, adenosine triphosphate; DNA, deoxyribonucleic acid; ECG, electrocardiogram; HER2, human epidermal growth factor receptor 2; IFN, interferon; KIT, stem cell factor receptor/CD117; LVD, left ventricular dysfunction; LVEF, left ventricular ejection fraction; NO, nitric oxide; PDGFRα, platelet-derived growth factor alpha; PRR, pattern-recognition receptor; RNA, ribonucleic acid; VEGF, vascular endothelial growth factor.
Radiation
Radiation is an effective cancer therapy, nevertheless it has serious cardiovascular side-effects, such that it has been termed radiation-induced heart disease (RIHD) and radiation-induced vascular disease (RIVD).20,21 These symptoms include pericarditis, cardiomyopathy, myocardial fibrosis, coronary artery disease, peripheral vascular disease, pericardial effusion, valvular disease, arrhythmias, and autonomic dysfunction.20,47 RIHD can have acute, subacute, and late presentation and may affect the entire cardiovascular system at any stage.21 Besides the well-known radiation effects to the heart, medium-sized to large-sized vessels demonstrate lipid accumulation, inflammation, thrombosis, increase in intimal thickness, and connective tissue after exposure.91,92
Cardiotoxicity, secondary to radiation, is increased in severity and incidence with increasing doses of radiation (i.e. dose dependent), volume of body area exposed, time since exposure, and adjuvant therapies.19 The cancer therapy-induced cardiotoxicity effects of radiation have been observed 15–30 years after completion of therapy.93 The location of exposure has also been reported to affect cardiotoxicity. For example, studies show that radiation over the left side of the chest, compared with the right, results in increased occurrence of cardiovascular pathology.94 In addition, radiation exposure of the mediastinum and neck causes higher rates of cerebrovascular disease and hypertension, which could be mediated by carotid baroreceptor damage.95,96 Known mechanisms of cardiotoxicity from radiation exposure comprise fibrosis, endothelial cell damage,20 and oxidative stress.48
Antiangiogenesis
Angiogenesis refers to the formation of new blood vessels, which can contribute to tumor growth and survival.97,98 There are two classes of antiangiogenesis therapies for the use of cancer therapeutics: (1) antibodies specific for VEGF (e.g. bevacizumab) and (2) small molecular tyrosine kinase inhibitors (TKI) against the VEGF receptor (e.g. sorafenib and sunitinib). Although VEGF contributes to the development of cancer via the formation of new blood vessels in tumors, it also has an important role in the normal physiologic function of endothelial and renal cell survival, vasodilation, and cardiac contractile function.99 Therefore, its inhibition has significant cardiovascular side effects. Specifically, VEGF inhibition induces conditions such as hypertension, thromboembolism, ischemia, cardiac contractile dysfunction, and heart failure.78 A retrospective analysis of clinical trials demonstrated elevated blood pressure within 4 weeks of initiating therapy with sunitinib.81 Mechanisms that contribute to these pathophysiological states include, inhibition of nitric oxide (NO) and prostacyclin, increased production of endothelin-1,79 oxidative stress, and cell apoptosis.80 It is important to note that the approval for the use of bevacizumab was revoked in 2014 for the treatment of breast cancer due to high incidence of heart failure.75 Interestingly, VEGF tyrosine kinase inhibition has an increased rate of cardiovascular toxicity, when compared with VEGF immunotherapy due to poor selectivity of the drugs and off-target effects.100
Tyrosine kinase inhibitors
Due to the ubiquitous nature of tyrosine kinase receptors, the class of TKI targets several different pathways. For example, sunitinib and sorafenib target VEGF, lapatinib targets epidermal growth factor receptor (EGFR) and HER2 (see below), and imatinib targets Bcr-Abl tyrosine kinase. Inhibition of each of these tyrosine kinase receptors exerts a unique cardiovascular pathology that includes heart failure,76,101,102 cardiomyopathy,73,74 hypertension,78 thromboembolism, and cardiac contractile dysfunction. In addition to mechanisms of VEGF inhibition-induced cardiovascular disease (see above), TKIs against VEGF have also been shown to disrupt mitochondrial metabolism, impair myocardial function, and result in myocardial apoptosis.81 These cardiovascular toxicity events have been shown to persist for years after therapy cessation.103
Human epidermal growth factor receptor type 2 immunotherapy
The use of monoclonal antibodies such as trastuzumab requires routine monitoring for cardiac function. Trastuzumab is targeted against HER2, a tyrosine kinase receptor that mediates cell growth and survival. This type of cancer therapy has been shown to induce cardiac contractile dysfunction, reduction in left ventricular function, heart failure, hypertension, and increased sympathetic tone.49,57,85–88 Inhibition of HER2 via trastuzumab is associated with the development of cardiotoxicity as early as 4–8 weeks after initiation of therapy, albeit reversible through treatment cessation.104,105 The cardiac myocyte HER2 receptor is activated when bound with neuregulin 1-β resulting in the promotion of protein synthesis, cell survival, and hypertrophy.106 The mechanisms of cardiotoxicity are hypothesized to occur through (1) disruption of cardiac repair and myocyte homeostasis and (2) increased sympathetic tone and activation of cardiac β-adrenergic receptors.107
Chemotherapeutics
Despite efficacy at reducing cancer, the use of anthracyclines is restricted by cardiotoxicity. Anthracyclines are a class of antibiotics that inhibit topoisomerase activity by intercalating between DNA base pairs, leading to DNA damage and eventual apoptosis.108 Cardiotoxicity occurs both after cumulative short- and long-term anthracycline exposure causing cardiac myocyte and endothelial cell injury.109–112 In a retrospective study of cancer patients treated with anthracyclines, the development of cardiotoxicity was positively correlated with patient age, treatment frequency, and dosage.59 Low dose, standard-therapy breast cancer treatment with anthracyclines resulted in up to 20% of patients developing cardiac systolic dysfunction within 6 months of treatment.113 Symptoms of cardiotoxicity associated with anthracycline use include cardiomyopathy, arrhythmia, acute myocarditis,57 heart failure,58 left ventricular dysfunction,59 and cardiac ischemia.60,61 Identified mechanisms that underlie anthracycline-induced cardiotoxicity include: (1) oxidative damage to cardiac myocytes accompanied by lipid peroxidation and mitochondrial dysfunction, (2) modulation of topoisomerase activity and DNA damage, (3) alterations in multidrug-resistant efflux proteins, and (4) decreased mesenchymal and circulating progenitor cells.62,112,114–116
Alkylating agents target rapidly growing cells for cell death by adding alkyl groups to DNA, thereby cross-linking the strands, resulting in the prevention of replication. As a consequence of off-target effects, this class of therapeutics also induces significant cardiotoxicity. Patients undergoing platinum-based drug treatments have been reported to develop hypertension, myocardial ischemia, thromboembolism, cerebrovascular disease, endothelial dysfunction, and coronary artery disease.18,117 Furthermore, patients treated with agents such as cyclophosphamide and ifosfamide have been observed to develop heart failure, reduction in left ventricular function, and arrhythmias.118,119 Nonetheless, precise mechanisms of cardiovascular toxicity are less clear. Studies have reported increased levels of intracellular adhesion molecule-1 (ICAM-1) and plasminogen activator and inhibitor type 1,53,120 as well as exacerbated oxidative stress-induced cardiac cell death.51 However, it has been shown that cisplatin is not completely excreted from the body and patients that exhibit late vascular toxicity also have measurable platinum serum levels years after completion of therapy, which could contribute to toxicity.121
Microtubule inhibitors target and disrupt microtubule structure and function, preventing cell separation during cell division, thereby inducing cell death. Microtubules not only assist with separation during cell division, they also pro-vide cytoplasmic structure. Microtubules exist dynamically between a monomeric and polymeric form through a constant state of polymerization and depolymerization. Paclitaxel is a microtubule inhibitor, which binds to tubulin and inhibits depolymerization. Patients undergoing treatment with paclitaxel have shown increased rates of arrhythmias,70 myocardial ischemia,69 and coronary spasm.49 The mechanism underlying this cardiovascular toxicity is largely unknown; however, paclitaxel has been shown to modulate calcium handling in cardiac myocytes. A study by Paul et al. (2000) demonstrated that depolymerization of microtubules increased calcium levels within the cytoplasm. There are also studies showing that disruption of the microtubule network affects cellular contraction. Chitaley and Webb (2002) demonstrated that enhanced microtubule depolymerization enhances aorta contraction in a Rho-kinase dependent manner.122 Nonetheless, the clinical implication of increased vascular contraction and whether it contributes to cardiovascular dysfunction in cancer survivors remains to be determined.
Antimetabolites have an increased incidence of cardiotoxicity that can occur early in cancer treatment and are proportional with dose and frequency.123 Antimetabolites interfere with mitosis by substituting metabolites necessary for DNA or RNA synthesis, thus damaging cells. Common cardiovascular side effects include cardiac ischemia,54,55 angina, disruption of cardiac electrical activity, and thrombosis.86 The underlying mechanism of these deleterious phenotypes has been proposed to be oxidative stress and endothelial cell damage.56
In summary, cancer therapy undoubtedly contributes to cardiovascular disease pathogenesis. There are multiple hypothesized mechanisms that have been postulated to result in cardiovascular toxicity; however, these mechanisms are broad and could be a deleterious side effect of cancer treatment, as opposed to the cause. We would like to offer an alternative hypothesis whereby cancer therapy induces cardiovascular toxicity through the action of DAMPs.
Damage-associated molecular patterns and cancer
Chronic inflammation is a distinct feature of both cardiovascular disease and cancer.9 However, the precise mechanisms that mediate this inflammation are only beginning to emerge. While infection is unequivocally linked to the pathogenesis of both cardiovascular disease124 and cancer,125 many patients with these diseases present sterile inflammation, or immune system activation in the absence of a pathogen. Although sterile inflammation has been proposed to arise through different mechanisms,126 DAMPs and the danger theory of immunity have led to a paradigm shift in the understanding of not only pathophysiology, but physiology also.10,11
DAMPs are endogenous molecules that have specific functional purposes inside cells and as such, are normally compartmentalized within membranes for performance of their various tasks. However, when these endogenous molecules are exposed to the extracellular environment, either by passive diffusion after membrane rupture or active secretion during stress, they unintentionally acquire additional immunogenic properties. These functions broadly include: the presentation of danger signals to other cells in a paracrine and endocrine manner, the activation of pattern-recognition receptors (PRRs) of the innate immune system, communication to the adaptive immune system and promotion of immunological memory, and finally the facilitation of tissue repair.127 Obviously, these functions are not all mutually exclusive and the ability of DAMPs to both promote and resolve inflammation is an evolutionarily conserved mechanism to restore immunological homeostasis. However, problems arise when the expression of DAMPs becomes excessive, chronic, and uncontrolled. We propose that disease progression ensues when the expression of DAMPs exceeds a ‘pathogenicity threshold’ (Figure 2).
Figure 2.

Hypothesized schematic of how damage-associated molecular patterns (DAMPs) contribute to the treatment and progression of cancer and cardiotoxicity. Due to the paradoxical contribution of DAMPs to cancer progression in the literature, we believe two key thresholds of DAMP expression exist. The first is the ‘anti-cancer’ threshold and this is where controlled amounts of DAMPs stimulate the immune system to mount an effective defense against the growing and potentially pathogenic tumor. This explains the benefits of immunogenic cell death (ICD) to cancer remission. The second threshold is the one of pathogenicity and this is when DAMP expression becomes so excessive and chronic that uncontrolled inflammation ensues, and this contributes to the progression of cancer, resistance to anti-cancer treatments, and cardiovascular disease.
DAMPs, damage-associated molecular patterns.
There are a large number of endogenous molecules that could potentially become DAMPs, which is dependent on the extracellular environment. Currently, DAMPs include a number of macromolecules composed of lipids, nucleic acids, and proteins from different sources such as extracellular matrix and cellular organelles. To add further complexity, macromolecules released into the extracellular environment surrounding stressed or decaying cells could be modified in such a way that their pathogenicity is either amplified or abrogated (e.g. oxidation).128 PRRs of the innate immune system (e.g. Toll-like receptors [TLRs]) are able to recognize distinct motifs within these macromolecules. Once these macromolecules are recognized, the innate immune system responds in a manner that is motif-specific and promotes a pro-inflammatory milieu that is needed to combat the danger (e.g. upon the recognition of viral pathogen-associated molecular patterns (PAMPs), the major defensive strategy employed by the host immune system is the activation of the interferon regulatory factors). Table 2 provides several examples of DAMPs observed to be released after cancer therapy, as well as their corresponding PRR. It should be noted that this list is by no means complete, as many other unknown molecules may fulfill the inclusion criteria of being DAMPs and PRR ligands, especially in the ever-changing tumorigenic microenvironment (e.g. neoantigens).
Table 2.
Secreted or released damage-associated molecular patterns that have been measured after various anti-cancer therapies.
| DAMP | Cancer type | Treatment | Author(s) | Corresponding PRR(s) |
|---|---|---|---|---|
| Actin | Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | DNGR-1 (CLEC9A) |
| Adenosine | Hairy cell leukemia | Pentostatin (2’-deoxycoformycin) | Johnston130 | A1, A2A, A2B, A3 |
| ATP | Bladder carcinoma | Photodynamic therapy | Garg et al.131 | P2X7, P2Y2 |
| Colorectal carcinoma and osteosarcoma | Mitoxantrone and oxaliplatin | Michaud et al.132 | ||
| Colorectal carcinoma and sarcoma | Various chemotherapeutic agents | Ghiringhelli et al.133 | ||
| Fibrosarcoma | Doxorubicin | Ma et al.134 | ||
| Cutaneous melanoma | Amino acid derivative LTX-401 | Eike et al.135 | ||
| T-cell leukemia | Ultraviolet light | Elliott et al.136 | ||
| Calreticulin | Bladder carcinoma | Photodynamic therapy | Garg et al.131 | CD91, scavenger receptors (LOX-1, SREC-1, and FEEL-1/CLEVER-1) |
| Bladder carcinoma | Photodynamic therapy | Garg et al.137 | ||
| Colorectal carcinoma | Doxorubicin | Obeid et al.138 | ||
| Colorectal carcinoma | Electrohyperthermia | Andocs et al.139 | ||
| Colorectal carcinoma and osteosarcoma | Mitoxantrone and oxaliplatin | Michaud et al.132 | ||
| Colorectal carcinoma, cutaneous melanoma, lung carcinoma, esophageal squamous cell carcinoma, and pancreatic carcinoma | Various chemotherapeutic agents | Yamamura et al.140 | ||
| Cytochrome c | Cutaneous melanoma | Amino acid derivative LTX-401 | Eike et al.135 | Unknown |
| Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | ||
| HSP60 | Squamous cell carcinoma | Photodynamic therapy | Korbelik et al.141 | CD91, scavenger receptors (LOX-1, SREC-1, & FEEL-1/CLEVER-1), TLR2, TLR4 |
| HSP70 | Bladder carcinoma | Photodynamic therapy | Garg et al.137 | |
| Colorectal carcinoma | Oxaliplatin and 5-fluorouracil | Fang et al.142 | ||
| Colorectal carcinoma | Electrohyperthermia | Ma et al.134 | ||
| Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | ||
| Prostate adenocarcinoma | Heating and UVC irradiation | Brusa et al.143 | ||
| Squamous cell carcinoma | Photodynamic therapy | Korbelik et al.141 | ||
| HSP90 | Colorectal carcinoma | Electrohyperthermia | Ma et al.134 | |
| Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | ||
| Myeloma cells | Bortezomib | Spisek et al.144 | ||
| GRP78 (BiP) | Squamous cell carcinoma | Photodynamic therapy | Korbelik et al.141 | |
| GP96 (GRP94) | Squamous cell carcinoma | Photodynamic therapy | Korbelik et al.141 | |
| HMGB1 | Colorectal carcinoma | Doxorubicin and linoleic acid | Luo et al.145 | RAGE. TIM3, TLR2, TLR4, TLR9 |
| Colorectal carcinoma | Electrohyperthermia | Ma et al.134 | ||
| Colorectal carcinoma | Oxaliplatin and 5-fluorouracil | Fang et al.142 | ||
| Colorectal carcinoma and osteosarcoma | Mitoxantrone and oxaliplatin | Michaud et al.132 | ||
| Colorectal carcinoma and thymoma cells | Doxorubicin and irradiation, respectively | Apetoh et al.146 | ||
| Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | ||
| Prostate adenocarcinoma | Heating and UVC irradiation | Brusa et al.143 | ||
| Prostate adenocarcinoma | Various chemotherapeutic agents | Zhou et al.147 | ||
| Cutaneous melanoma | Amino acid derivative LTX-401 | Eike et al.135 | ||
| Thymoma cells | X-rays | Apetoh et al.148 | ||
| IL-1α/β | Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | IL-1R |
| Peroxiredoxin-1 | Lung squamous cell carcinoma/adenocarcinoma | Photodynamic therapy | Tracy et al.129 | CD14, TLR4 |
| S100A8/A9 | Prostate adenocarcinoma | Phorbol 12-myristate,13-acetate | Hermani et al.149 | RAGE |
| Uric acid | Thymoma cells | Etoposide and cyclophosphamide | Hu et al.150 | CD14, TLR2, TLR4 |
ATP, adenosine triphosphate; BiP, binding immunoglobulin protein; CD, cluster of differentiation; DAMP, damage-associated molecular pattern; FEEL-1/CLEVER-1, Fasciclin EGF-like/common lymphatic endothelial and vascular endothelial receptor-1; GP96, glycoprotein 96; GRP, glucose-regulated protein; HMGB1, high mobility group box 1; HSP, heat shock protein; IFN, interferon; IL, interleukin; IL-1R, interleukin-1 receptor; LOX-1, lectin-type oxidized LDL receptor 1; PRR, pattern-recognition receptor; P2X7, purinergic receptor P2X, ligand-gated ion channel 7; P2Y2, purinergic receptor P2Y, G-protein coupled 2; RAGE, receptor for advanced glycation end products; SREC-1, scavenger receptor class F member 1; TIM3, T cell/transmembrane, immunoglobulin, and mucin; TLR, Toll-like receptor; UVC, ultraviolet C.
The participation of DAMPs as the mediators of the inflammation in most cases presumes that cell death is the initiating mechanism of their release.151 However, determining whether cell death primarily drives pathophysiology or is a secondary bystander is difficult. Although there are many forms of cell death,152 necrosis was originally thought of as the primary source of pro-inflammatory DAMPs due to disintegration of the plasma membrane and passive release of intracellular constituents into the extracellular environment.153 However, we have come to learn that apoptosis can also be immunostimulatory, as a result of the programed release of immunogenic molecules,127,154 and necroptosis further refines the ability of cells to rapidly present DAMPs to the immune system with the regulated and controlled breakdown of the plasma membrane.155 Cell stress or injury can also fuel the secretion of DAMPs127 and this emphasizes that cell death is not an absolute precursor to participation of DAMPs in pathophysiology.
The participation of DAMPs in cancer is seemingly contradictory. On one hand, DAMPs have been proposed to play a beneficial role in cancer therapy by interacting with the immune system and promoting an ‘anti-cancer vaccine effect’ (Figure 2), even in the absence of a traditional adjuvant.156–159 On the other hand, DAMPs could contribute to the progression of cancer160–167 and promote resistance to cancer treatments.15,147 The former is made possible due to the ability of certain cancer therapies to induce immunogenic cell death (ICD) that is associated with the emission of DAMPs from dying cancer cells. DAMPs can then be trafficked by signaling pathways, which are instigated and regulated by a complex interplay between endoplasmic reticulum stress, reactive oxygen species, and certain metabolic/biosynthetic processes.158 The ultimate response of DAMPs in cancer (i.e. antitumorigenic or protumorigenic) may depend on a number of different factors,156 such as:
The histopathology of the cancer (and therefore its anatomical location).
The type of cell death modality that is triggered by ICD, and the biochemical processes activated (this, in itself, may also depend on a number of different and varying factors).
The types and abundance of immune cells present to phagocytose debris.
Finally, whether a cancer antigen is recognized or not.
In summation, the conflicting contributions of DAMPs in cancer have revealed a Janus face.157,159 Nonetheless, given the well-documented role of DAMPs in exacerbating inflammation in cardiovascular disease,168 we believe that DAMPs, irrespective of their contribution to cancer treatment and progression, could serve as a novel facilitator of cardiotoxicity during and following cancer therapy.49 DAMPs in cancer could arise from a number of different sources, including (paradoxically) tumor growth and remodeling,160–167 as well as carcinogenic environmental toxins.169
Damage-associated molecular patterns and cardiovascular disease
While it has been reviewed in more depth elsewhere,168 there is ample evidence that increased levels of DAMPs are associated with cardio-vascular disease in human populations. For example, hypertensive patients have higher plasma levels of mitochondrial DNA, high mobility group box 1 (HMGB1), heat shock protein (HSP) 60 and HSP70, or fibrinogen, and each of these is capable of activating distinct PRRs that then start the immune-response cascade.168 Additionally, DAMPs such as HMGB1, advanced glycation end products (AGEs), hyaluronan, oxidized low-density lipoprotein, and uric acid are present in higher levels in the circulation or the plaques in patients with atherosclerosis and S100 proteins are increased in stroke.170,171 Consequently, PRRs such as TLRs seem to be chronically activated in cardiovascular disease, including atherosclerosis, cardiomyopathy, hypertension and cerebrovascular disease.12,168,172–175
There are also strong indications from basic science studies that release of DAMPs is causative for cardiovascular disease. Thus, experimental administration of DAMPs has detrimental effects on cardiovascular parameters and inhibition of DAMPs, or their respective PRRs, improves cardiovascular disease phenotypes and outcomes.168 HMGB1 administration increased myocardial infarct size and promoted the development of microvascular thrombosis.176 Our group demonstrated that mitochondrial DNA infusion leads to hypertension in a pre-eclampsia rodent model.177
Additionally, treatment with TLR4 or TLR9 inhibitors decreased blood pressure in spontaneous hypertensive rat (SHR), and treatment with the TLR4 inhibitor eritoran decreased myocardial ischemia-reperfusion injury;12 TLR4 and TLR2 deficient mice are protected from doxorubicin-induced cardiomyopathy.178,179 Genetic deficiency in various TLRs or their common downstream signaling partner MyD88 protects apolipoprotein E (apoE) knock-out mice from atherosclerosis.180
Hypothesis
As discussed above, there is a clear link between cancer therapy and cardiovascular disease. While it is logical to surmise that the traditional cardiovascular disease risk factors (e.g. obesity, dyslipidemia, insulin resistance, and tobacco use) contribute to cancer therapy-induced cardiotoxicity (especially given that these are also risk factors to cancer itself), correlational analysis does not support this notion.4,5 Therefore, the clinical and basic science data summarized in this review strongly support the notion that cancer therapy directly causes cardiovascular toxicity that would not have occurred in its absence. The mechanisms by which specific cancer therapies or radiotherapy lead to cardiovascular disease is in some cases clearly demonstrated, for instance interference with endothelial NO signaling in the case of VEGF receptor inhibitors. In the majority of remaining cases, however, the processes by which cancer therapy causes cardiovascular disease are unclear. We believe that DAMPs and innate immune-system activation are the missing contributing factors for the development of cancer therapy-induced cardiotoxicity (Figure 3).
Figure 3.
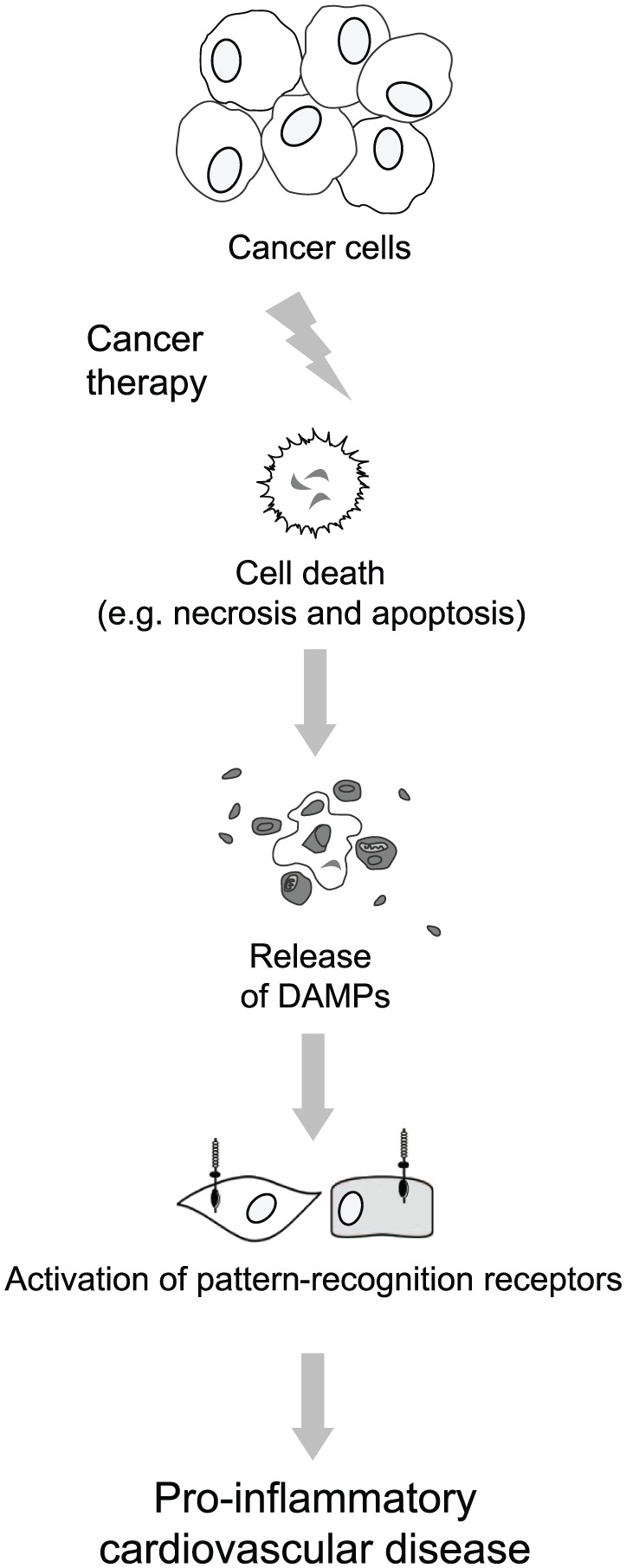
Danger signaling in cardio-oncology. The nature of cancer therapy is to reduce tumor size by causing sudden and rapid cancer-cell death. Because damage-associated molecular patterns (DAMPs) are released from dead and dying cells (via necrosis or apoptosis), this results in the release of DAMPs from cells during cancer therapy. DAMPs activate the immune system through activation of pattern-recognition receptors from cell types within the cardiovascular system (e.g. endothelial cells, vascular smooth muscle); we hypothesize that the overactivation of this system results in a pro-inflammatory cardiovascular disease following cancer therapy.
DAMPs, damage-associated molecular patterns.
In support of this hypothesis, chemotherapeutic agents and radiotherapy are effective against cancer because they induce cell death in rapidly dividing cell populations. As a consequence of sudden cell death in a large number of cells simultaneously, there is a rapid and massive release of various DAMPs that enter the systemic circulation and induce an inflammatory response through activation of the innate immune system. As discussed above, both in vivo and in vitro treatments with chemotherapeutic agents or radiation induce release of DAMPs (Table 2). An increase in circulating DAMPs post-chemotherapy could contribute to the increased prevalence of thrombosis in cancer patients.181 Thrombosis has a physiological role in immune defense, where monocytes respond to PAMPs and DAMPs by releasing tissue factor and initiating coagulation pathways.182 Therefore, an increased presence of circulating DAMPs could play a role in the increased presence of thrombotic events in these patients. It has also been shown that chemo- and radio-therapy causes activation of innate immune receptors, such as PRRs.146
In a perfect illustration of the Janus face effect, DAMP release and PRR activation following administration of chemotherapy may be simultaneously beneficial for cancer treatment and detrimental for other systems and organs, including the cardiovascular system. Thus, ICD can drive both immune-mediated tumor suppression and pro-inflammatory cytokine-mediated tissue injury via DAMPs. Similarly, immune cells, and in particular, T cells, also present a paradox in the context of the pathophysiology of cardio-oncology. While promoting T-cell expansion may be beneficial in diminishing cancer progression and tumorigenesis, they are also known to promote cardiovascular disease,183 and specifically, hypertension and its associated hallmarks.184
The therapeutic potential of adding agonists for TLR9 (CpG DNA), TLR3 (poly I:C), or TLR4 (endotoxin analogs) for a synergistic effect to radiotherapy or chemotherapeutics was recently evaluated and clear anti-tumor effects have been ascribed to this approach.185 On the other hand, long-term consequences of TLR ligand administration on any cardiovascular parameters are only beginning to be investigated.186 Data from our group and others would suggest that high amounts of DAMPs, whether released from tumor cell death following cancer therapy or administered therapeutically, might have a negative impact on the cardiovascular system.
Potential value of damage-associated molecular patterns in cardio-oncology
There are continuing advances in cancer therapeutics; however, the challenge now exists of developing avenues allowing these treatments to be efficacious without significant cost. The prevalence of cancer therapy-induced cardiotoxicity has led to systematic monitoring and the need for early identification of biomarkers for high-risk patients.187,188 Currently, circulating cardiac troponins (TnI, TnT) are considered the gold-standard marker for cardiac injury and are used as a diagnostic adjuvant to echocardiograms and other diagnostic modalities. TnI is sensitive and specific for myocardial injury allowing for early myocardial damage detection prior to any clinical detection through physical examination or imaging, especially in anthracycline-based chemotherapy regimens.189 Other studies have also looked at the usefulness of TnI and TnT as surrogate markers for myocardial damage with the use of anti-VEGF TKI chemotherapeutics.18,190,191 Nonetheless, cardiac troponins may not be the only molecules released during cardiovascular damage after an insult such as chemotherapy or radiation. Theoretically, a wide array of circulating DAMPs could be measured in cancer patients prior to, during, and after treatment. Therefore, DAMPs present an opportunity for identifying and treating cancer therapy-induced cardiotoxicity. For example, obvious therapeutic targets for DAMPs include antagonism of the PRR activated by a specific DAMP, or direct ligand neutralization, thereby reducing the inflammation that promotes cardiovascular disease. Given the beneficial effects of DAMPs in some cancers, perhaps specific PRR antagonism in cardiovascular tissues is warranted.156–159
In conclusion, the use of DAMPs as a diagnostic adjuvant (i.e. biomarker) or therapeutic drug could be a novel approach to decrease cardiovascular morbidity and prevent premature mortality from cardiovascular toxicity for the millions of patients that have successfully outlived their initial cancer diagnosis. After all, what is the point of tolerating the toxicity of cancer-therapy to survive cancer if you subsequently succumb to cardiovascular disease?
Acknowledgments
Special thanks to Theodora Szasz for her contributions to the outline and hypothesis section of the paper.
Footnotes
Funding: This work was supported by the National Institutes of Health (grant number HL-134604, Program Project Grant 2017-22).
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Nicole S. Klee, Department of Physiology, Medical College of Georgia at Augusta University, 1120 15th Street, Augusta, GA 30912, USA.
Cameron G. McCarthy, Department of Physiology, Medical College of Georgia at Augusta University, Augusta, GA, USA
Patricia Martinez-Quinones, Departments of Physiology and Surgery, Medical College of Georgia at Augusta University, Augusta, GA, USA.
R. Clinton Webb, Department of Physiology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
References
- 1. American Cancer Society. Cancer treatment and survivorship: facts and figures 2016-2017. Atlanta 2016, https://www.cancer.gov/publications/dictionaries/cancer-terms?CdrID=44004.
- 2. Wickramasinghe CD, Nguyen KL, Watson KE, et al. Concepts in cardio-oncology: definitions, mechanisms, diagnosis and treatment strategies of cancer therapy-induced cardiotoxicity. Future Oncol 2016; 12: 855–870. [DOI] [PubMed] [Google Scholar]
- 3. Okwuosa TM, Anzevino S, Rao R. Cardiovascular disease in cancer survivors. Postgrad Med J 2017; 93: 82–90. [DOI] [PubMed] [Google Scholar]
- 4. Landy DC, Miller TL, Lopez-Mitnik G, et al. Aggregating traditional cardiovascular disease risk factors to assess the cardiometabolic health of childhood cancer survivors: an analysis from the Cardiac Risk Factors in Childhood Cancer Survivors Study. Am Heart J 2012; 163: 295–301.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mulrooney DA, Armstrong GT, Huang S, et al. Cardiac outcomes in adult survivors of childhood cancer exposed to cardiotoxic therapy: a cross-sectional study. Ann Intern Med 2016; 164: 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 2009; 78: 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Granger JP. An emerging role for inflammatory cytokines in hypertension. Am J Physiol Heart Circ Physiol 2006; 290: H923–H924. [DOI] [PubMed] [Google Scholar]
- 8. Dzielak DJ. The immune system and hypertension. Hypertension 1992; 19(Suppl. 1): I36–I44. [DOI] [PubMed] [Google Scholar]
- 9. Koene RJ, Prizment AE, Blaes A, et al. Shared risk factors in cardiovascular disease and cancer. Circulation 2016; 133: 1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991–1045. [DOI] [PubMed] [Google Scholar]
- 11. Matzinger P. The danger model: a renewed sense of self. Science 2002; 296: 301–305. [DOI] [PubMed] [Google Scholar]
- 12. McCarthy CG, Wenceslau CF, Goulopoulou S, et al. Circulating mitochondrial DNA and toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 2015; 107: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCarthy CG, Webb RC. The toll of the gridiron: damage-associated molecular patterns and hypertension in American football. FASEB J 2016; 30: 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han Y, Liao X, Gao Z, et al. cTnI exacerbates myocardial ischemia/reperfusion injury by inducing adhesion of monocytes to VECs via TLR4/NF-kappaB-dependent pathway. Clin Sci 2016; 130: 2279–2293. [DOI] [PubMed] [Google Scholar]
- 15. Liu L, Yang M, Kang R, et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011; 7: 112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berardi R, Caramanti M, Savini A, et al. State of the art for cardiotoxicity due to chemotherapy and to targeted therapies: a literature review. Crit Rev Oncol Hematol 2013; 88: 75–86. [DOI] [PubMed] [Google Scholar]
- 17. Bonita R, Pradhan R. Cardiovascular toxicities of cancer chemotherapy. Semin Oncol 2013; 40: 156–167. [DOI] [PubMed] [Google Scholar]
- 18. Cameron AC, Touyz RM, Lang NN. Vascular complications of cancer chemotherapy. Can J Cardiol 2016; 32: 852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams MJ, Hardenbergh PH, Constine LS, et al. Radiation-associated cardiovascular disease. Crit Rev Oncol Hematol 2003; 45: 55–75. [DOI] [PubMed] [Google Scholar]
- 20. Taunk NK, Haffty BG, Kostis JB, et al. Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Front Oncol 2015; 5: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weintraub NL, Jones WK, Manka D. Understanding radiation-induced vascular disease. J Am Coll Cardiol 2010; 55: 1237–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaiser J. When less is more: some cancer therapies may work better if doses are timed to thwart tumor evolution. Science 2017; 355: 1144–1146.28302821 [Google Scholar]
- 23. Gopal S, Miller KB, Jaffe IZ. Molecular mechanisms for vascular complications of targeted cancer therapies. Clin Sci 2016; 130: 1763–1779. [DOI] [PubMed] [Google Scholar]
- 24. Salami J, Crews CM. Waste disposal – an attractive strategy for cancer therapy. Science 2017; 355: 1163–1167. [DOI] [PubMed] [Google Scholar]
- 25. Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med 2016; 14: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. National Cancer Institute. Cancer treatment: side effects. National Cancer Institute: National Institues of Health, 2015. [Google Scholar]
- 27. National Cancer Institute. NCI dictionary of cancer terms: definition of cardiotoxocity, 2015, https://www.cancer.gov/publications/dictionaries/cancer-terms?CdrID=44004
- 28. Bloom MW, Hamo CE, Cardinale D, et al. Cancer therapy-related cardiac dysfunction and heart failure: part 1: definitions, pathophysiology, risk factors, and imaging. Circ Heart Fail 2016; 9(1): e002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.0, 2010. [Google Scholar]
- 30. Colevas AD, Setser A. The NCI Common Terminology Criteria for Adverse Events (CTCAE) v3.0 is the new standard for oncology trials. J Clin Oncol 2004; 22(14): 6098. [Google Scholar]
- 31. US Food and Drug Administration (FDA). Doxorubicin hydrochloride liposome injection for intravenous infusion, https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/203263lbl.pdf (1995, accessed 24 March 2017).
- 32. Gasowski J, Piotrowicz K. Breast cancer, age, and hypertension: a complex issue. Hypertension 2012; 59: 186–188. [DOI] [PubMed] [Google Scholar]
- 33. Hooning MJ, Botma A, Aleman BM, et al. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J Natl Cancer Inst 2007; 99: 365–375. [DOI] [PubMed] [Google Scholar]
- 34. Dores H, Abecasis J, Correia MJ, et al. Detection of early sub-clinical trastuzumab-induced cardiotoxicity in breast cancer patients. Arq Bras Cardiol 2013; 100: 328–332. [PubMed] [Google Scholar]
- 35. Plana JC, Galderisi M, Barac A, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2014; 27: 911–939. [DOI] [PubMed] [Google Scholar]
- 36. Daher IN, Daigle TR, Bhatia N, et al. The prevention of cardiovascular disease in cancer survivors. Tex Heart Inst J 2012; 39: 190–198. [PMC free article] [PubMed] [Google Scholar]
- 37. Adams MJ, Lipshultz SE. Pathophysiology of anthracycline- and radiation-associated cardiomyopathies: implications for screening and prevention. Pediatr Blood Cancer 2005; 44: 600–606. [DOI] [PubMed] [Google Scholar]
- 38. Carver JR, Shapiro CL, Ng A, et al. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol 2007; 25: 3991–4008. [DOI] [PubMed] [Google Scholar]
- 39. Lenneman CG, Sawyer DB. Cardio-oncology: an update on cardiotoxicity of cancer-related treatment. Circ Res 2016; 118: 1008–1020. [DOI] [PubMed] [Google Scholar]
- 40. Hunt SA. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2006; 47: 1503–1505. [DOI] [PubMed] [Google Scholar]
- 41. Kalam K, Marwick TH. Role of cardioprotective therapy for prevention of cardiotoxicity. Eur J Cancer 2013; 49: 2900–2909. [DOI] [PubMed] [Google Scholar]
- 42. Swain SM, Whaley FS, Gerber MC, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol 1997; 15: 1318–1332. [DOI] [PubMed] [Google Scholar]
- 43. Seicean S, Seicean A, Plana JC, et al. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J Am Coll Cardiol 2012; 60: 2384–2390. [DOI] [PubMed] [Google Scholar]
- 44. Desai CS, Martin SS, Blumenthal RS. Non-cardiovascular effects associated with statins. BMJ 2014; 349: g3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Centers for Disease Control and Prevention NCfHS. Underlying Cause of Death 1999-2013 on CDC WONDER Online Database: Centers for Disease Control and Prevention, National Center for Health Statistics; 2015 [updated 2016]. Data are from the Multiple Cause of Death Files, 1999-2013, as compiled from data provided by the 57 vital statistics jurisdictions through the Vital Statistics Cooperative Program, http://wonder.cdc.gov/ucd-icd10.html
- 46. Page IH. The mosaic theory of arterial hypertension—its interpretation. Perspect Biol Med 1967; 10: 325–333. [DOI] [PubMed] [Google Scholar]
- 47. Groarke JD, Tanguturi VK, Hainer J, et al. Abnormal exercise response in long-term survivors of Hodgkin lymphoma treated with thoracic irradiation: evidence of cardiac autonomic dysfunction and impact on outcomes. J Am Coll Cardiol 2015; 65: 573–583. [DOI] [PubMed] [Google Scholar]
- 48. Sardaro A, Petruzzelli MF, D’Errico MP, et al. Radiation-induced cardiac damage in early left breast cancer patients: risk factors, biological mechanisms, radiobiology, and dosimetric constraints. Radiother Oncol 2012; 103: 133–142. [DOI] [PubMed] [Google Scholar]
- 49. Moslehi JJ. Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med 2016; 375: 1457–1467. [DOI] [PubMed] [Google Scholar]
- 50. Tiersten A, Wo J, Jacobson C, et al. Cardiac toxicity observed in association with high-dose cyclophosphamide-based chemotherapy for metastatic breast cancer. Breast 2004; 13: 341–346. [DOI] [PubMed] [Google Scholar]
- 51. Mythili Y, Sudharsan PT, Sudhahar V, et al. Protective effect of DL-alpha-lipoic acid on cyclophosphamide induced hyperlipidemic cardiomyopathy. Eur J Pharmacol 2006; 543: 92–96. [DOI] [PubMed] [Google Scholar]
- 52. Nagi MN, Al-Shabanah OA, Hafez MM, et al. Thymoquinone supplementation attenuates cyclophosphamide-induced cardiotoxicity in rats. J Biochem Mol Toxicol 2011; 25: 135–142. [DOI] [PubMed] [Google Scholar]
- 53. Nuver J, De Haas EC, Van Zweeden M, et al. Vascular damage in testicular cancer patients: a study on endothelial activation by bleomycin and cisplatin in vitro. Oncol Rep 2010; 23: 247–253. [PubMed] [Google Scholar]
- 54. Van Cutsem E, Hoff PM, Blum JL, et al. Incidence of cardiotoxicity with the oral fluoropyrimidine capecitabine is typical of that reported with 5-fluorouracil. Ann Oncol 2002; 13: 484–485. [DOI] [PubMed] [Google Scholar]
- 55. Labianca R, Beretta G, Clerici M, et al. Cardiac toxicity of 5-fluorouracil: a study on 1083 patients. Tumori 1982; 68: 505–510. [DOI] [PubMed] [Google Scholar]
- 56. Polk A, Vistisen K, Vaage-Nilsen M, et al. A systematic review of the pathophysiology of 5-fluorouracil-induced cardiotoxicity. BMC Pharmacol Toxicol 2014; 15: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bowles EJ, Wellman R, Feigelson HS, et al. Risk of heart failure in breast cancer patients after anthracycline and trastuzumab treatment: a retrospective cohort study. J Natl Cancer Inst 2012; 104: 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 2003; 97: 2869–2879. [DOI] [PubMed] [Google Scholar]
- 59. Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 1979; 91: 710–717. [DOI] [PubMed] [Google Scholar]
- 60. Schwarzer S, Eber B, Greinix H, et al. Non-Q-wave myocardial infarction associated with bleomycin and etoposide chemotherapy. Eur Heart J 1991; 12: 748–750. [PubMed] [Google Scholar]
- 61. Durkin WJ, Pugh RP, Solomon J, et al. Treatment of advanced lymphomas with bleomycin (NSC-125066). Oncology 1976; 33: 140–145. [DOI] [PubMed] [Google Scholar]
- 62. Menna P, Salvatorelli E, Minotti G. Anthracycline degradation in cardiomyocytes: a journey to oxidative survival. Chem Res Toxicol 2010; 23: 6–10. [DOI] [PubMed] [Google Scholar]
- 63. Octavia Y, Tocchetti CG, Gabrielson KL, et al. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 2012; 52: 1213–1225. [DOI] [PubMed] [Google Scholar]
- 64. Lyu YL, Kerrigan JE, Lin CP, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 2007; 67: 8839–8846. [DOI] [PubMed] [Google Scholar]
- 65. Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012; 18: 1639–1642. [DOI] [PubMed] [Google Scholar]
- 66. Miya T, Fujikawa R, Fukushima J, et al. Bradycardia induced by irinotecan: a case report. Jpn J Clin Oncol 1998; 28: 709–711. [DOI] [PubMed] [Google Scholar]
- 67. Ozkan HA, Bal C, Gulbas Z. Assessment and comparison of acute cardiac toxicity during high-dose cyclophosphamide and high-dose etoposide stem cell mobilization regimens with N-terminal pro-B-type natriuretic peptide. Transfus Apher Sci 2014; 50: 46–52. [DOI] [PubMed] [Google Scholar]
- 68. Cubbon RM, Lyon AR. Cardio-oncology: concepts and practice. Indian Heart J 2016; 68(Suppl. 1): S77–S85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Subar M, Muggia FM. Apparent myocardial ischemia associated with vinblastine administration. Cancer Treat Rep 1986; 70: 690–691. [PubMed] [Google Scholar]
- 70. Rowinsky EK, Eisenhauer EA, Chaudhry V, et al. Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol 1993; 20(4 Suppl. 3): 1–15. [PubMed] [Google Scholar]
- 71. Howarth FC, Calaghan SC, Boyett MR, et al. Effect of the microtubule polymerizing agent taxol on contraction, Ca2+ transient and L-type Ca2+ current in rat ventricular myocytes. J Physiol 1999; 516: 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moslehi JJ, Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol 2015; 33: 4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Duran JM, Makarewich CA, Trappanese D, et al. Sorafenib cardiotoxicity increases mortality after myocardial infarction. Circ Res 2014; 114: 1700–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kerkela R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 2006; 12: 908–916. [DOI] [PubMed] [Google Scholar]
- 75. Choueiri TK, Mayer EL, Je Y, et al. Congestive heart failure risk in patients with breast cancer treated with bevacizumab. J Clin Oncol 2011; 29: 632–638. [DOI] [PubMed] [Google Scholar]
- 76. Ran HH, Zhang R, Lu XC, et al. Imatinib-induced decompensated heart failure in an elderly patient with chronic myeloid leukemia: case report and literature review. J Geriatr Cardiol 2012; 9: 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Atallah E, Durand JB, Kantarjian H, et al. Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood 2007; 110: 1233–1237. [DOI] [PubMed] [Google Scholar]
- 78. Bair SM, Choueiri TK, Moslehi J. Cardiovascular complications associated with novel angiogenesis inhibitors: emerging evidence and evolving perspectives. Trends Cardiovasc Med 2013; 23: 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lankhorst S, Saleh L, Danser AJ, et al. Etiology of angiogenesis inhibition-related hypertension. Curr Opin Pharmacol 2015; 21: 7–13. [DOI] [PubMed] [Google Scholar]
- 80. Semeniuk-Wojtas A, Lubas A, Stec R, et al. Influence of tyrosine kinase inhibitors on hypertension and nephrotoxicity in metastatic renal cell cancer patients. Int J Mol Sci 2016; 17: 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007; 370: 2011–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol 2009; 53: 2231–2247. [DOI] [PubMed] [Google Scholar]
- 83. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016; 16: 131–144. [DOI] [PubMed] [Google Scholar]
- 84. Sonnenblick M, Rosin A. Cardiotoxicity of interferon. A review of 44 cases. Chest 1991; 99: 557–561. [DOI] [PubMed] [Google Scholar]
- 85. Ewer MS, Lippman SM. Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J Clin Oncol 2005; 23: 2900–2902. [DOI] [PubMed] [Google Scholar]
- 86. Lenneman CG, Abdallah WM, Smith HM, et al. Sympathetic nervous system alterations with HER2+ antagonism: an early marker of cardiac dysfunction with breast cancer treatment? Ecancermedicalscience. 2014; 8: 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hedhli N, Huang Q, Kalinowski A, et al. Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation 2011; 123: 2254–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pentassuglia L, Sawyer DB. The role of Neuregulin-1beta/ErbB signaling in the heart. Exp Cell Res 2009; 315: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Scappaticci FA, Skillings JR, Holden SN, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst 2007; 99: 1232–1239. [DOI] [PubMed] [Google Scholar]
- 90. Advani RH, Hong F, Horning SJ, et al. Cardiac toxicity associated with bevacizumab (Avastin) in combination with CHOP chemotherapy for peripheral T cell lymphoma in ECOG 2404 trial. Leuk Lymphoma 2012; 53: 718–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Russell NS, Hoving S, Heeneman S, et al. Novel insights into pathological changes in muscular arteries of radiotherapy patients. Radiother Oncol 2009; 92: 477–483. [DOI] [PubMed] [Google Scholar]
- 92. Fajardo LF. Is the pathology of radiation injury different in small vs large blood vessels? Cardiovasc Radiat Med 1999; 1: 108–110. [DOI] [PubMed] [Google Scholar]
- 93. Jaworski C, Mariani JA, Wheeler G, et al. Cardiac complications of thoracic irradiation. J Am Coll Cardiol 2013; 61: 2319–2328. [DOI] [PubMed] [Google Scholar]
- 94. McGale P, Darby SC, Hall P, et al. Incidence of heart disease in 35,000 women treated with radiotherapy for breast cancer in Denmark and Sweden. Radiother Oncol 2011; 100: 167–175. [DOI] [PubMed] [Google Scholar]
- 95. Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood 2011; 117: 412–418. [DOI] [PubMed] [Google Scholar]
- 96. Souza V, Silva E, Ribeiro M, et al. Hypertension in patients with cancer. Arq Bras Cardiol 2015; 104: 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990; 82: 4–6. [DOI] [PubMed] [Google Scholar]
- 98. Dome B, Hendrix MJ, Paku S, et al. Alternative vascularization mechanisms in cancer: athology and therapeutic implications. Am J Pathol 2007; 170: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hoeben A, Landuyt B, Highley MS, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 2004; 56: 549–580. [DOI] [PubMed] [Google Scholar]
- 100. Hall PS, Harshman LC, Srinivas S, et al. The frequency and severity of cardiovascular toxicity from targeted therapy in advanced renal cell carcinoma patients. JACC Heart Fail 2013; 1: 72–78. [DOI] [PubMed] [Google Scholar]
- 101. Richards CJ, Je Y, Schutz FA, et al. Incidence and risk of congestive heart failure in patients with renal and nonrenal cell carcinoma treated with sunitinib. J Clin Oncol 2011; 29: 3450–3456. [DOI] [PubMed] [Google Scholar]
- 102. Khakoo AY, Kassiotis CM, Tannir N, et al. Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer 2008; 112: 2500–2508. [DOI] [PubMed] [Google Scholar]
- 103. Steingart RM, Bakris GL, Chen HX, et al. Management of cardiac toxicity in patients receiving vascular endothelial growth factor signaling pathway inhibitors. Am Heart J 2012; 163: 156–163. [DOI] [PubMed] [Google Scholar]
- 104. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med 2007; 357: 39–51. [DOI] [PubMed] [Google Scholar]
- 105. Ewer MS, Vooletich MT, Durand JB, et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J Clin Oncol 2005; 23: 7820–7826. [DOI] [PubMed] [Google Scholar]
- 106. Khouri MG, Douglas PS, Mackey JR, et al. Cancer therapy-induced cardiac toxicity in early breast cancer: addressing the unresolved issues. Circulation 2012; 126: 2749–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sysa-Shah P, Tocchetti CG, Gupta M, et al. Bidirectional cross-regulation between ErbB2 and beta-adrenergic signalling pathways. Cardiovasc Res 2016; 109: 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs 1997; 54(Suppl. 4): 1–7. [DOI] [PubMed] [Google Scholar]
- 109. Steinherz LJ, Steinherz PG, Tan CT, et al. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 1991; 266: 1672–1677. [PubMed] [Google Scholar]
- 110. Cardinale D, Colombo A, Lamantia G, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol 2010; 55: 213–220. [DOI] [PubMed] [Google Scholar]
- 111. Shan K, Lincoff AM, Young JB. Anthracycline-induced cardiotoxicity. Ann Intern Med 1996; 125: 47–58. [DOI] [PubMed] [Google Scholar]
- 112. Horenstein MS, Vander Heide RS, L’Ecuyer TJ. Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol Genet Metab 2000; 71: 436–444. [DOI] [PubMed] [Google Scholar]
- 113. Geisberg CA, Abdallah WM, da Silva M, et al. Circulating neuregulin during the transition from stage A to stage B/C heart failure in a breast cancer cohort. J Card Fail 2013; 19: 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vejpongsa P, Yeh ET. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol 2014; 64: 938–945. [DOI] [PubMed] [Google Scholar]
- 115. McCaffrey TA, Tziros C, Lewis J, et al. Genomic profiling reveals the potential role of TCL1A and MDR1 deficiency in chemotherapy-induced cardiotoxicity. Int J Biol Sci 2013; 9: 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. De Angelis A, Piegari E, Cappetta D, et al. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation 2010; 121: 276–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Haugnes HS, Wethal T, Aass N, et al. Cardiovascular risk factors and morbidity in long-term survivors of testicular cancer: a 20-year follow-up study. J Clin Oncol 2010; 28: 4649–4657. [DOI] [PubMed] [Google Scholar]
- 118. Gottdiener JS, Appelbaum FR, Ferrans VJ, et al. Cardiotoxicity associated with high-dose cyclophosphamide therapy. Arch Intern Med 1981; 141: 758–763. [PubMed] [Google Scholar]
- 119. Quezado ZM, Wilson WH, Cunnion RE, et al. High-dose ifosfamide is associated with severe, reversible cardiac dysfunction. Ann Intern Med 1993; 118: 31–36. [DOI] [PubMed] [Google Scholar]
- 120. Vaughn DJ, Palmer SC, Carver JR, et al. Cardiovascular risk in long-term survivors of testicular cancer. Cancer 2008; 112: 1949–1953. [DOI] [PubMed] [Google Scholar]
- 121. Brouwers EE, Huitema AD, Beijnen JH. Long-term platinum retention after treatment with cisplatin and oxaliplatin. BMC Clin Pharmacol 2008; 8: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chitaley K, Webb RC. Microtubule depolymerization facilitates contraction of rat aorta via activation of Rho-kinase. Vascul Pharmacol 2002; 38: 157–161. [DOI] [PubMed] [Google Scholar]
- 123. de Forni M, Malet-Martino MC, Jaillais P, et al. Cardiotoxicity of high-dose continuous infusion fluorouracil: a prospective clinical study. J Clin Oncol 1992; 10: 1795–1801. [DOI] [PubMed] [Google Scholar]
- 124. Desvarieux M, Demmer RT, Jacobs DR, Jr., et al. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens 2010; 28:1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Plummer M, de Martel C, Vignat J, et al. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health 2016; 4: e609–e616. [DOI] [PubMed] [Google Scholar]
- 126. Liston A, Masters SL. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 2017; 17: 208–214. [DOI] [PubMed] [Google Scholar]
- 127. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol 2015; 6: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Speer T, Rohrer L, Blyszczuk P, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013; 18; 38: 754–768. [DOI] [PubMed] [Google Scholar]
- 129. Tracy EC, Bowman MJ, Henderson BW, et al. Interleukin-1alpha is the major alarmin of lung epithelial cells released during photodynamic therapy to induce inflammatory mediators in fibroblasts. Br J Cancer 2012; 107: 1534–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Johnston JB. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk Lymphoma 2011; 52(Suppl. 2): 43–45. [DOI] [PubMed] [Google Scholar]
- 131. Garg AD, Krysko DV, Verfaillie T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31: 1062–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Michaud M, Martins I, Sukkurwala AQ, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334: 1573–1577. [DOI] [PubMed] [Google Scholar]
- 133. Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15: 1170–1178. [DOI] [PubMed] [Google Scholar]
- 134. Ma Y, Adjemian S, Mattarollo SR, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013; 38: 729–741. [DOI] [PubMed] [Google Scholar]
- 135. Eike LM, Mauseth B, Camilio KA, et al. The cytolytic amphipathic beta(2,2)-amino acid LTX-401 induces DAMP release in melanoma cells and causes complete regression of B16 melanoma. PLoS One 2016; 11: e0148980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461: 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Garg AD, Krysko DV, Vandenabeele P, et al. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother 2012; 61: 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61. [DOI] [PubMed] [Google Scholar]
- 139. Andocs G, Meggyeshazi N, Balogh L, et al. Upregulation of heat shock proteins and the promotion of damage-associated molecular pattern signals in a colorectal cancer model by modulated electrohyperthermia. Cell Stress Chaperones 2015; 20: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Yamamura Y, Tsuchikawa T, Miyauchi K, et al. The key role of calreticulin in immunomodulation induced by chemotherapeutic agents. Int J Clin Oncol 2015; 20: 386–394. [DOI] [PubMed] [Google Scholar]
- 141. Korbelik M, Sun J, Cecic I. Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res 2005; 65: 1018–1026. [PubMed] [Google Scholar]
- 142. Fang H, Ang B, Xu X, et al. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol Immunol 2014; 11: 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Brusa D, Migliore E, Garetto S, et al. Immunogenicity of 56 degrees C and UVC-treated prostate cancer is associated with release of HSP70 and HMGB1 from necrotic cells. Prostate 2009; 69: 1343–1352. [DOI] [PubMed] [Google Scholar]
- 144. Spisek R, Charalambous A, Mazumder A, et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 2007; 109: 4839–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Luo Y, Chihara Y, Fujimoto K, et al. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur J Cancer 2013; 49: 741–751. [DOI] [PubMed] [Google Scholar]
- 146. Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 2007; 220: 47–59. [DOI] [PubMed] [Google Scholar]
- 147. Zhou J, Chen X, Gilvary DL, et al. HMGB1 induction of clusterin creates a chemoresistant niche in human prostate tumor cells. Sci Rep 2015; 5: 15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13w: 1050–1059. [DOI] [PubMed] [Google Scholar]
- 149. Hermani A, De Servi B, Medunjanin S, et al. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res 2006; 312 : 184–197. [DOI] [PubMed] [Google Scholar]
- 150. Hu DE, Moore AM, Thomsen LL, et al. Uric acid promotes tumor immune rejection. Cancer Res 2004; 64: 5059–5062. [DOI] [PubMed] [Google Scholar]
- 151. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008; 8: 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Miyake Y, Yamasaki S. Sensing necrotic cells. Adv Exp Med Biol 2012; 738: 144–152. [DOI] [PubMed] [Google Scholar]
- 154. Davidovich P, Kearney CJ, Martin SJ. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol Chem 2014; 395: 1163–1171. [DOI] [PubMed] [Google Scholar]
- 155. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 2013; 38: 209–223. [DOI] [PubMed] [Google Scholar]
- 156. Garg AD, Martin S, Golab J, et al. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ 2014; 21: 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 2016; 35: 5931–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Krysko DV, Garg AD, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12: 860–875. [DOI] [PubMed] [Google Scholar]
- 159. Krysko O, Love Aaes T, Bachert C, et al. Many faces of DAMPs in cancer therapy. Cell Death Dis 2013; 4: e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bettum IJ, Vasiliauskaite K, Nygaard V, et al. Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Lett 2014; 344: 28–39. [DOI] [PubMed] [Google Scholar]
- 161. Ghavami S, Rashedi I, Dattilo BM, et al. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol 2008; 83: 1484–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. He Y, Zha J, Wang Y, et al. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res 2013; 73: 629–639. [DOI] [PubMed] [Google Scholar]
- 163. Ichikawa M, Williams R, Wang L, et al. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res 2011; 9: 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Jovanovic IP, Pejnovic NN, Radosavljevic GD, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer 2014; 134: 1669–1682. [DOI] [PubMed] [Google Scholar]
- 165. Liu X, Zhu L, Lu X, et al. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem Biophys Res Commun 2014; 453: 486–492. [DOI] [PubMed] [Google Scholar]
- 166. Maywald RL, Doerner SK, Pastorelli L, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA 2015; 112: E2487–E2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Sack U, Walther W, Scudiero D, et al. Novel effect of antihelminthic niclosamide on S100A4-mediated metastatic progression in colon cancer. J Natl Cancer Inst 2011; 103: 1018–1036. [DOI] [PubMed] [Google Scholar]
- 168. McCarthy CG, Goulopoulou S, Wenceslau CF, et al. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 2014; 15; 306: H184–H196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Heijink IH, Pouwels SD, Leijendekker C, et al. Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol 2015; 52: 554–562. [DOI] [PubMed] [Google Scholar]
- 170. Foerch C, Wunderlich MT, Dvorak F, et al. Elevated serum S100B levels indicate a higher risk of hemorrhagic transformation after thrombolytic therapy in acute stroke. Stroke 2007; 38: 2491–2495. [DOI] [PubMed] [Google Scholar]
- 171. Land WG. The role of damage-associated molecular patterns (DAMPs) in human diseases: part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ Med J 2015;15(2):e157– e170. [PMC free article] [PubMed] [Google Scholar]
- 172. Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med 2007; 4: 444–454. [DOI] [PubMed] [Google Scholar]
- 173. Montesinos MC, Shaw JP, Yee H, et al. Adenosine A(2A) receptor activation promotes wound neovascularization by stimulating angiogenesis and vasculogenesis. Am J Pathol 2004; 164: 1887–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lin YC, Chang YM, Yu JM, et al. Toll-like receptor 4 gene C119A but not Asp299Gly polymorphism is associated with ischemic stroke among ethnic Chinese in Taiwan. Atherosclerosis. 2005; 180: 305–309. [DOI] [PubMed] [Google Scholar]
- 175. Thomas JA, Haudek SB, Koroglu T, et al. IRAK1 deletion disrupts cardiac Toll/IL-1 signaling and protects against contractile dysfunction. Am J Physiol Heart Circ Physiol 2003; 285: H597–H606. [DOI] [PubMed] [Google Scholar]
- 176. Ito T, Kawahara K, Nakamura T, et al. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost 2007; 5: 109–116. [DOI] [PubMed] [Google Scholar]
- 177. Goulopoulou S, Matsumoto T, Bomfim GF, et al. Toll-like receptor 9 activation: a novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin Sci 2012; 123: 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Riad A, Bien S, Gratz M, et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur J Heart Fail 2008; 10: 233–243. [DOI] [PubMed] [Google Scholar]
- 179. Nozaki N, Shishido T, Takeishi Y, et al. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation 2004; 110: 2869–2874. [DOI] [PubMed] [Google Scholar]
- 180. Hosseini H, Li Y, Kanellakis P, et al. Toll-like receptor (TLR)4 and MyD88 are essential for atheroprotection by peritoneal B1a B Cells. J Am Heart Assoc 2016; 5:e002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Prandoni P. Antithrombotic strategies in patients with cancer. Thromb Haemost 1997; 78: 141–144. [PubMed] [Google Scholar]
- 182. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 2013; 13: 34–45. [DOI] [PubMed] [Google Scholar]
- 183. Jones DP, True HD, Patel J. Leukocyte trafficking in cardiovascular disease: insights from experimental models. Mediators Inflamm 2017; 2017 DOI: 10.1155/2017/9746169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007; 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Gonzalez-Reyes S, Marin L, Gonzalez L, et al. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer 2010; 10: 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Goulopoulou S, McCarthy CG, Webb RC. Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol Rev 2016; 68: 142–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Kilickap S, Barista I, Akgul E, et al. cTnT can be a useful marker for early detection of anthracycline cardiotoxicity. Ann Oncol 2005; 16: 798–804. [DOI] [PubMed] [Google Scholar]
- 188. Wollert KC, Kempf T, Wallentin L. Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin Chem 2017; 63: 140–151. [DOI] [PubMed] [Google Scholar]
- 189. Cardinale D, Sandri MT, Colombo A, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 2004; 109: 2749–2754. [DOI] [PubMed] [Google Scholar]
- 190. Hurwitz HI, Douglas PS, Middleton JP, et al. Analysis of early hypertension and clinical outcome with bevacizumab: results from seven phase III studies. Oncologist 2013; 18: 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Nakaya A, Kurata T, Yokoi T, et al. Retrospective analysis of bevacizumab-induced hypertension and clinical outcome in patients with colorectal cancer and lung cancer. Cancer Med 2016; 5: 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]