

Abstract
Regulatory T cells mediated suppression serves pivotal mechanism of negative-regulation of immune mediated inflammation. Type 1 regulatory T cells (Tr1 cells) are an important subset of CD4+ T cells that help to control excessive inflammatory responses and maintain tolerance. One way in which Tr1 cells mediate an anti-inflammatory role is via their production of interleukin 10 (IL-10), which dampens the function of both antigen-presenting cells and antigen-specific effector T cells. Additionally, Tr1 cells are capable of killing effector and myeloid cells through the Perforin-Granzyme B pathway. In vitro differentiated Tr1 cells suppress development of autoimmune tissue inflammation upon adoptive transfer in vivo. Here we describe the in vitro differentiation of naïve murine CD4+ T cells to IL-10 producing Tr1 cells by using IL-27 stimulation.
Keywords: T cell differentiation, CD4, IL-27, Tr1, IL-10
INTRODUCTION
Tr1 cells are a class of regulatory T cells that predominantly produce IL-10 and have been found to be primarily associated with suppressing tissue inflammation in autoimmunity and graft vs. host disease. IL-10, a potent anti-inflammatory cytokine, suppresses T cell response especially Th1 cell mediated responses. Tr1 cells secrete minimal amounts of IL-4 and downregulate GATA3 expression, which distinguish them from Th2 cells. They also lack IL-17 and express lower levels of RORC, which distinguish them from Th17. Furthermore, unlike CD25+Foxp3+ regulatory T cells, Tr1 have lower IL-2 secretion, and high levels of IFN-γ secretion. Tr1 cells may transiently express FOXP3 upon activation, but it is not expressed persistently and never reaches the high levels characteristic of Tregs. Thus, Tr1 cells can suppress inflammation and produce IL-10, but unlike Tregs they do not require constitutive expression of FoxP3 for their generation and immunosuppressive functions.
We (Awasthi, et al., 2007) and others (Fitzgerald, et al., 2007, Stumhofer, et al., 2007) have demonstrated that IL-27, a heterodimeric cytokine of IL-12 family, either alone or in combination with TGF-β induced the differentiation of Tr1 cells. Similar to its other family members, IL-27 is produced by cells of the innate immune system. Our preliminary data suggests that IL-27 is produced from innate immune cells (macrophages, DCs and tissue resident macrophages). IL-27 consists of two subunits, p28 and the Epstein-Barr virus-induced gene 3 (EBI3) product. When given in vivo, IL-27 suppresses development of effector T cells and inhibits development of autoimmunity. The IL-27 receptor is a heterodimer that is composed of a specific IL-27R subunit, also called WSX-1, and the gp130 subunit that is shared with the IL-6 receptor family. IL-27 was initially described to enhance initial Th1 commitment but accumulating data suggests that IL-27 has dominant anti-inflammatory properties. Thus, analysis of IL-27R deficient mice revealed increased inflammation in an infectious disease model and increased neuroinflammation in experimental autoimmune encephalomyelitis (EAE). Lack of IL-27 signaling resulted in an increased Th1/Th17 response that could account for enhanced tissue inflammation. Indeed, we reported that IL-27 together with TGF-β can induce IL-10 producing regulatory T cells that have Tr1-like properties. IL-27 alone can also induce IL-10-producing Tr1 cells but in the absence of TGF-β, the cells coproduce large quantities of IFN-γ with IL-10. Consistent with this observation, IL-27R (WSX-1-/-) deficient mice have a defect in generating IL-10 producing Tr-1 cell. Thus, IL-27 might also be necessary to control exaggerated immunopathology partly by regulating induction of Tr1 cells. We have developed a protocol that allows Tr1 cell differentiation in vitro with IL-27, which is detailed here.
<Note> All experiments must be approved by the appropriate institutional and or national review boards.
BASIC PROTOCOL 1: Generation of IL-10 producing CD4 T cells (Tr1 cells) in vitro
In this protocol, murine spleen and lymph node cells are isolated, enriched for total CD4 T cells, and purified for CD44lowCD62Lhigh naïve CD4 T cells. After purification of naïve CD4 T cells are cultured on a plate bound anti-CD3 and anti-CD28 antibodies in the presence of recombinant IL-27 protein for 3 to 4 days. During their differentiation, Tr1 cells express 3 different waves of genes that coordinately act to induce differentiation of IL-10 producing Tr1 cells.
Materials
C57BL/6 mice (e.g., The Jackson Laboratory)
Phosphate buffered saline (pH 7.4)
Clone medium (APPENDIX 1)
Magnetic cell sorting (MACS) beads for CD4+ cell purification (CD4 (L3T4) MicroBeads, mouse; Miltenyi Biotec (#130-049-201))
MACS buffer (APPENDIX 2)
70 um cell strainer
Fluorescent conjugated anti-mouse CD44 (clone: IM7), CD62L (clone: MEL-14), and CD4 (clone: RM4-5) antibody
Anti-mouse CD3 (clone: 145-2C11) and CD28 (clone: 37.51) antibody
Recombinant IL-27 protein (e-bioscience # 14-8271-63)
Corning® Costar® 96 well flat bottom cell culture plate (Sigma (#CLS3595))
Protocol steps
#1 preparation of single cell suspension of lymphocytes from spleen and lymph nodes
Isolate spleen and lymph nodes (subiliac, axillary, mandibular, and parotid), mince cells and pass cellular suspension through 70 micron cell strainer into cold (4°C) DPBS in a sterile manner.
Optional: Lyse red blood cell (RBC) using ACK (Life Technologies (#A10492-01))
<Note> RBC lysis is not necessary as red blood cells will be eliminated in step #2.
#2 enrichment of CD4+ cells using Magnetic cell sorting
Count the number of live cells (e.g. trypan blue exclusion dye), spin down the cells at 1400rpm for 5 minutes at 4°C and replace DPBS with MACS buffer. Add appropriate concentrations of anti-CD4 MACS beads calculated based on the number of cells following the manufacturer’s protocol. The volumes of MACS buffer and beads are calculated based on the number of cells in the preparation (usually 1 × 108 mononuclear cells are obtained from one mouse by this procedure). Mix single cell suspension with anti-CD4 MACS beads and incubate for 10 to 15 minutes on ice before washing them by cold MACS buffer. Filter cellular suspension using 70micron cell strainer to remove cellular aggregates and debris. At this point, CD4+ T cells can be isolated using either manual MACS columns or an automated MACS separator. Wash the collected enriched CD4+ T cells once with cold DPBS and resuspend them into clone medium and keep on ice for further processing.
#3 purification of naïve CD4+ cells using FACS sorting
Stain MACS purified CD4+ T cells with anti-CD44, anti-CD62L, and anti-CD4 mouse monoclonal antibodies in clone medium for 10-15 minutes at 4°C. Use a cell sorter by making following gate as mentioned (fig.1) to isolate CD4+CD62LhiCD44lo naïve CD4+ T cells and verify the purity after sorting (the purity needs to be over 95%). Sorted naïve CD4+ T cells cells can be stored in culture medium on ice until starting their differentiation into Tr1 cells in a few hours.
Fig 1. Gating method for sorting naïve CD4 cells by FACS.
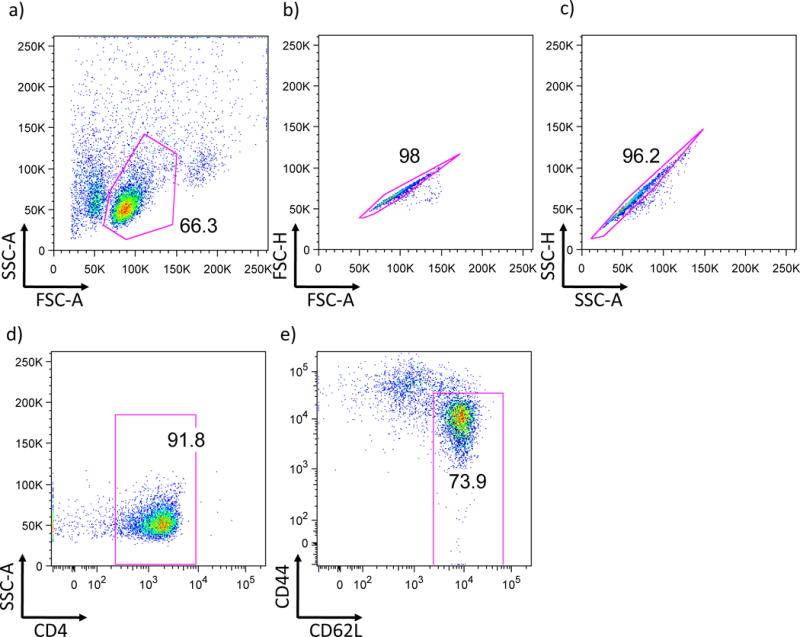
a) A flowcytometric scheme using forward scatter count (FSC) and side scatter count (SSC) to identify the cell population is shown. b and c) Excluding doublet cells by taking the population showing linear appearance in FSC/SSC-Area (A) and FSC/SSC-Height (H). d) After exclusion of doublets, single cells are further gated by CD4 expression. e) CD4+ cells are further analyzed by the expression of CD44 and CD62L.
#4 cell culture for Tr1 differentiation
Coat the cell culture grade flat bottom plate with anti-CD3 (2ug/ml) and anti-CD28 (2ug/ml) mouse monoclonal antibodies diluted in sterile PBS for 2-4 hours at 37°C or overnight at 4°C. Remove the coating buffer and add 100ul of culture medium with or without IL-27 (50 ng/ml) (Tr1 and Th0 conditions, respectively) into the plate to avoid drying the plate. When activating and differentiating naïve CD4+ T cells into 96 well plate, add 100ul of cell suspension of 1 × 106/ml of naïve CD4 T cells per well (so that the final concentration of IL-27 is 25ng/ml) to start Tr1 differentiation. Culture cells at 37°C for up to 4 days.
Differentiation of Tr1 cells can also be done in 24 and 6 well plates as per the requirements of further downstream experiments.
Optional: Tr1 differentiation can also be performed with soluble anti-CD3 (1 ug/ml) with irradiated syngenic APCs instead of plate bound system described above.
SUPPORT PROTOCOL 1: Characterization of Tr1 cells
A number of assays can be used to determine the differentiation of Tr1 cells. Tr1 cells produce regulatory cytokine IL-10 (signature cytokine that defines Tr1 cells) that induces resolution of tissue inflammation by multiple mechanisms. They also have intrinsic gene expression profile that can distinguish them from other effector and regulatory T cells. Accordingly, the methods of functional analysis of Tr1 cells, as well as of transcriptional profiling regulatory T cells is given in the protocol. Each method can be used based on the specific needs of the investigator.
Cytokine expression and production
As a mainstay of Tr1 characterization, quantitative PCR analysis, flow cytometric analysis and Enzyme-Linked ImmunoSorbent Assay (ELISA) are straightforward methods to assess expression and production of IL-10. Maximal stimulation of T cells using phorbol 12-myristate 13- acetate (PMA) and ionomycin allows to analyze IL-10 producing cells by intracellular cytokine staining. Sandwich ELISA, using anti-mouse IL-10 antibodies, is a quantitative method for analysis of IL-10 secreted by Tr1 cells.
Gene expression profiling of Tr1 cells
Tr1 cells have differential gene expression dynamics that regulate linage specific gene profile inducing IL-10. Some of the transcriptomic approaches for deeper analysis of Tr1 cells are described.
Materials
RNAeasy mini kit (Qiagen (#74134)
iScript cDNA synthesis kit (BioRad (#1708891)
TaqMan® Fast Universal PCR Master Mix (Life Technologies (#4366072)
Taqman probe IL-10 (Life Technologies (Mm00439614_m1)
PMA (Sigma (#P8139)) (stock 0.1mg/ml)
Ionomycin (Sigma (#I0634) (stock 1mg/ml)
Monensin (BD GolgiStop™ (#554724))
Corning® 96 well V bottom plate
Fixation/Permeabilization Solution Kit (BD bioscience (#554714)
Fluorescent conjugated anti-mouse IL-10 and IFN-γ antibody
Corning® 96 well EIA/RIA plate
Tween® 20 (Sigma (#P1379))
10% BSA Diluent/Blocking Solution (KPL solution: KPL (#50-61-10))
Recombinant IL-10 for ELISA standard (Biolegend (#575809)
Purified rat anti-mouse IL-10 antibody (capture antibody (BD Pharmingen (#551215)))
Biotin labeled rat anti-mouse IL-10 antibody (secondly antibody (BD Pharmingen (#554423)))
Avidin peroxidase (AP) (stock 2.9mg/ml (Sigma (#A7419))
3,3′,5,5′-Tetramethylbenzidine (TMB) buffer (KPL (#52-00-00))
sulfuric acid stop solution (H3PO4) (KPL (#50-85-04))
Protocol steps
Cytokine expression and production
#1 Expression of IL-10 mRNA in Tr1 cells
After the in vitro differentiation of Tr1 cells, determine the mRNA expression of IL-10 by Taqman PCR. Briefly, harvest the cells at 48-72 hours for preparing RNA using RNAeasy kit (Qiagen). Reversely transcribe the mRNA into cDNA by cDNA synthesis kit (BioRad). Measure the expression of IL-10 using IL-10 Taqman probe and appropriate buffer mix (Life Technologies).
#2 Intracellular cytokine staining
After in vitro differentiation of Tr1 cells for 3 to 4 days as described, collect culture medium supernatant for following ELISA analysis. Cells are then re-suspended in culture medium containing PMA (0.1ug/ml), Ionomycin (1ug/ml), and GolgiStop for 4 hours at 37°C. After the stimulation, wash the cells with PBS and transfer them into V bottom 96 well plate or FACS tubes.
Spin down cells at 1400rpm for 5 minutes and remove PBS, resuspend cells into PBS and add 7-amino-actinomycin. D (7AAD) or Propidium Iodide to identify dead cells and incubate for 5-10 minutes on ice. Wash the cells once with PBS and resuspend cells in fixation buffer (BD bioscience) and incubate for 5-10 minutes at room temperature or 20-30 minutes on ice. Next, pellet the cells by spinning down them at 1400rpm for 5minutes, remove fixation buffer and add cell permeabilization buffer (BD bioscience) premixed with antibodies against IL-10 and IFN-γ for 20-30 minutes on ice. It is recommended to continue staining procedure in dark to obtain the best results.
Once staining is complete, wash the cells once with permeabilization buffer and resuspend the washed cells pellet into PBS. Analyze the frequency of IL-10 and IFN-γ producing cells by flow cytometry (fig.2).
Fig 2. Intracellular cytokine staining analysis of Tr1 cells by FACS.
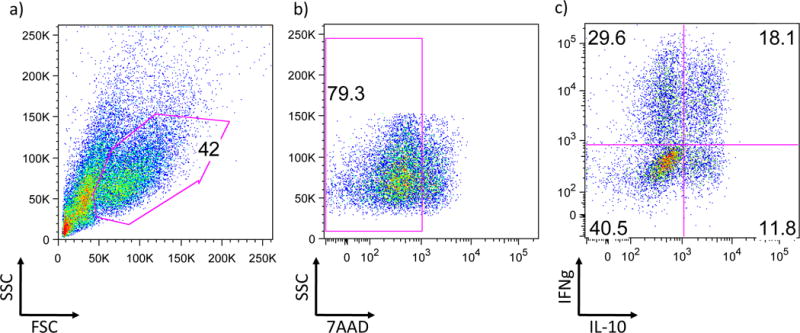
a) A flowcytometric scheme using forward scatter count (FSC) and side scatter count (SSC) to identify the cell population is shown. b) Excluding dead cells by appropriate marker (7AAD in this case). c) After dead cell exclusion live cells are further analyze by the expression of IFNg and IL-10. About 50% of IL-10 positive cells also co-express IFNg.
#3 ELISA analysis
Detection of the absolute amounts of IL-10 produced in culture medium supernatant by ELISA. Detailed protocol is shown in this manual (e.g. U11.2). Sandwich ELISA using primary and detection antibodies is good for analyzing small amount of cytokine. Enzymatic reaction by HRP and TMB stopped by sulfuric acid is capable of reading at 450nm.
Three days method is shown as follows:
- Day 1: Coat plate by capture antibodies
- Dilute capture antibodies in coating buffer (2 ug/ml of buffer (0.1M NaHPO4 pH 9.0)) and add 100ul/well in a 96 well Corning® 96 well EIA/RIA. Seal the plate and incubate overnight at 4°C.
- Day 2: Blocking and standards
- Wash plate 3 times (Washing buffer (PBS with 0.5% Tween® 20)) and block plate by adding 200ul of blocking buffer (1% BSA (10% KPL solution)). Seal plate and incubate at room temperature for 2hr.
- Prepare samples and standards (6400pg/ml as the top; standard proteins need to be stock in -80°C until use). After washing plate 3 times, add 100ul of Standards to first two wells. Add 50ul of medium to the bottom ones and make serial dilutions. Add samples by appropriate dilution and leave in 4°C fridge overnight.
- Day 3: Addition of secondary antibodies and detection of positive cells:
- Wash plate 3 times and add secondary antibodies. Dilute secondary antibodies in 10% KPL solution (1ug/ml of buffer). Add 50ul/well and shake for 1-1.5hr at room temperature.
- Add 50ul/well Avidin peroxidase (25ul AP in 25ml of 10% KPL buffer for 5 plates) and incubate at room temperature for 30-40 minutes. After washing 3 times, add 100ul of substrate solution (TMB) per well and wait for the standard color change in Row G. Add 100ul of stop solution (1M H3PO4) per well. Read plates on ELISA microplate reader using 450nm wavelength.
Gene expression profiling of Tr1 cells
As described above, we induced the differentiation of naive CD4+ T cells into Tr1 cells using IL-27, and measured transcriptional profile using microarray at 72 hours with appropriate normalization. As controls, we measured the mRNA profile for cells that were activated without the addition of differentiating cytokines (Th0), at the same time-point. This type of comparison gives us 329 genes that are unique to Tr1 differentiation and exclude all T cell activation markers. Fig3 demonstrates a selected set of differentially expressed genes that distinguish Tr1 cells from Th0 cells. A methods for performance of microarray studies using cDNA derived from purified cells are described in Current Protocols in Molecular Biology (). Gene profiling may also be performed using RNAseq technology.
Fig 3. Genome-wide expression profiles of Tr1 differentiation.
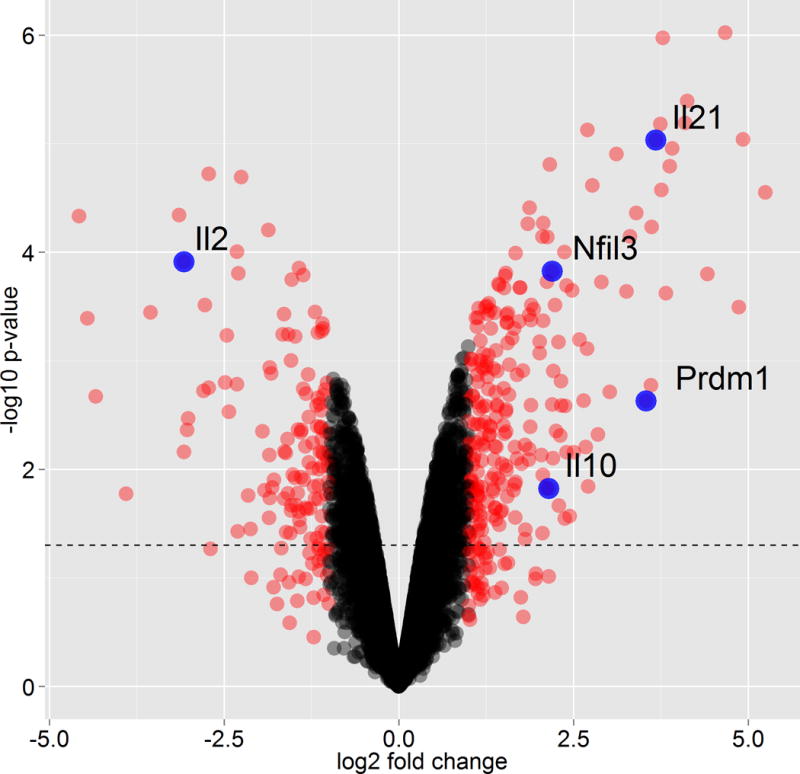
Comparison between Tr1 polarizing conditions (IL-27) and control activated Th0 cells at 72 hours time point. Shown are volcano plot for the log2 fold-change (x-axis) versus -log10 p-value (y-axis, representing the probability that the gene is differentially expressed). Black dotted line marks p-value 0.05 and red dots marks genes with fold change higher or lower than two.
REAGENTS AND SOLUTIONS
APPENDIX 1: 10% clone media
| DMEM | 500 ml |
| Sodium pyruvate | 7.5 ml (100mM) |
| L-Glutamine | 5 ml (200mM) |
| Pen/Strep | 5 ml (10,000U/ml) |
| Non-essential amino acids | 5 ml (MEM Non-Essential Amino Acids Solution (100×)) |
| Fetal bovine serum | 50 ml |
| Arginine Asparagine | 5 ml (L-Arginine 1.16g, L-Asparagine 0.36g/ 100ml w/ddH2O) |
| Folic acid | 5 ml (Folic acid 0.06g, 1N NaOH 0.5ml/ 100ml w/ddH2O) |
| Vitamins | 5 ml (MEM vitamin mix solusion (100×) |
| 2-mercaptoethanol | 2 ml (of pre-diluted stock) |
| Gentamicin | 1 ml (50mg/ml) |
2-mercaptoethanol pre-diluted stock:
13.1 μl of 14.2 M 2-mercaptoethanol into 15 ml of DPBS.
APPENDIX 2: MACS buffer
| DPBS (pH 7.4) | 500ml |
| Bovine serum albumin | 2.5g |
| 0.5M EDTA | 2ml |
COMMENTARY
Background Information
Immune tolerance is one of the critical features of the immune system, which control tissue inflammation in autoimmunity and help in maintaining homoeostasis in the immune system. Although various mechanisms are known to induce immune tolerance, regulatory T cells play dominant role in maintaining immune homeostasis primarily by T cell tolerance. In 1997, Ronocarlo and colleagues identified that chronic stimulation of T cells in the presence of IL-10 induced a distinct type of regulatory T cells called Tr1 cells, which predominantly produced IL-10 and TGFb1 (Groux, et al., 1997). Tr1 cells suppressed antigen-specific T cells responses and prevented T cells-mediated colitis. Though both Tregs and Tr1 cells produced high amounts of IL-10, they differed in their Foxp3 expression. Tr1 cells have shown to produce specific cytokine pattern (IL-10, TGF-β, IFN-γ, IL-5, and low amounts of IL-2) but without any expression of Foxp3. In addition to their specific cytokines pattern, specific surface markers and genetic signature of Tr1 cells were also identified. CD49 and lymphocyte activation gene-3 (Lag-3) are predominantly expressed on Tr1 cells and are critical for their immune-suppressive functions (Gagliani, et al., 2013). Similarly, C-Maf, Ahr and Prdm1 were identified as critical transcription factors that are required for the induction and development of Tr1 cells.
Although chronic activation of naïve CD4+ T cells in the presence of IL-10 promoted the generation of Tr1 cells the protocol did not allow expansion of large quantities of Tr1 for functional or mechanistic analysis. Because of this, additional methodologies had to be developed for the generation of Tr1 cells.
A number of protocols were developed these included using specific subsets of antigen presenting cells (APCs) to induce generation of Tr1 cells. Immature DCs (iDCs), plasmacytoid DCs (pDC) and IL-10-treated tolerogenic DCs were shown to preferentially induce the generation of Tr1 cells. In addition, immunosuppressive drugs such as vitamin D3 and dexamethasone were shown to induce the development of Tr1 cells. Each of these protocols had limitations.
Using IL-27 as an in vitro differentiation factor provided a better method by which Tr1 cells can be generated in vitro. IL-27-induced generation of Tr1 cells is faster and more efficient as compared to IL-10-induced generation of Tr1 cells. IL-27 is known to enhance proliferation of naïve T cells vigorously while IL-10 suppresses T cells proliferation. This makes IL-27 as a superior differentiation factor for Tr1 cells over IL-10 and allows generation of large quantities of Tr1 cells even starting with small number of T cells. Moreover, only one time activation is sufficient to generate Tr1 cells by IL-27 while IL-10-mediated generation of Tr1 cells requires repetitive activation of T cells. Therefore, IL-27 can generate Tr1 cells in short period of time (48-72 hours) whereas IL-10-mediated generation of Tr1 cells requires longer time (3 weeks).
Critical Parameters
To obtain the best results of T cell activation and differentiation for Th1 and Th2, T cells are generally cultured for 4-5 days or even longer to obtain effector phenotypes. However, keeping Tr1 cells longer than 4 days in culture might result in the loss of viable cell numbers and for their ability to maintain cytokine production since IL-27 limits IL-2 production (Villarino, et al., 2006) which sustain and maintain T cell survival and growth. Thus, to maintain Tr1 cells in culture for longer-term periods culturing cells with exogenous IL-2 might be worthwhile.
IL-27 is a heterodimeric cytokine, and as such, its recombinant form is especially sensitive to freeze thaw cycles, which should be strictly avoided. Furthermore, recombinant IL-27 stock should not be stored for prolonged periods of time.
Time of analysis is also critical. IL-10 mRNA is induced at 24 hours post cell stimulation and peaks at about 36 hours following activation with IL-27, but it cannot be detected by intracellular staining or in culture media until about 48 hours post stimulation. Optimal protein levels of IL-10 are detected at 72-96 hours after Tr1 polarization. Supernatants obtained at 72 hours after activation should be stored frozen at −20C for further use in ELISA.
Troubleshooting
IL-10 is usually easily detectable in the supernatants, but intracellular staining for the cytokine poses a bigger challenge. If IL-10 detection proves problematic using flow cytometry, one can try to decrease PMA concentration during cell restimulation down to 20ng/ml. Additionally, anti-CD3 concentration during Tr1 polarization can be increased up to 10ug/ml and IL-27 concentration can also be increased to 50ng/ml. Furthermore, use of IL-10 reporter mice (both IL-10-GFP and IL-10-Thy1.1) mice, which develop a very strong signal would allow to overcome this challenging step.
Anticipated Results
Typically, in vitro culture with IL-27 to generate Tr1 cells results in a population containing 15% to 30% IL-10 producing cells and about 30% IFN-γ-expressing cells (as assessed by intracellular staining). If IL-10 reporter mice are used, one should expect 40% to 60% of IL-10 positive cells after 3 days of polarization. The amount of IL-10 detected in the culture supernatants is variable and dependent on the starting cell density. Typically, if starting with 1×105 cells in a 96-well plate, one should expect the amount of IL-10 to be between 10 and 15ng/ml after 3 days in culture.
Time Considerations
Generation and analysis of Tr1 cells can be completed in 4 days. Polarized cells can be analyzed after 3 days of culture. The intracellular cytokine staining protocol usually takes about 5 hours. Detection of IL-10 by ELISA typically takes 3 days. However, the first two steps (coating plates with capture antibodies and blocking) can be prepared in parallel to save time. Time needed for RNA extraction, reverse transcription and qPCR will vary greatly depending on the number of samples. However, if handling a small number of samples, one can expect to complete it in 3 hours.
Acknowledgments
This work was supported by Grants from NIH (NS076410, AI073748, and NS045937 to V.K. Kuchroo). N. Chihara was supported by postdoctoral fellowship for research abroad from the Japan Society for the Promotion of Science. A. Awasthi was supported by intermediate fellowship from the Wellcome Trust India Alliance.
LITERATURE CITED
- Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nature immunology. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CJ, Gran B, Ciric B, Rostami A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nature immunology. 2007;8:1372–1379. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, Guo B, Herbert DR, Bulfone A, Trentini F, Di Serio C, Bacchetta R, Andreani M, Brockmann L, Gregori S, Flavell RA, Roncarolo MG. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nature medicine. 2013;19:739–746. doi: 10.1038/nm.3179. [DOI] [PubMed] [Google Scholar]
- Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O’Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nature immunology. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- Villarino AV, Stumhofer JS, Saris CJ, Kastelein RA, de Sauvage FJ, Hunter CA. IL-27 limits IL-2 production during Th1 differentiation. Journal of immunology. 2006;176:237–247. doi: 10.4049/jimmunol.176.1.237. [DOI] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]