

Abstract
Inspired by previously discovered enhanced analgesic efficacy between soluble epoxide hydrolase (sEH) and phosphodiesterase 4 (PDE4) inhibitors, we designed, synthesized and characterized 21 novel sEH/PDE4 dual inhibitors. The best of these displayed good efficacy in in vitro assays. Further pharmacokinetic studies of a subset of 4 selected compounds led to the identification of a bioavailable dual inhibitor N-(4-methoxy-2-(trifluoromethyl)benzyl)-1-propionylpiperidine-4-carboxamide (MPPA). In a lipopolysaccharide induced inflammatory pain rat model, MPPA rapidly increased in the blood (Tmax = 30 min; Cmax = 460 nM) after oral administration of 3 mg/kg and reduced inflammatory pain with rapid onset of action correlating with blood levels over a time course of 4 hours. Additionally, MPPA does not alter self-motivated exploration of rats with inflammatory pain or the withdrawal latency in control rats.
TOC image

Introduction
Multi-target ligands are designed to improve treatment of complex diseases1. Target combinations are selected to improve the therapeutic impact on one or several physiological disorders. For example, phosphodiesterase 4 (PDE4) is the major enzyme degrading cAMP into 5-AMP and thus, PDE4 inhibition increases cAMP levels which subsequently leads to a down-regulation of multiple inflammatory mediators.2 PDE4 is therefore a valuable target in the treatment of inflammatory diseases such as psoriasis, psoriatic arthritis (PsA),2 chronic obstructive pulmonary diseases (COPD)3 and asthma.4 Additionally, cAMP levels in neurons are shown to be correlated with symptoms of neuronal disorders such as depression,5 schizophrenia6 and Alzheimer’s disease.7 Nevertheless, the historical challenge of targeting PDE4 for therapy is the limited therapeutic index of PDE4 inhibitors due to vasculitis and severe dose limiting side effects such as headache, gastric hyper secretion emesis, and nausea.3,8–10 Separately from PDE4, soluble epoxide hydrolase (sEH) inhibition has beneficial physiological effects in animal models of inflammation,11 pain,12 hypertension13 and depression.14 The sEH metabolizes endogenous cytochrome P450s (CYP450) derived epoxy-fatty acids (EpFAs) such as epoxyeicosatrienoic acids (EETs) from arachidonic acid.13 Inhibition of sEH elevates EpFA levels, which have anti-hypertensive, anti-inflammatory, and analgesic properties.6
In a previous study sEH inhibitors and PDE4 inhibitors were shown separately to increase plasma levels of natural anti-inflammatory analgesic epoxy fatty acids. The same study showed that co-administration of PDE4 and sEH inhibitors resulted in an enhanced analgesic effect compared to the individual treatments.15 This enhanced analgesia of the combination motivated us to develop a bioavailable sEH/PDE4 dual inhibitor. The development of poly-pharmacological agents requires the combination of at least two pharmacophores in one molecule. To achieve this goal we followed a medicinal chemistry driven approach,16–18 where the essential structural moieties of known ligands were identified and combined in one novel molecular design, which subsequently was evaluated and re-designed in an iterative process. The pharmacokinetics (PK) and biological properties of the best compounds were evaluated in an animal model. In the future these tools will enable us to investigate the benefits of the dual ligand strategy in analgesia.3,8–10
Results
Dual ligand design and structural development
The design of dual modulators requires a combination of the pharmacophores from two inhibitors into one novel molecule. The choice of template inhibitors is essential, and was based on potency, water solubility, bioavailability and accessibility of space surrounding the inhibitors within the target binding-pocket. The initial dual inhibitor design was inspired by the sEH inhibitor GSK 2256294 and the PDE4 inhibitor Rolipram (Scheme 1). GSK 2256294 was chosen due to its subnanomolar potency on recombinant human and rat sEH and its successful phase I clinical trial.19,20 Rolipram was selected based on its high in vivo efficacy against PDE4, and its previous usage in combination with a sEH inhibitor against pain.15 The efficacy of PDE4 inhibitors, such as in Roflumilast, that have more bulky functional groups in place of a pyrrolidone, as well as cocrystal structures of PDE4 with the dialkoxyphenyl family of inhibitors21 suggest that the PDE4 active site is spacious enough to accommodate larger substituents on the dialkoxy phenyl ring of Rolipram. Therefore, the 2-, 4-substituted benzyl amide moiety was abstracted from GSK 2256294 and combined with the 3-(cyclopentyloxy)-4-methoxybenzene moiety from Rolipram, leading to the 1st dual inhibitor design (Scheme 1). Within this initial dual inhibitor design, the 2-trifluoromethyl-4-methoxy substitution of the benzyl amide (R1, R2 in Scheme 1) was evaluated by replacement. Interestingly, the initial design has a core structure similar to Apremilast, an FDA approved PDE4 inhibitor (PDE4I) (in red, Scheme 1). This similarity inspired the second dual inhibitor design, in which the 3-(cyclopentyloxy)-4-methoxy substitution of the central benzene moiety was evaluated by replacement. After discarding the 3-(cyclopentyloxy)-4-methoxy benzene substitution, the derivatives maintained PDE4 inhibition. This finding led to the assumption that the 4-methoxy benzyl amide moiety might be sufficient as a PDE4I pharmacophore. Therefore, structural fragments were introduced in the design improving the pharmacokinetic properties of the dual inhibitors. TPPU is an in house potent sEHI with good bioavailability.22 To increase structural diversity and improve bioavailability the 1-(piperidin-1-yl)propan-1-one fragment of TPPU was combined with the 2-trifluoro-4-methoxy benzyl amide moiety of the previous design, creating the third and final dual inhibitor design. Several N-substitutions of the piperidine fragment were evaluated within this structural class.
Scheme 1.

Development of sEH/PDE4 dual inhibitor design.
Synthesis
Scheme 2 shows the synthetic pathways toward sEH/PDE4 dual inhibitors. A central method was the amide synthesis. In general a benzoic acid derivative or a piperidine carboxylic acid derivative was activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and a catalytic amount of 4-dimethylaminopyridine under dry conditions, followed by the addition of a benzylamine derivative to produce compounds 1-11, 14 and 19-21.16 Benzoic acids used for the synthesis of compounds 14 and 19-20 were prepared as described below. To introduce the 2-diflurormethoxy substitution, methyl 4-hydroxy benzoate and sodium chloro difluoro acetate were reacted under basic conditions and gave, after decarboxylation, the 4-(difluoromethoxy)benzoic acid methyl ester (12).23 After ester hydrolysis the substituted benzoic acid (13) was used for the final amide synthesis step. For the preparation of the 3-(cyclopentyloxy)benzoic acid (17) and 3-(cyclopentyloxy)-4-fluorobenzoic acid (18), 4-fluoro substituted and non-substituted 3-hydroxy methyl benzoates were reacted with cyclopentyl bromide under basic conditions in presence of a catalytic amount of potassium iodide to generate the cyclopentyloxy substituted intermediates 15 and 16.4 Again after ester hydrolysis the benzoic acid derivatives 17 and 18 were used for the final amide coupling step. The synthesis of the piperidine containing inhibitors started with preparation of the intermediate 21. Deprotection of the amine was performed with trifluoroacetic acid, delivering 22.24 Two methods were used for the following final synthetic step, each depending on the required substitution. To introduce the amide substitution, appropriate acid chlorides were purchased or generated with oxalylchloride and further reacted with 22 under dry basic conditions to produce 23-25.25 To synthesize the ethyl and propyl substituted piperidine derivatives (26, 27), 22 was reacted with ethyl iodide or propyl iodide under basic conditions.
Scheme 2.
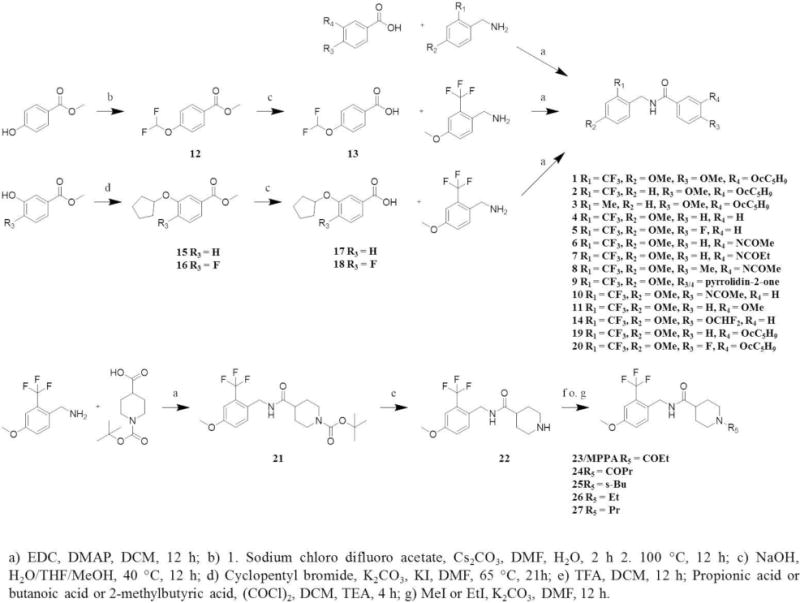
Synthetic routes for the production of sEH/PDE4 dual inhibitors
Inhibition of sEH and PDE4 activities
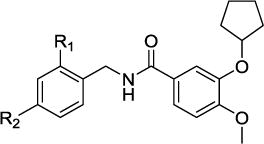
The in vitro profile of the initial dual inhibitor 1 showed an IC50 on the human sEH at a subnanomolar concentration (0.6 ± 0.1 nM), a cAMP increase of more than 200% compared to Rolipram at 1 μM and a moderate water solubility of 37.5 μM. Replacing the 2-trifluoromethyl-4-methoxy benzyl substitution by simpler 2-trifluoromethyl and 2-methyl benzyl substituents resulted in a significant drop in potency on both targets, while the moderate water solubility stayed constant.
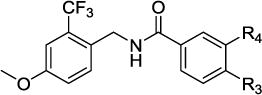
Within the second structural class (Group 2) of dual sEH/PDE4 inhibitors the substitution pattern at the central benzene moiety was investigated. The first compound of this class, 4, contained a non-substituted central benzene fragment, resulting in a slight decrease of potency on both targets. Nevertheless, the sEH IC50 was in the low one digit nanomolar range and the cAMP increase was 160 ± 12% of Rolipram at 1 μM, while the water solubility improved up to 100 μM. Introduction of the fluorine substitution at position 4 of the benzene ring (5) only affected the PDE4 potency by limiting the cAMP increase to the level of Rolipram at 1 μM. Introduction of the N-acetamide substitution motif of the PDE4 inhibitor Apremilast in the meta position of the benzene moiety did not improve the potency of the compound (6) compared to the non-substituted inhibitor 4. Shifting the acetamide substitution in the para position (10), elongating the substituent to propionamide (7) or introducing an additional methyl group in para position (8) decreased inhibition on both targets as well as water solubility. Thus, the Apremilast inspired design did not improve the dual inhibitors. A para methoxy benzene substitution (11) also did not improve target inhibition compared to the non-substituted inhibitor (4), while the para difluoromethoxy benzene substituted inhibitor 14 even decreased water solubility to 10 μM. The meta cyclopentoxy benzene substitution in dual inhibitor 19 improved the sEH inhibition compared to the non-substituted inhibitor 4, but water solubility decreased by one order of magnitude and cAMP increase relative to Rolipram at 1 μM slightly decreased too. For inhibitor 20, the meta cyclopentyloxy benzene substitution was combined with a para fluoro benzene substituent in order to increase metabolic stability. The additional para fluoro substituent did not change sEH inhibition (IC50 0.4 nM), improved the cAMP increase relative to Rolipram at 1 μM and restored the water solubility to 100 μM. The third structural class (Group 3) of dual sEH/PDE4 inhibitors enfolds in different N-substitution of the piperidine moiety. The dual inhibitor 22 contains a non-substituted piperidine, which led to a significant loss in sEH inhibitory potency (IC50 370 ± 170 nM), a higher cAMP increase than Rolipram at 1 μM and an improved water solubility of 1 mM. Ethyl and propyl substituted modulators (26, 27) similarly had low sEH inhibitory potencies and high water solubility, but only 60% cAMP increase compared to Rolipram at 1 μM. Dual inhibitor 23/MPPA with a propionamide piperidine substitution showed a low, one digit nanomolar sEH IC50, 200% cAMP increase compared to Rolipram at 1 μM and 100 μM water solubility. Increasing the alkyl chain of the N-substitution to butyramide (24) and sec-butylamide (25) decreased sEH inhibition and even more dramatically the water solubility.
The in vitro screening process shown in Table 1, revealed 4 compounds with both adequate potency on both targets and suitable water solubility. To better characterize those 4 compounds (1, 19, 20 and 23/MPPA), the melting point and IC50 value for human and rat recombinant enzymes were measured and compared to the single target model ligands (TPPU and Rolipram). Previous studies have shown a difference in potency of sEH inhibition comparing human and rat sEH.26 Table 2 shows a drop in potency on the rsEH compared to hsEH for compound 1, 19 and 20 by 3 fold or more. 23/MPPA demonstrated a comparatively much lower potency (75-fold) on the rsEH than on the hsEH.
Table 1.
Evaluation of dual sEH/PDE4 inhibitors in vitro activity
| |||||
|---|---|---|---|---|---|
|
| |||||
| ID | R1 | R2 | hsEH IC50a [nM] |
cAMP increaseb (% of Rolipram) |
w.s.c [μM] |
| Rolipram | – | – | >100 000 | 100 | 750 |
| GSK2256294 | – | – | 0.027 | – | – |
| 1 | CF3 | OMe | 0.6 ± 0.1 | 240 ± 14 | 37.5 |
| 2 | CF3 | – | 15 ± 3 | 180 ± 12 | 37.5 |
| 3 | Me | – | 180 ± 40 | 200 ± 11 | 37.5 |
| |||||
|---|---|---|---|---|---|
|
| |||||
| ID | R3 | R4 | hsEH IC50a [nM] |
cAMP increaseb (% of Rolipram) |
w.s.c [μM] |
| 4 | – | – | 2 ± 0.9 | 160 ± 12 | 100 |
| 5 | F | – | 2.8 ± 0.4 | 110 ± 13 | 100 |
| 6 | – | NHCOMe | 2.9 ± 1.1 | 140 ± 15 | 100 |
| 7 | – | NHCOEt | 3.7 ± 1 | 150 ± 18 | 50 |
| 8 | Me | NHCOMe | 13 ± 2 | 140 ± 15 | 20 |
| 9 | pyrrolidin-2-one | 3 ± 2 | 130 ± 25 | 100 | |
| 10 | NHCOMe | – | 44 ± 5 | 100 ± 19 | 50 |
| 11 | OMe | – | 1 ± 0.2 | 160 ± 17 | 100 |
| 14 | OCHF2 | – | 1.5 ± 0.4 | 150 ± 14 | 10 |
| 19 | – | cyclopentyloxy | 0.4 ± 0.1 | 147 ± 9 | 10 |
| 20 | F | cyclopentyloxy | 0.4 ± 0 | 222 ± 8 | 100 |

| ||||
|---|---|---|---|---|
|
| ||||
| ID | R5 | hsEH IC50a [nM] |
cAMP increaseb (% of Rolipram) |
w.s.c [μM] |
| TPPUD | – | 3.7 | 40 ± 10 | 200 |
| 21 | BOC | 1.7 ± 0.7 | 100 ± 12 | 100 |
| 22 | – | 370 ± 170 | 138 ± 5 | 1000 |
| 23/MPPA | COEt | 2.1 ± 0.5 | 200 ± 19 | 100 |
| 24 | COPr | 4 ± 2 | 110 ± 16 | 50 |
| 25 | s-Bu | 7 ± 3 | 160 ± 11 | 0.5 |
| 26 | Et | 109 ± 16 | 60 ± 10 | 1000 |
| 27 | n-Pr | 190 ± 20 | 60 ± 14 | 1000 |
In vitro evaluation of sEH/PDE4 dual inhibitors Group 1: 1-3, Group 2: 4-11, 14, 19-20 and Group 3: 21-27.
The sEH IC50 values are recorded on recombinant human sEH protein (hsEH) using MNPC as substrate at 5 μM concentration32;
PDE4 inhibition is evaluated by cAMP increase induced by 1 μM concentration of the dual inhibitors in live human embryonic kidney (HEK) cells, transfected with a PKA biosensor and visualized relative to Rolipram at 1 μM;
water solubility (w.s.) is measured in sodium phosphate buffer (0.1 M, pH 7.4) with 1% DMSO by precipitation. All experiments were performed in triplicates. Results are average ± standard deviation of at least three measurements.
Table 2.
In vitro evaluation for laboratory species
| ID | IC50(hsEH)a [nM] |
IC50(rsEH)b [nM] |
EC50 (cAMP increase)c [nM] |
Melting pointd (°C) |
|---|---|---|---|---|
| TPPUd | 3.7 | 2.8 | – | 226 |
| Rolipram | >100 000 | – | 340 ± 0.06 | 127 |
| 1 | 0.6 ± 0.1 | 14 ± 1 | 1.9 ± 0.03 | 144 |
| 19 | 0.4 ± 0.1 | 1.3 ± 0.2 | 128 ± 0.05 | 106 |
| 20 | 0.4 ± 0 | 1.7 ± 0.2 | 3.9 ± 0.06 | 99 |
| 23/MPPA | 2.1 ± 0.5 | 150 ± 16 | 8.1 ± 0.05 | 133 |
IC50 evaluation of dual inhibitors.
The sEH IC50 on recombinant human sEH (hsEH);
The sEH IC50 on recombinant rat sEH (rsEH);
IC50 evaluated on cAMP increase in live HEK cells, transfected with a PKA biosensor; All experiments were performed in triplicates;
Melting points were measured on an automated melting point system from Stanford Research Systems (OptiMelt-MPA100);
Literature values.26 Results are average ± standard deviation of at least three measurements.
More than 20 PDE4 isoforms are known today27,30, we decided therefore to use a cell based activity assay for our screening. This provides the benefit of a broad PDE4 evaluation and a better estimate towards the later in vivo effect of our compound. We additionally tried to evaluate one of our most potent compounds (23) on recombinant PDE4 enzyme and were therefore forced to pick one out of 20 isoforms. We chose PDE4B1, but did sadly not find inhibition up 10 μM. Out of commercial reasons we did not screen the remaining 19 PDE4 isoforms.
Pharmacokinetics
To identify the optimal candidate for use in vivo, the pharmacokinetics of 4 dual compounds were determined with oral administration by cassette dosing. Compounds 1, 19, 20 and 23/MPPA were administered at 0.3 mg/kg by oral gavage to 4 rats. Compound 1 was detected at 0.5 h with a 3 ± 2.2 nM blood concentration, while compound 20 was not observed above the limit of detection in the blood (Table S2). Compound 19 did not increase over single digit nM blood concentration over 12 h. 23/MPPA reached the highest blood concentration of 60±13 nM and was observed in blood for several hours. Therefore, 23/MPPA having the best observed pharmacokinetics of the in vivo cassette compounds was chosen for further in vivo evaluation and will be referred to as MPPA. Later single administration of MPPA was also evaluated for PK parameters (Table S3). A 3 mg/kg dose that demonstrated in vivo efficacy resulted in a 460 ± 16 nM blood concentration (n=3) at 0.5 h and remained in the blood several hours. The blood concentration of the 3 mg/kg dose is 3-fold above the rat sEH IC50 and stays above the inhibitory concentration for about 2 h. Single administration of 30 and 100 mg/kg (each n=1) dosed to rats resulted in concentrations reaching 1,060 nM and 1,730 nM respectively. Thus, MPPA is absorbed quickly after oral administration and therefore has potential as a rapid acting pharmaceutical agent.
In vivo Efficacy
The efficacy of MPPA was evaluated in an LPS induced inflammatory pain model. For this assay the compound or vehicle control were administered by oral gavage followed immediately by an intraplantar injection of lipopolysaccharide (LPS) to one hindpaw and measured over a time course for their thermal withdrawal latency (Figure 1). Baseline scores were assessed before the begin of the assay and normalized to 100%. The LPS results in lower than baseline scores that continue to decrease over the first several hours indicating a painful state. Oral administration of 3 mg/kg MPPA resulted in significantly increased thermal withdrawal latencies (interpreted as pain relief) compared to the vehicle control. The rapid onset of anti-inflammatory analgesia at 30 min correlates directly with the increase of MPPA in the blood (Tmax = 30 min) and decreases with the clearance from the blood after 4 hours. Interestingly, though hypoalgesia is apparent at the initial time point in the LPS assay, MPPA did not alter nociceptive thresholds in naïve animals. The dual inhibitor MPPA had no effect on withdrawal latency in naïve animals (Figure S4) while the PDE4 inhibitor Rolipram at a similar dose (3 mg/kg) significantly increased withdrawal latency. This effect of Rolipram in the absence of enhanced pain perception suggests a central nervous system activity. In addition to efficacy testing, we also observed spontaneous locomotion in the treated animals. Alteration of a normal pain response and sedation can be serious side effects. In LPS treated rats, MPPA administration did not decrease exploration in an open field assay compared to the vehicle. This contrasted with Rolipram which altered locomotion in the open field assay (Figure S5). This may indicate that the sEH/PDE4 dual inhibitor MPPA does not have effects similar to Rolipram. However, future investigations of MPPA in causing known side effects of classical PDE4Is such as vasculitis, headache, emesis and nausea will be required.
Figure 1.

MPPA is efficacious against LPS induced inflammatory pain. Baseline thermal withdrawal latencies were determined for each rat before administration of compounds. After baseline determination, MPPA and the vehicle control were administered by oral gavage immediately before a 50 μl intraplantar injection of LPS in saline (arrow) to induce inflammatory pain in male rats. The efficacy of MPPA peaked at 30 min post LPS injection and was largely dissipated by 4 hours. The efficacy at 3 mg/kg was statistically significant compared to vehicle control. Scores are the mean ± SEM reported as percent of baseline (baseline scores normalized to 100%) calculated as the score *100/baseline score (Mann-Whitney Rank Sum Test, T = 448.000 n(small)= 24 n(big)= 36 *p≤0.001, n=6-9/group).
Discussion
In this study, 21 dual sEH/PDE4 inhibitors were synthesized and their in vitro potencies were assessed. The dual ligands with the most potential from each Group (1-3) were selected based on potency on both targets and water solubility. The pharmacokinetic properties of these selected compounds (1, 19, 20, MPPA) revealed good blood concentrations of 1-(piperidin-1-yl)propan-1-one fragment, which was anticipated in the design process. In comparison, the cyclopentyloxy substituted benzyl fragments resulted in lower blood concentrations. Surprisingly, the addition of fluorine on the cyclopentyloxy benzyl fragment diminished the lowered these even further. MPPA, containing the 1-(piperidin-1-yl)propan-1-one fragment, was chosen out of the PK cassette and further investigated. The dose escalation revealed a fast absorption of MPPA combined with a rapid clearance. At a 3 mg/kg dose, the blood concentration of MPPA in rats reached levels above the IC50 of both targets but deceased substantially after 4 hours. The efficacy evaluation showed a direct correlation between compound blood concentration and analgesic efficacy. Therefore, a future goal of this study is to identify labile positions in the structure of MPPA and substitute those with more metabolically stable moieties while maintaining potency on the targets. Finally, MPPA did not alter nociceptive responses in naïve rats compared to Rolipram. Nevertheless the dual inhibitor developed will need to be further investigated towards major potential side effects known from PDE inhibition to ensure the safety of the compound.
Conclusion
In conclusion, this study presents a proof of concept for the sEH/PDE4 dual inhibitor approach on analgesia and therefore adds to the concept of previous poly-pharmacological tools targeting sEH in combination with other enzymes.16,18,31 Nevertheless, several experiments will have to be performed in the future to clarify the therapeutic improvements of the dual inhibitor. Further in vivo experiments, including ferret or other emetic models are necessary to define the side effects caused by the dual inhibitor in comparison to classical PDEIs.
Despite this, the dual ligand MPPA opens the path to future investigations of sEH/PDE4 target interactions and the crosstalk between cAMP and EpFA15. In addition, MPPA has the potential to impact further pathological conditions in addition to inflammatory pain, such as depression and COPD.3,5,14,20 The efficacy was limited by the rapid elimination of the compound suggesting that the next generation design of the dual inhibitor should focus on improving metabolic stability, while maintaining the rapid absorption and activity. Still, MPPA represents a new potential approach for pain management as well as a tool for investigating the biology of pain.
EXPERIMENTAL SECTION
Chemistry
General
All reagents and solvents were purchased from commercial suppliers and were used directly without further purification. All reactions were carried out at room temperature unless otherwise specified. Reactions were monitored by thin-layer chromatography (TLC) on Merck F254 silica gel 60 aluminum sheets, and spots were revealed with UV light (254 mm), potassium permanganate or Ninhydrin stains. 1H NMR spectra were recorded on a 400 MHz Bruker Avance III HD Nanobay Spectrometer with deuterated chloroform (CDCl3; δ = 7.24 ppm) or deuterated dimethyl sulfoxide (DMSO-d6) containing TMS as internal standard. 13C NMR spectra were recorded on a Bruker Avance III HD Nanobay spectrometer at 100 MHz. The percent purity of the inhibitors reported in this manuscript were determined by HPLC-UV using Agilent 1200 series HPLC system equipped with Phenomenex Luna2 C18 reverse phase column (C18, 4.6 mm × 150 mm, 5 μm) coupled with Agilent G1314 UV–vis detector (detection at 200, 210, 254 and 360) with solvent gradient acetonitrile/water 20 to 95% over 15 min and they are expressed as percent total OD at 254 and 360 nm. The purity of all final compounds was above 95%. HRMS spectra were recorded on Thermo Electron LTQ-Orbitrap XL Hybrid MS in ESI mode.
General synthetic procedure A for compounds 1-11, 14 and 19-21 shown on the example 3-(cyclopentyloxy)-4-methoxy-N-(4-methoxy-2-(trifluoromethyl)benzyl) (1)
0.2 g (0.85 mmol) of 3-(cyclopentyloxy)-4-methoxybenzoic acid, 160 mg (1 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 20 mg (0.17 mmol) 4-(dimethylamino)pyridine were dissolved in 10 ml dichloromethane and stirred under a nitrogen atmosphere for 1 h. Subsequently, 160 μl (0.93 mmol) (4-methoxy-2-(trifluoromethyl)phenyl)methanamine was added to the mixture. The reaction was further stirred for 12 h. After completion of the reaction, dichloromethane was removed under reduced pressure. The residue was dissolved in 20 ml of ethyl acetate and washed with aqueous sodium hydroxide solution (2 M, 3 × 20 mL), aqueous hydrochloric acid solution (2 M, 3 × 20 mL) and brine (20 mL). The organic solvent was dried over magnesium sulfate and removed under reduced pressure. The crude product was purified by recrystallization from ethyl acetate and hexane mixture. The pure product remained as a white solid.
General synthetic method B to produce compounds 13, 17 and 18 shown with the example 4-(difluoromethoxy)benzoic acid (13)
0.47 g (2.3 mmol) methyl 4-(difluoromethoxy)benzoate (12) and 0.47 g (12 mmol) sodium hydroxide were stirred in a mixture of 10 ml of tetrahydrofuran, 10 ml of methanol and 5 ml of water under a nitrogen atmosphere at 40 °C for 12 h. The organic solvents were removed under reduced pressure and the remaining aqueous phase was acidified through dropwise addition of concentrated hydrochloric acid solution. The precipitated product was filtered, dried and used in the next step without further purification.
General synthetic method C for compounds 15 and 16, shown with the synthesis of methyl 3-(cyclopentyloxy)benzoate (15)
1 g (6.6 mmol) of methyl 3-hydroxybenzoate, 22 mg (0.13 mmol) of potassium iodide and 1.4 g (9.9 mmol) of potassium carbonate are stirred in 6.5 ml of dimethylformamide at 65 °C. Subsequently 0.92 ml (8.6 mmol) of bromocyclopentane was added dropwise. The reaction was further stirred for 21 h, cooled down to rt and diluted with 50 ml dichloromethane, washed with aqueous sodium hydroxide solution (2 M, 3 × 50 mL) and brine (50 mL). The organic solvent was dried over magnesium sulfate and evaporated under reduced pressure. The product was used in the next step without further purification.
General synthetic method D to produce compounds 23/MPPA and 24, shown on the example N-(4-methoxy-2-(trifluoromethyl)benzyl)-1-propionylpiperidine-4-carboxamide 23/MPPA
28 μL (0.38 mmol) of propionic acid and 40 μL (0.47 mmol) of oxalyl chloride were stirred in 1 mL of dry dichloromethane under a nitrogen atmosphere for 1 h, to generate propionyl chloride. In a separate flask 0.1 g (0.32 mmol) of N-(4-methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide 22 and 80 μL (0.5 mmol) triethylamine were stirred in 2 ml of dry dichloromethane at 0 °C under a nitrogen atmosphere. The produced propionyl chloride solution was now added slowly to the amine containing reaction flask and the whole mixture was stirred for 4 h. The reaction was diluted with 7 mL of dichloromethane, washed with hydrochloric acid aqueous solution (2 M, 3 × 10 mL), sodium hydroxide aqueous solution (2 M, 3 × 10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate and evaporated under reduced pressure. The remaining crude material was purified by flash column chromatography (acetone/ethyl acetate = 50:50).
General synthetic method E to produce compounds 26 and 27, illustrated with the synthesis of 1-ethyl-N-(4-methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide 26
0.15 g (0.48 mmol) of N-(4-methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide 22, 0.13 g (0.95 mmol) of potassium carbonate and 38 μL (0.48 mmol) ethyl iodide were stirred in 3.2 mL of dimethylformamide for 24 h. After the reaction was completed, the pure product was precipitated with the addition of 2 M sodium hydroxide aqueous solution.
3-(Cyclopentyloxy)-4-methoxy-N-(4-methoxy-2-(trifluoromethyl)benzyl) (1)
Yield: 0.18 g (50%). 1H NMR (400 MHz, Chloroform-d) δ 7.86 – 6.25 (m, 6H), 4.89 – 4.81 (m, 1H), 4.74 (d, J = 6.1, 1.3 Hz, 2H), 3.89 (s, 3H), 3.85 (s, 3H), 2.33 – 1.20 (m, 8H); 13C-NMR (DMSO-d6) δ 166.7, 157.1, 151.3, 147, 128.2, 128, 127.8, 126.9, 126.4, 124.1, 121.3, 118.2, 115.1, 112.6, 109.3, 80.1, 55.2, 54.1, 42.7, 31.7, 30.6, 24.1, 23.7; HRMS: measured [M+Cl]− 374.1523 (calculated: 374.1521).
3-(Cyclopentyloxy)-4-methoxy-N-(2-(trifluoromethyl)benzyl)benzamide (2)
Yield: 0.16 (49%). 1H NMR (400 MHz, Chloroform-d) δ 7.90 – 6.47 (m, 7H), 4.88 – 4.84 (m, 1H), 4.82 (d, J = 6.4 Hz, 2H), 3.89 (s, 3H), 2.08 – 1.50 (m, 8H); 13C-NMR (DMSO-d6) δ 168.3, 156.1, 148.3, 147.5, 127.2, 127, 126.9, 126.7, 126.5, 123.4, 120.3, 119.2, 116.3, 112.1, 108.3, 79.1, 53.1, 41.7, 33.7, 31.4, 25.1, 23.1; HRMS: measured [M+Cl]− 428.1239 (calculated: 428,1237).
3-(Cyclopentyloxy)-4-methoxy-N-(2-methylbenzyl)benzamide (3)
Yield: 0.15 (62%).1H NMR (400 MHz, Chloroform-d) δ 7.63 – 6.07 (m, 7H), 4.93 – 4.81 (m, 1H), 4.65 (d, J = 5.4 Hz, 2H), 3.89 (s, 3H), 2.40 (s, 3H), 2.08 – 1.54 (m, 8H); 13C-NMR (DMSO-d6) δ 167.3, 155.1, 147.3, 146.5, 128.1, 128, 127.9, 125.7, 120.8, 119.1, 117.3, 113.1, 109.3, 79.5, 52.1, 42.7, 31.7, 30.4, 24.1, 22.1, 19.1; HRMS: measured [M+Cl]− 458.1344 (calculated: 458.1342).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)benzamide (4)
Yield 0.11 g (34%). 1H NMR (400 MHz, Chloroform-d) δ 7.36 (m, 8H), 6.40 (s, 1H), 3.7 (s, 3H); 13C-NMR (DMSO-d6) δ 166.1, 158.1, 150.2, 146, 127.8, 127.6, 127.3, 126.9, 126.1, 123.1, 120.3, 119.2, 116.1, 112.6, 109.1, 53.1, 40.7, HRMS: measured [M+HCOO]− 354.0950 (calculated: 354.0953).
4-Fluoro-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (5)
Yield: 0.21 g (66%). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (t, J = 5.8 Hz, 1H), 8.26 – 6.91 (m, 7H), 4.58 (d, J = 5.7 Hz, 2H), 3.82 (s, 3H); 13C-NMR (DMSO-d6) δ 168.1, 155.1, 148.2, 147, 130.8, 129.6, 129.7,128.9, 128.1, 126.1, 125.1, 120.1, 118.2, 117.1, 113.6, 107.1, 51.1, 40.5, HRMS: measured [M+HCOO]− 372.0856 (calculated: 372.0854).
3-Acetamido-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (6)
Yield: 0.23 mg (56%). 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.94 (t, J = 5.8 Hz, 1H), 8.24 – 6.44 (m, 7H), 4.52 (d, J = 5.6 Hz, 2H), 3.77 (s, 3H), 2.00 (s, 3H); 13C-NMR (DMSO-d6) δ 166.9, 159.1, 151.2, 145, 141.3, 128.6, 127.4, 127.3, 127.1, 126. 8, 122.1, 121.3, 118.3, 117.1, 112.6, 109.7, 55.8, 42.6; HRMS: measured [M−H]− 365.1114 (calculated: 365.1113).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)-3-propionamidobenzamide (7)
Yield: 0.14 (41%). 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.04 (t, J = 5.9 Hz, 1H), 8.13 – 7.01 (m, 7H), 4.63 (d, J = 5.6 Hz, 2H), 3.88 (s, 3H), 2.40 (q, J = 7.5 Hz, 2H), 1.15 (t, J = 7.5 Hz, 3H); 13C-NMR (DMSO-d6) δ 167.9, 155.1, 150.2, 146, 142.3, 129.6, 128.5, 128.3, 128.2, 128, 121.1, 119.3, 118.1, 116.1, 110.6, 109.7, 56.8, 41.6, 10.1; HRMS: measured [M+HCOO]− 425.1320 (calculated: 425.1321).
3-Acetamido-N-(4-methoxy-2-(trifluoromethyl)benzyl)-4-methylbenzamide (8)
Yield: 0.1 g (29%). 1H NMR (400 MHz, DMSO-d6) δ 9.43 (s, 1H), 8.95 (t, J = 5.8 Hz, 1H), 8.32 – 6.80 (m, 6H), 4.57 (d, J = 5.6 Hz, 2H), 3.82 (s, 3H), 2.25 (s, 3H), 2.08 (s, J = 2.7 Hz, 3H). 13C-NMR (DMSO-d6) δ 166, 158.1, 152.2, 146, 142.3, 128.3, 128.1, 128, 127.8, 121.1, 120.3, 119.3, 118.1, 112.7, 109.9, 56.8, 40.6, 17.3; HRMS: measured [M+HCOO]− 435.1321 (calculated: 435.1321).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)-2-oxoindoline-6-carboxamide (9)
Yield: 0.14 (44%). 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 9.00 (t, J = 5.8 Hz, 1H), 7.79 – 6.95 (m, 6H), 4.57 (d, J = 5.6 Hz, 2H), 3.82 (s, 3H), 3.55 (s, 2H); 13C-NMR (DMSO-d6) δ 176.7, 167.8, 155.1, 147.2, 141, 127, 126.9, 126.8, 126.6, 126.1, 124, 123.5, 120.1, 119.7, 118.1, 113.6, 109.6, 52.1, 42.7, 36.5; HRMS: measured [M+HCOO]− 409.1008 (calculated: 409.1007).
4-Acetamido-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (10)
Yield: 0.16 (50%). 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.95 (t, J = 5.8 Hz, 1H), 8.14 – 7.11 (m, 7H), 4.63 (d, J = 5.6 Hz, 2H), 3.87 (s, 3H), 2.14 (s, 3H); 13C-NMR (DMSO-d6) δ 166.3, 157.1, 152.2, 147, 142.3, 128.6, 128.5, 128.3, 127.9, 123.1, 122.3, 119.3, 118.1, 112.8, 109.3, 56.8, 41.6; HRMS: measured [M−H]− 365.1111 (calculated: 365.1113).
4-Methoxy-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (11)
Yield: 0.16 g (48%). 1H NMR (400 MHz, DMSO-d6) δ 8.88 (t, J = 5.8 Hz, 1H), 8.17 – 7.73 (m, 2H), 7.57 – 7.33 (m, 1H), 7.32 – 7.14 (m, 2H), 7.09 – 6.92 (m, 2H), 4.57 (d, J = 5.7 Hz, 2H), 3.83 (s, 3H), 3.82 (s, 3H); 13C-NMR (DMSO-d6) δ 167.7, 156.1, 150.3, 148, 129.2, 128, 127.9, 127.7, 127.4, 123.1, 120.3, 119.2, 116.1, 113.6, 109.1, 56.2, 53.1, 41.7; HRMS: measured [M+HCOO]− 384.1055 (calculated: 384.1057).
Methyl 4-(difluoromethoxy)benzoate (12)
1 g (6.6 mmol) methyl 4-hydroxybenzoate, 4.3 g (13 mmol) cesium carbonate and 2.5 g (16 mmol) sodium chloro difluoro acetate were dissolved in 48 ml dimethylformamide and 7 ml water under a nitrogen atmosphere. The mixture was first stirred for 15 min at rt and subsequently heated to 100 °C for 2 h. After completion of the reaction, the mixture was diluted with 50 mL of water and the product extracted with 100 mL of dichloromethane. The organic phase was dried over magnesium sulfate and evaporated under reduced pressure. The product was used in the next step without further purification. Yield: 0.47 g (35%). 1H NMR (400 MHz, DMSO-d6) δ 8.02 (d, J = 8.9 Hz, 2H), 7.40 (s, 1H), 7.30 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H).
4-(Difluoromethoxy)benzoic acid (13)
Yield: 0.18 (41%). 1H NMR (400 MHz, DMSO-d6) δ 13.02 (s, 1H), 8.00 (d, J = 8.8 Hz, 2H), 7.39 (s, 1H), 7.32 – 7.25 (m, 2H).
4-(Difluoromethoxy)-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (14)
Yield: 0.11 g (37%). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (t, J = 5.7 Hz, 1H), 8.05 – 7.92 (m, 2H), 7.47 – 7.43 (m, 1H), 7.36 (s, 1H), 7.32 – 7.26 (m, 2H), 7.26 – 7.20 (m, 2H), 4.58 (d, J = 5.6 Hz, 2H), 3.82 (s, 3H); 13C-NMR (DMSO-d6) δ 167.8, 167.3, 167.1, 166.9, 157.1, 152.3, 148.3, 128.2, 128.1, 128, 127.9, 127.6, 123.2, 121.3, 119.1, 117.1, 112.6, 109.1, 50.1, 42.7; HRMS: measured [M−H]− 374.0813 (calculated: 374.0811).
Methyl 3-(cyclopentyloxy)benzoate (15)
Yield: 1.1 g (76%). 1H NMR (400 MHz, DMSO-d6) δ 7.85 – 7.03 (m, 4H), 4.87 (tt, J = 5.9, 2.5 Hz, 1H), 3.84 (s, 3H), 2.16 – 1.42 (m, 8H).
Methyl 3-(cyclopentyloxy)-4-fluorobenzoate (16)
Yield: 1 g (70%). 1H NMR (400 MHz, DMSO-d6) δ 8.00 – 6.91 (m, 3H), 5.18 – 4.68 (m, 1H), 3.85 (s, 3H), 2.20 – 1.37 (m, 8H).
3-(Cyclopentyloxy)benzoic acid (17)
Yield: 0.4 g (85%). 1H NMR (400 MHz, DMSO-d6) δ 12.95 (s, 1H), 7.92 – 6.80 (m, 4H), 5.02 – 4.59 (m, 1H), 2.12 – 1.48 (m, 8H).
3-(Cyclopentyloxy)-4-fluorobenzoic acid (18)
Yield: 0.8 g (71%). 1H NMR (400 MHz, DMSO-d6) δ 13.06 (s, 1H), 7.95 – 6.87 (m, 3H), 4.99 – 4.90 (m, 1H), 2.02 – 1.54 (m, 8H).
3-(Cyclopentyloxy)-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (19)
Yield: 0.1 g (30%). 1H NMR (400 MHz, DMSO-d6) δ 9.00 (t, J = 5.8 Hz, 1H), 7.79 – 6.89 (m, 7H), 4.96 – 4.81 (m, 1H), 4.57 (d, J = 5.7 Hz, 2H), 3.82 (s, 3H), 2.04 – 1.47 (m, 8H); 13C-NMR (DMSO-d6) δ 167.1, 156.2, 151.1, 148, 128.4, 128.2, 128.1, 127.9, 127.2, 126.5, 123.9, 120.3, 119.2, 116.1, 113.6, 109.1, 80.2, 53.1, 41.7, 30.7, 30.6, 24.1, 23.7; HRMS: measured [M+HCOO]− 438.1527 (calculated: 438.1528).
3-(Cyclopentyloxy)-4-fluoro-N-(4-methoxy-2-(trifluoromethyl)benzyl)benzamide (20)
Yield: 0.1 (33%). 1H NMR (400 MHz, DMSO-d6) δ 9.04 (t, J = 5.8 Hz, 1H), 7.82 – 7.09 (m, 7H), 4.95 (tt, J = 5.9, 2.4 Hz, 1H), 4.59 (d, J = 5.6 Hz, 2H), 3.82 (s, 3H), 2.12 – 1.50 (m, 8H);); 13C-NMR (DMSO-d6) δ 166.9, 166.4; 155.2, 155.3, 152.1, 147, 128.5, 128.3, 128.2, 128, 127.5, 122.9, 120.1, 119.2, 117.1, 112.6, 109.7, 80.6, 52.7, 40.7, 30.6, 30, 24.3, 23.9; HRMS: measured [M−H]− 410.1379 (calculated: 410.1377).
tert-Butyl 4-((4-methoxy-2-(trifluoromethyl)benzyl)carbamoyl)piperidine-1-carboxylate (21)
Yield: 1.1 g (42%). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (t, J = 5.8 Hz, 1H), 7.69 – 6.86 (m, 3H), 4.35 (d, J = 5.6 Hz, 2H), 3.95 (d, J = 13.2 Hz, 2H), 3.82 (s, 3H), 2.71 (d, J = 24.0 Hz, 2H), 2.42 – 2.31 (m, 1H), 1.70 (d, J = 13.2 Hz, 2H), 1.45 (d, J = 4.2 Hz, 2H), 1.40 (s, 9H); 13C-NMR (DMSO-d6) δ 173.9, 159.7, 158.8, 129.2, 126.5, 126.3,126.2, 125.8, 123.8, 117.5, 109.2, 79.8, 55.8, 45.7, 45.2, 41.5, 38.9, 29.7, 29.6, 28.5, 28.3, 28.1; HRMS: measured [M+HCOO]− 461.1896 (calculated: 461.1895).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide (22)
1.3 g (3 mmol) tert-Butyl 4-((4-methoxy-2-(trifluoromethyl)benzyl)carbamoyl)piperidine-1-carboxylate 21 and 2.8 mL (36 mmol) of trifluoroacetic acid were stirred in 16 mL of dichloromethane at 0 °C for 2 h. The organic phase was diluted with 4 mL dichloromethane and extracted with hydrochloric acid aqueous solution (10 %, 3 × 20 mL). The aqueous layer was basified through the addition of solid sodium hydroxide pellets. From the basic aqueous layer the product was extracted with ethyl acetate (3 × 60 mL). The organic phase was dried over magnesium sulfate and evaporated. The remaining product was used in the next step without further purification. Yield: 0.73 g (77%). 1H NMR (400 MHz, DMSO-d6) δ 8.27 (t, J = 5.8 Hz, 1H), 7.81 – 6.84 (m, 3H), 4.34 (d, J = 5.7 Hz, 2H), 3.82 (s, 3H), 2.99 (dt, J = 12.3, 3.4 Hz, 2H), 2.66 – 2.45 (m, 3H), 1.76 – 1.32 (m, 4H); 13C-NMR (DMSO-d6) δ 172.8, 158.7, 157.8, 128.4, 128.2, 128.1, 127.8, 125.3, 124.7, 123.9, 117.9, 109.1, 56.8, 44.7, 43.2, 41.7, 39.9; 29.4, 29.3; HRMS: measured [M+H]+ 317.1493 (calculated: 317.1492).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)-1-propionylpiperidine-4-carboxamide 23/MPPA
Yield: 70 mg (63%). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (t, J = 5.8 Hz, 1H), 7.63 – 6.99 (m, 3H), 4.35 (d, J = 6.0 Hz, 2H), 3.82 (s, 3H), 3.10 – 2.88 (m, 2H), 2.73 – 2.63 (m, 3H), 2.39 – 2.23 (m, 2H), 1.88 – 1.64 (m, 2H), 1.34 – 1.19 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H); 13C-NMR (DMSO-d6) δ 176.9, 173.8, 156.5, 128.5, 128.2, 127.9, 127.8, 125.1, 124.8, 123.4, 118.7, 109.9, 56.9, 43.7, 42.2, 40.9, 38.9; 28.9, 28.8, 26.1, 10.2; HRMS: measured [M+FA]− 417.1633 (calculated: 417.1633).
1-Butyryl-N-(4-methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide (24)
Yield: 0.05 g (37%). 1H NMR (400 MHz, DMSO-d6) δ 8.35 (t, J = 5.8 Hz, 1H), 7.53 – 7.04 (m, 3H), 4.35 (d, J = 5.9 Hz, 2H), 3.94 – 3.85 (m, 1H), 3.82 (s, 3H), 3.01 (t, J = 12.7 Hz, 2H), 2.71 – 2.55 (m, 1H), 2.28 (td, J = 7.3, 3.1 Hz, 2H), 1.73 (t, 2H), 1.59 – 1.44 (m, 3H), 1.42 – 1.27 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H); 13C-NMR (DMSO-d6) δ 176.7, 175.8, 155.5, 128.4, 128.2, 128, 127.9, 124.1, 124, 122.4, 119.7, 109.3, 54.9, 42.7, 41.2, 40.7, 37.9; 29.9, 28.8, 27.1, 19.2, 13.2; HRMS: measured [M−H]− 385.0268 (calculated: 385.0267).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)-1-(2-methylbutanoyl)piperidine-4-carboxamide (25)
0.1 g (0.32 mmol) of N-(4-methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide (22) and 44 μL (0.32 mmol) triethylamine were stirred in 5 mL dry dichloromethane at 0 °C under a nitrogen atmosphere. Subsequently, 39 μL (0.32 mmol) of 2-methylbutanoyl chloride was added and the reaction mixture was stirred for 4 h. The reaction was diluted with 5 mL of dichloromethane and washed with hydrochloric acid aqueous solution (2 M, 3 × 10 mL), sodium hydroxide aqueous solution (2 M, 3 × 10 mL) and once with 10 mL brine. The organic phase was dried over magnesium sulfate and removed under reduced pressure. The crude material was purified by flash column chromatography (acetone/ethyl acetate = 50:50). Yield: 82 mg (65%). 1H NMR (400 MHz, DMSO-d6) δ 8.37 (t, J = 5.8 Hz, 1H), 7.49 – 7.09 (m, 3H), 4.39 – 4.48 (m, 1H), 4.37 – 4.34 (m, 2H), 4.07 – 3.95 (m, 1H), 3.82 (s, 3H), 3.1 – 2.96 (m, 1H), 2.71 (h, J = 6.7 Hz, 1H), 2.58 (t, J = 11.9 Hz, 1H), 2.47 (dd, J = 11.5, 3.9 Hz, 1H), 1.83 – 1.7 (m, 2H), 1.62 – 1.21 (m, 2H), 0.97 (t, J = 7.7 Hz, 3H), 0.81 (q, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6) δ 176.9, 175.7, 155.4, 128.3, 128.1, 128, 127.8, 125.2, 125.1, 123.4, 119.1, 109.2, 53.9, 42.5, 41, 40.2, 39.2, 37.9; 29.7, 27.5, 17.3, 10.2; HRMS: measured [M+HCOO]− 445.1947 (calculated: 445.1949).
1-Ethyl-N-(4-methoxy-2-(trifluoromethyl)benzyl)piperidine-4-carboxamide (26)
Yield: 40 mg (24%). %). 1H NMR (400 MHz, DMSO-d6) δ 8.13 (t, J = 5.7 Hz, 1H), 7.66 – 6.43 (m, 3H), 4.12 (d, J = 5.7 Hz, 2H), 3.72 (s, 3H), 3.31 (dt, J = 12.1, 3.3 Hz, 2H), 2.67 – 2.48 (m, 3H), 1.77 – 1.42 (m, 4H), 3.02 – 2.98 (m, 2H), 0.95 – 1.16 (m, 3H); 13C-NMR (DMSO-d6) δ 172.8, 158.7, 157.8, 128.2, 128.1, 127.8, 127.7, 125.3, 124.7, 123.9, 117.9, 109.1, 56.8, 49.9, 44.7, 43.2, 41.7, 39.9; 29.4, 29.3, 13.3; HRMS: measured [M+H]+ 345.1814 (calculated: 345.1815).
N-(4-Methoxy-2-(trifluoromethyl)benzyl)-1-propylpiperidine-4-carboxamide (27)
Yield: 0.06 g (35%). 1H NMR (400 MHz, DMSO-d6) δ 8.9 (t, J = 5.7 Hz, 1H), 7.55 – 6.41 (m, 3H), 4.3 (d, J = 5.8 Hz, 2H), 3.75 (s, 3H), 3.21 (dt, J = 12.2, 2.9 Hz, 2H), 2.77 – 2.38 (m, 3H), 1.76 – 1.41 (m, 4H), 2.51 – 2.39 (m, 2H), 1.46 – 1.31 (m, 2H), 0.93 – 0.79 (m, 3H) ; 13C-NMR (DMSO-d6) δ 173.8, 157.7, 156.8, 129.2, 128.6, 128.5, 128.3, 128.1, 125.7, 123.8, 117.6, 109.4, 59.3, 55.8, 47.9, 43.7, 42.2, 41.9, 39.7; 29.1, 21.2, 12.3; HRMS: measured [M+H]+ 359.1970 (calculated: 359.1971).
Biochemistry
sEH activity assay
The assay was performed as previously described.32 All hsEH and rsEH IC50 values were determined by a fluorescence-based assay system in a 96-well format. Non-fluorescent MNPC cyano(6-methoxy-naphthalen-2-yl)methyl trans-[(3-phenyloxiran-2-yl)methyl] carbonate was used as the assay substrate at a concentration of 5 μM. This substrate is hydrolyzed by the sEH to the fluorescent 6-methoxynaphthaldehyde.32 The formation of the product was measured (λem = 330 nm, λex = 465 nm) by a Molecular Device M-2 plate reader. All measurements were performed in triplicate.
PDE4 activity cell assay
HEK293 cell culture and transfection
HEK293 cells were cultured in DMEM (Gibco life technologies) containing 10% FBS, 100 units/ml penicillin-streptomycin, and 2 mM L-glutamine. Cell transfections were performed with the PIE Transfection reagent (Qiagen) according to manufacturer’s instructions.
Förster Resonance Energy Transfer (FRET)
Measurements to track PKA activity. HEK293 cells were transfected with PM-AKAR3 plasmid DNA (a plasma membrane-targeted PKA activity reporter) according to methods described previously.28 Images were acquired using a Zeiss AXIO inverted fluorescence microscope (Carl Zeiss micrscopy, LLC, Thornwood, NY) with a 40× oil-emersion objective lens and a charge-coupled device camera controlled by Metafluor software (Molecular Devices, Sunnyvale, CA). FRET was recorded by exciting the donor fluorophore at 430-455 nm and measuring emission fluorescence with two filters (475DF40 for cyan and 535DF25 for yellow). Images were subjected to background subtraction, and were acquired every 30 seconds with exposure time of 200 ms. The donor/acceptor FRET ratio was calculated and normalized to the ratio value of baseline. The binding of cAMP to AKAR3 increases YFP/CFP FRET ratio with increasing concentration.27,33
PDE4B1 activity assay
PDE4B1 IC50 for rat and human were conducted by BPS Bioscience according to standard procedures. MPPA in a range of 0.001 – 10 μM and Rolipram, as a reference compound, were assessed for IC50.
Water solubility approximation
Solutions of the compound under investigation were prepared in 0.1 M PBS buffer at pH 7.4 and 1% DMSO and placed in a 96-well transparent flat bottom microtiter plate. Precipitation of the compounds turbidity was measured at 650 nm using a microplate reader (Infinite M200).
Pharmacology and Behavior
Pharmacokinetic study in rat
All the animal experiments were performed according to the protocols approved by the Animal Use and Care Committee of University of California Davis. Male Sprague-Dawley rats (n=4, 8 weeks old, 250-300 g) were used in the pharmacokinetic study of sEH/PDE4 dual inhibitors. Inhibitors were dissolved in 100% polyethylene glycol 300 to form a clear solution. A cassette of four inhibitors (inhibitor 1, 19, 20 and 23; 0.3 mg/kg of each inhibitor, 0.9-1.2 mL per body weight) in solution was given by oral gavage. Whole blood (10 μL) was collected with a pipette from the tail vein punctured by a lancet at 0, 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48 and 72 hours after oral dosing with the inhibitor. Each blood sample was immediately transferred to a tube containing 100 μL of water with 0.1% EDTA and mixed and stored at −80°C until analysis. The blood samples were processed and sEH/PDE4 dual inhibitor concentrations determined according to the previously reported method by Liu et al.34
Evaluation of analgesic efficacy in rat pain model
A thermal withdrawal latency (Hargreaves’s) assay was performed as previously described.15 Thermal withdrawal thresholds (TWL) were determined before dosing to determine a baseline score. After baseline determination, 500 μL of 100% PEG300 vehicle or 3 mg/kg MPPA were oral gavaged. Immediately post oral gavage, 50 μl of a 0.2 μg/ml solution of lipopolysaccharide (LPS) from Escherichia coli (0111:B4; Sigma Aldrich L2630) in saline was injected intraplantar in one hind paw (ipsilateral paw). The rats were then assessed for TWL over a time course of 4 hours (0.5, 1, 2 and 4 hrs). The ipsilateral TWLs were measured 5 times per rat per time point and scores are reported as an average of a group of rats (n=6-9).
Open field assay
Each rat was placed in an open top acrylic box with a floor area size of 40 cm × 40 cm and walls 30 cm high marked with a 16 square grid. The exploration of the box was observed over 2 min. The score represents the sum of the number of squares the rat passed with both of its hind paws plus the number of vertical rears. Individual rats are compared to themselves (pre- and post-treatment) to calculate the percent of baseline (pre-treatment) scores and were then averaged.
Statistics
Statistical analysis for the PDE4 cell assay was performed using GraphPad Prism 6 software (La Jolla, CA). Results are shown as mean ± SEM and were analyzed by one-way ANOVA with post hoc Bonferroni’s multiple compression test. Behavioral nociceptive assay data were analyzed using SigmaPlot 11.0 for Windows (Systat Software Inc., San Jose, CA). The applied statistical methods are reported in the figure legend with p values ≤ 0.05 considered significant.
Supplementary Material
Acknowledgments
This work was supported, in part, by National Institutes of Health grants: NIEHS R01 ES002710 and NIEHS/Superfund Research Program P42 ES004699 and Grants NIEHS T32ES007059, NIH 5T32DC008072-05 and 4T32HL086350-09 (to K.W.). A personal thanks is directed at the Services for International Students and Scholars (SISS) and within this organization to Simone Kueltz. EP thanks the German Research Foundation (DFG; Sachbeihilfe PR1405/2-2; Heisenberg-Professur PR1405/4-1; SFB 1039 Teilprojekt A07) for financial support.
Abbreviations
- % of Bl
percent of baseline
- 5-AMP
5 adenosine monophosphate
- cAMP
cyclic adenosine monophosphate
- Cmax
the maximum (or peak) serum concentration that a drug achieves in a specified compartment
- COPD
chronic obstructive pulmonary diseases
- Ctrl
control
- CYP450
cytochrome P450
- DHETs
dihydroxy eicosatrienoic acid
- DMSO
Dimethylsulfoxide
- EETs
epoxyeicosatrienoic acids
- ESI
Electrospray ionization
- FBS
Fetal bovine serum
- FDA
Food and Drug Administration
- FRET
Förster Resonance Energy Transfer
- GSK
Glaxosmithkline
- HPLC
High-performance liquid chromatography
- HRMS
High-resolution mass spectrometry
- hsEH
human soluble epoxide hydrolase
- IC50
the concentration of an inhibitor where the response (or binding) is reduced by half
- LPS
lipopolysaccharide
- mg/kg
mg compound per kg bodyweight
- PD
pharmacodynamics
- PDE4
phosphodiesterase 4
- PDE4I
phosphodiesterase 4 inhibitor
- PEG300
Polyethylene Glycol 300
- PK
pharmacokinetic
- PKA
protein kinase A
- PO
per oral
- PsA
psoriatic arthritis
- rsEH
rat soluble epoxide hydrolase
- rt
room temperature
- sEH
soluble epoxide hydrolase
- sEHI
soluble epoxide hydrolase inhibitor
- Tmax
the amount of time that a drug is present at the maximum concentration in blood
- TWL
Thermal withdrawal threshold
- ws
water solubility
Footnotes
Supporting Information
The results for PDE4 screening value tables and figures, Pharmacokinetics table, Naïve control TWLs and open field assay are given in supporting information as well as the molecular formula strings. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Reddy AS, Zhang S. Polypharmacology: Drug Discovery for the Future. Expert Rev Clin Pharmacol. 2013;6(1):41–47. doi: 10.1586/ecp.12.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Aso M, Montesinos MC, Mediero A, Wilder T, Schafer PH, Cronstein B. Apremilast, a Novel Phosphodiesterase 4 (PDE4) Inhibitor, Regulates Inflammation through Multiple cAMP Downstream Effectors. Arthritis Res Ther. 2015;17:249. doi: 10.1186/s13075-015-0771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown WM. Treating COPD with PDE 4 Inhibitors. Int J Chron Obstruct Pulmon Dis. 2007;2(4):517–533. [PMC free article] [PubMed] [Google Scholar]
- 4.Ashton MJ, Cook DC, Fenton G, Karlsson JA, Palfreyman MN, Raeburn D, Ratcliffe AJ, Souness JE, Thurairatnam S, Vicker N. Selective Type IV Phosphodiesterase Inhibitors as Antiasthmatic Agents. The Syntheses and Biological Activities of 3-(Cyclopentyloxy)-4-Methoxybenzamides and Analogues. J Med Chem. 1994;37(11):1696–1703. doi: 10.1021/jm00037a021. [DOI] [PubMed] [Google Scholar]
- 5.Fujita M, Richards EM, Niciu MJ, Hines CS, Pike VW, Jr, Z CA. cAMP Signaling in Brain Is Decreased in Unmedicated Depressed Patients and Increased by Treatment with a Selective Serotonin Reuptake Inhibitor. Mol Psychiatry. 2017;22(5):754–759. doi: 10.1038/mp.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandon NJ. Uncovering the Function of Disrupted in Schizophrenia 1 through Interactions with the cAMP Phosphodiesterase PDE4: Contributions of the Houslay Lab to Molecular Psychiatry. Cell Signal. 2016;28(7):749–752. doi: 10.1016/j.cellsig.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 7.Gurney ME, D’Amato EC, Burgin AB. Phosphodiesterase-4 (PDE4) Molecular Pharmacology and Alzheimer’s Disease. Neurotherapeutics. 2015;12(1):49–56. doi: 10.1007/s13311-014-0309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dal Piaz V, Giovannoni MP. Phosphodiesterase 4 Inhibitors, Structurally Unrelated to Rolipram, as Promising Agents for the Treatment of Asthma and Other Pathologies. Eur J Med Chem. 2000;35(5):463–480. doi: 10.1016/s0223-5234(00)00179-3. [DOI] [PubMed] [Google Scholar]
- 9.Rock EM, Benzaquen J, Limebeer CL, Parker LA. Potential of the Rat Model of Conditioned Gaping to Detect Nausea Produced by Rolipram, a Phosphodiesterase-4 (PDE4) Inhibitor. Pharmacol Biochem Behav. 2009;91(4):537–541. doi: 10.1016/j.pbb.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Prickaerts J, Heckman PRA, Blokland A. Investigational Phosphodiesterase Inhibitors in Phase I and Phase II Clinical Trials for Alzheimer’s Disease. Expert Opin Investig Drugs. 2017;26(9):1033–1048. doi: 10.1080/13543784.2017.1364360. [DOI] [PubMed] [Google Scholar]
- 11.Mclellan GJ, Aktas Z, Hennes-beean E, Kolb AW, Larsen V, Schmitz EJ, Clausius HR, Yang J, Hwang SH, Morisseau C, Inceoglu B, Hammock BD, Brandt CR. Induced Uveitis in the Rabbit. J Ocul Biol. 2017;4(1):1–17. doi: 10.13188/2334-2838.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner K, Inceoglu B, Hammock BD. Soluble Epoxide Hydrolase Inhibition, Epoxygenated Fatty Acids and Nociception. Prostaglandins Other Lipid Mediat. 2011;96(1–4):76–83. doi: 10.1016/j.prostaglandins.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morisseau C, Hammock BD. Impact of Soluble Epoxide Hydrolase and Epoxyeicosanoids on Human Health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren Q, Ma M, Ishima T, Morisseau C, Yang J, Wagner KM, Zhang J. Gene Deficiency and Pharmacological Inhibition of Soluble Epoxide Hydrolase Confers Resilience to Repeated Social Defeat Stress. Proc Natl Acad Sci U S A. 2016;113(13):E1944–E1952. doi: 10.1073/pnas.1601532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD. Analgesia Mediated by Soluble Epoxide Hydrolase Inhibitors Is Dependent on cAMP. Proc Natl Acad Sci USA. 2011;108(12):5093–5097. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blöcher R, Lamers C, Wittmann SK, Merk D, Hartmann M, Weizel L, Diehl O, Brüggerhoff A, Boß M, Kaiser A, Schader T, Göbel T, Grundmann M, Angioni C, Heering J, Geisslinger G, Wurglics M, Kostenis E, Brüne B, Steinhilber D, Schubert-Zsilavecz M, Kahnt AS, Proschak E. N-Benzylbenzamides: A Novel Merged Scaffold for Orally Available Dual Soluble Epoxide Hydrolase/Peroxisome Proliferator-Activated Receptor Gamma Modulators. J Med Chem. 2016;59(1):61–81. doi: 10.1021/acs.jmedchem.5b01239. [DOI] [PubMed] [Google Scholar]
- 17.la Buscató E, Blöcher R, Lamers C, Klingler F-M, Hahn S, Steinhilber D, Schubert-Zsilavecz M, Proschak E. Design and Synthesis of Dual Modulators of Soluble Epoxide Hydrolase and Peroxisome Proliferator-Activated Receptors. J Med Chem. 2012;55(23):10771–10775. doi: 10.1021/jm301194c. [DOI] [PubMed] [Google Scholar]
- 18.Meirer K, Glatzel D, Kretschmer S, Wittmann S, Hartmann M, Blöcher R, Angioni C, Geisslinger G, Steinhilber D, Hofmann B, Fürst R, Proschak E. Design, Synthesis and Cellular Characterization of a Dual Inhibitor of 5-Lipoxygenase and Soluble Epoxide Hydrolase. Molecules. 2016;22(1):E45. doi: 10.3390/molecules22010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Podolin PL, Bolognese BJ, Foley JF, Long E, Peck B, Umbrecht S, Zhang X, Zhu P, Schwartz B, Xie W, Quinn C, Qi H, Sweitzer S, Chen S, Galop M, Ding Y, Belyanskaya SL, Israel DI, Morgan BA, Behm DJ, Marino JP, Kurali E, Barnette MS, Mayer RJ, Booth-Genthe CL, Callahan JF. In Vitro and in Vivo Characterization of a Novel Soluble Epoxide Hydrolase Inhibitor. Prostaglandins Other Lipid Mediat. 2013;104–105:25–31. doi: 10.1016/j.prostaglandins.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, Newby DE, Wilkinson IB, Tal-Singer R, Mayer RJ, Cheriyan J. Pharmacokinetics, Pharmacodynamics and Adverse Event Profile of GSK2256294, a Novel Soluble Epoxide Hydrolase Inhibitor. Br J Clin Pharmacol. 2016;81(5):971–979. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillette S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim SH, Schlessinger J, Zhang KYJ. Structural Basis for the Activity of Drugs That Inhibit Phosphodiesterases. Structure. 2004;12(12):2233–2247. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Lee KSS, Liu JY, Wagner KM, Pakhomova S, Dong H, Morisseau C, Fu SH, Yang J, Wang P, Ulu A, Mate CA, Nguyen LV, Hwang SH, Edin ML, Mara AA, Wulff H, Newcomer ME, Zeldin DC, Hammock BD. Optimized Inhibitors of Soluble Epoxide Hydrolase Improve in Vitro Target Residence Time and in Vivo Efficacy. J Med Chem. 2014;57(16):7016–7030. doi: 10.1021/jm500694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown A, Rawson D, Storer R, Swain A. Chemical Compounds. 007868 A2. US Patent. 2012
- 24.Anandan SK, Webb HK, Chen D, Wang YX, Aavula BR, Cases S, Cheng Y, Do ZN, Mehra U, Tran V, Vincelette J, Waszczuk J, White K, Wong KR, Zhang LN, Jones PD, Hammock BD, Patel DV, Whitcomb R, MacIntyre DE, Sabry J, Gless R. 1-(1-Acetyl-Piperidin-4-Yl)-3-Adamantan-1-Yl-Urea (AR9281) as a Potent, Selective, and Orally Available Soluble Epoxide Hydrolase Inhibitor with Efficacy in Rodent Models of Hypertension and Dysglycemia. Bioorganic Med Chem Lett. 2011;21(3):983–988. doi: 10.1016/j.bmcl.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Z, Pissarnitski DA, Josien HB, Bara TA, Clader JW, Li H, McBriar MD, Rajagopalan M, Xu R, Terracina G, Hyde L, Song L, Zhang L, Parker EM, Osterman R, Buevich AV. Substituted 4-Morpholine N-Arylsulfonamides as Gamma-Secretase Inhibitors. Eur J Med Chem. 2016;124:36–48. doi: 10.1016/j.ejmech.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 26.Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-Aryl-3-(1-Acylpiperidin-4-Yl)urea Inhibitors of Human and Murine Soluble Epoxide Hydrolase: Structure-Activity Relationships, Pharmacokinetics, and Reduction of Inflammatory Pain. J Med Chem. 2010;53(19):7067–7075. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soto D, De Arcangelis V, Zhang J, Xiang YK, Liu S, Li Y, Kim S, Fu Q, Parikh D, Sridhar B, Shi Q, Zhang X, Guan Y, Chen X, Xiang YK. Phosphodiesterases Coordinate cAMP Propagation Induced by Two Stimulatory G Protein-Coupled Receptors in Hearts. Circ Res. 2009;109(17):6578–6583. doi: 10.1073/pnas.1117862109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bian H, Zhang J, Wu P, Varty LA, Jia Y, Mayhood T, Hey JA, Wang P. Differential Type 4 cAMP-Specific Phosphodiesterase (PDE4) Expression and Functional Sensitivity to PDE4 Inhibitors among Rats, Monkeys and Humans. Biochem Pharmacol. 2004;68(11):2229–2236. doi: 10.1016/j.bcp.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Halpin DMG. ABCD of the Phosphodiesterase Family: Interaction and Differential Activity in COPD. Int J Chron Obstruct Pulmon Dis. 2008;3(4):543–561. doi: 10.2147/copd.s1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved Expression and Functions of PDE4 in Rodent and Human Heart. Basic Res Cardiol. 2011;106:249–262. doi: 10.1007/s00395-010-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang SH, Wagner KM, Morisseau C, Liu J-Y, Dong H, Wecksler AT, Hammock BD. Synthesis and Structure-Activity Relationship Studies of Urea-Containing Pyrazoles as Dual Inhibitors of Cyclooxygenase-2 and Soluble Epoxide Hydrolase. J Med Chem. 2011;54(8):3037–3050. doi: 10.1021/jm2001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent Substrates for Soluble Epoxide Hydrolase and Application to Inhibition Studies. Anal Biochem. 2005;343(1):66–75. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res. 2016;119(8):931–943. doi: 10.1161/CIRCRESAHA.116.308964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JY, Lin YP, Qiu H, Morisseau C, Rose TE, Hwang SH, Chiamvimonvat N, Hammock BD. Substituted Phenyl Groups Improve the Pharmacokinetic Profile and Anti-Inflammatory Effect of Urea-Based Soluble Epoxide Hydrolase Inhibitors in Murine Models. Eur J Pharm Sci. 2013;48(4–5):619–627. doi: 10.1016/j.ejps.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.