

SUMMARY
Although EZH2 enzymatic inhibitors have shown anti-tumor effects in EZH2-mutated lymphoma and ARID1A-mutated ovarian cancer, many cancers do not respond because EZH2 can promote cancer independently of its histone methyltransferase activity. Here we identify ZRANB1 as the EZH2 deubiquitinase. ZRANB1 binds, deubiquitinates, and stabilizes EZH2. Depletion of ZRANB1 in breast cancer cells results in EZH2 destabilization and growth inhibition. Systemic delivery of ZRANB1 small interfering RNA (siRNA) leads to marked antitumor and antimetastatic effects in preclinical models of triple-negative breast cancer (TNBC). Intriguingly, a small-molecule inhibitor of ZRANB1 destabilizes EZH2 and inhibits the viability of TNBC cells. In patients with breast cancer, ZRANB1 levels correlate with EZH2 levels and poor survival. These findings suggest the therapeutic potential for targeting the EZH2 deubiquitinase ZRANB1.
In Brief
Many cancer cells are sensitive to depletion of EZH2 but resistant to EZH2 inhibitors, due to EZH2’s enzyme-independent cancer-promoting function. Zhang et al. identify ZRANB1 as an EZH2 deubiquitinase and a potential anticancer target.
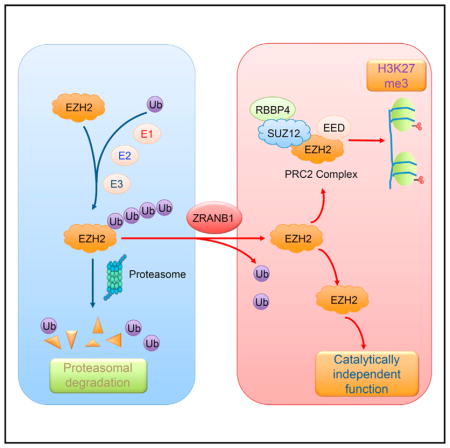
INTRODUCTION
EZH2, the catalytic component of the Polycomb repressive complex 2 (PRC2), silences gene transcription by methylating histone H3 at lysine 27 (Czermin et al., 2002; Müller et al., 2002). EZH2 is mutated or highly expressed in many types of cancer, including lymphoma (Morin et al., 2010), melanoma (Bachmann et al., 2006), prostate cancer (Varambally et al., 2002), ovarian cancer (Lu et al., 2010), and breast cancer (Kleer et al., 2003). Experimentally, overexpression of EZH2 has been shown to promote cell proliferation, tumorigenesis, and metastasis (Bracken et al., 2003; Chang et al., 2011; Kleer et al., 2003; Min et al., 2010). Conversely, depletion of EZH2 leads to growth inhibition. For example, knockdown of EZH2 in a triple-negative breast cancer (TNBC) cell line, MDA-MB-231, suppressed tumor growth and metastasis in xenograft models (Gonzalez et al., 2009; Moore et al., 2013). The cancer-promoting function of EZH2 is also supported by genetically engineered mouse models. For instance, transgenic overexpression of EZH2 or its gain-of-function mutant in mice led to hyperplasia and accelerated Myc- or Bcl2-induced lymphomagenesis (Béguelin et al., 2013; Berg et al., 2014) and Erbb2-induced mammary tumorigenesis (Gonzalez et al., 2014; Li et al., 2009). Moreover, expression of a lymphoma-derived hyperactivating mutant of EZH2 from the endogenous locus in mouse B cells or melanocytes caused high-penetrance lymphoma or melanoma, respectively (Souroullas et al., 2016). These findings have prompted intensive efforts to develop EZH2 inhibitors.
Two recently developed, highly specific EZH2 enzymatic inhibitors, GSK126 and EPZ-6438, are currently in clinical trials for treating lymphomas (Kim and Roberts, 2016). Although these EZH2 inhibitors have shown antitumor effects in lymphoma cells with enzyme-activating mutations of EZH2 (Knutson et al., 2012; McCabe et al., 2012; Qi et al., 2012) and in ovarian cancer cells with inactivating mutations of ARID1A (Bitler et al., 2015), certain cancer cells are resistant to the enzymatic inhibition of EZH2 but sensitive to the genetic depletion of EZH2, suggesting that the tumor-promoting function of EZH2 depends on its catalytic and non-catalytic activity. Indeed, independently of its histone methyltransferase activity, EZH2 can promote cancer by stabilizing the PRC2 (Kim et al., 2015) or by acting as a transcriptional coactivator of androgen receptor (Xu et al., 2012), estrogen receptor (Shi et al., 2007), β-catenin (Shi et al., 2007), and nuclear factor κB (NF-κB) (Lee et al., 2011). Consequently, destroying EZH2 protein should be more effective than EZH2 inhibitors in targeting cancers that are dependent on EZH2’s non-catalytic activity.
The highly conserved, 76-amino acid polypeptide ubiquitin is added to protein substrates through a multi-step process starting with ubiquitin activation by a ubiquitin-activating enzyme (E1), followed by its transfer to a lysine residue on the substrate, which is mediated by ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3) (Glickman and Ciechanover, 2002; Pickart, 2001). Ubiquitin contains seven lysines. Whereas lysine 63 (K63)-linked polyubiquitination alters the substrate’s subcellular localization, affects its activity, and modulates its interaction with other proteins (Chen and Sun, 2009; Pickart and Fushman, 2004), all non-K63 ubiquitin linkages can target proteins for degradation via the proteasome (Xu et al., 2009). Ubiquitination is reversed by deubiquitinating enzymes (DUBs, or deubiquitinases), a group of proteases that remove monoubiquitin or poly-ubiquitin chains from the substrate (Wilkinson, 1997; Xiao et al., 2016). EZH2 protein is subject to ubiquitin-dependent degradation by several E3 ligases, including β-TrCP, SMURF2, and FBW7 (Jin et al., 2017; Sahasrabuddhe et al., 2015; Yu et al., 2013); however, the deubiquitinase that reverses this ubiquitination is unknown. Here we identify an ovarian tumor protease (OTU) family member, ZRANB1 (also known as Trabid), as an EZH2 deubiquitinase and a potential therapeutic target in cancer.
RESULTS
ZRANB1 Regulates EZH2 Protein Level
GSK126 has been shown to inhibit the growth of EZH2-mutated lymphoma (McCabe et al., 2012) and ARID1A-mutated ovarian cancer (Bitler et al., 2015). To evaluate the effect of GSK126 on breast cancer cell proliferation, we treated two TNBC cell lines, BT549 and a lung-metastatic subline (LM2) of MDA-MB-231 cells (Minn et al., 2005), with this compound. A previously reported GSK126-sensitive human lung cancer cell line (Kim et al., 2015), A549, was used as a positive control. In the presence of 8 μM GSK126, A549 cells showed an approximately 80% reduction in the number of viable cells in a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figure S1A), whereas BT549 and LM2 cells showed a modest reduction and no reduction, respectively (Figures S1B and S1C). In all three cell lines, 2 μM GSK126 treatment completely abrogated histone H3 lysine 27 (H3K27) trimethylation (H3K27me3), the marker of EZH2 activity (Figures S1A–S1C), despite no anti-proliferative effect on LM2 cells. On the other hand, small hairpin RNA (shRNA)-mediated silencing of EZH2 expression inhibited the proliferation of LM2 cells (Figure S1D), suggesting that LM2 cells are responsive to knockdown of EZH2 but refractory to enzymatic inhibition of EZH2.
We reasoned that targeting the EZH2 DUB could lead to the destabilization of EZH2. To identify the EZH2 DUB, we first screened for EZH2-interacting DUBs using a panel of 46 DUBs fused to a triple-epitope tag, SFB (S-protein, FLAG tag, and streptavidin-binding peptide) (Zhang et al., 2013). We co-transfected each SFB-tagged DUB with MYC-tagged EZH2 into HEK293T cells, pulled down the DUBs with S-protein beads, and detected physical association of EZH2 with six DUBs: USP22, USP39, USP44, USP49, USP53, and ZRANB1 (Figure 1A). Reciprocally, each of these six SFB-tagged DUBs could be pulled down by MYC-EZH2, but not by MYC-GFP (Figure S2A). To determine whether the six EZH2-interacting DUBs affect EZH2 ubiquitination and protein levels, we transfected them individually into HEK293T cells, and we found that USP22 and ZRANB1 decreased the polyubiquitination of EZH2 (Figure S2B); however, only ZRANB1 upregulated endogenous EZH2 protein (Figure S2C). Conversely, silencing ZRANB1 expression in MDA-MB-231 cells by two independent small interfering RNAs (siRNAs) (2 and 5) reduced endogenous EZH2 protein levels by 70% and 60%, respectively (Figure 1B). Moreover, overexpression of ZRANB1 in HEK293T cells increased endogenous EZH2 and H3K27 trimethylation levels, which could be reversed by co-expression of ZRANB1 siRNA (Figure 1C). It should be noted that knockdown of ZRANB1 did not change mRNA levels of EZH2 (Figure S2D) or other PRC2 components, EED, RBBP4, and SUZ12 (Figure S2E). We conclude from these data that ZRANB1 is a positive regulator of EZH2 protein, but not EZH2 mRNA. Furthermore, purified His-ZRANB1 could bind to purified GST-EZH2 under cell-free conditions (Figure 1D), suggesting that ZRANB1 may directly regulate EZH2.
Figure 1. ZRANB1 Regulates EZH2 Protein Level.

(A) Six of 46 DUBs physically associate with EZH2. Each SFB-tagged DUB was co-transfected with MYC-tagged EZH2 into HEK293T cells, followed by pull-down with S-protein beads and immunoblotting with antibodies against FLAG and MYC.
(B) Immunoblotting of EZH2, ZRANB1, and GAPDH in MDA-MB-231 cells transfected with ZRANB1 siRNA or a scramble control (Scr).
(C) Immunoblotting of EZH2, H3K27me3, H3, and FLAG-ZRANB1 in HEK293T cells transfected with SFB-ZRANB1 and ZRANB1 siRNA, alone or in combination.
(D) Left panel: purified GST-EZH2 was incubated with purified His-ZRANB1, followed by pull-down with nickel beads and immunoblotting with antibodies against EZH2 and ZRANB1. Right panel: purified recombinant proteins were analyzed by SDS-PAGE and Coomassie blue staining.
(E) Immunoblotting of EZH2, SUZ12, EED, ZRANB1, H3K27me1, H3K27me2, H3K27me3, H3, and β-actin in two independent clones of ZRANB1-knockout HEK293A cells generated by CRISPR-Cas9.
(F) Co-immunoprecipitation (coIP) of endogenous EZH2 with endogenous ZRANB1, SUZ12, and EED. EZH2 was immunoprecipitated from HEK293T cells, followed by immunoblotting with antibodies against ZRANB1, EZH2, SUZ12, and EED.
(G) Heatmap of 63 genes that were identified by RNA-seq analysis to be commonly upregulated (49 genes) or commonly downregulated (14 genes) in ZRANB1-knockdown and EZH2-knockdown LM2 cells. Threshold values are as follows: corrected p value < 0.05 and absolute log2 fold change (log2FC) > 0.7. Genes are shown in rows and samples are shown in columns. n = 3 samples per group.
(H) Venn diagrams of all genes that were identified by RNA-seq analysis to be upregulated (upper panel) or downregulated (lower panel) in ZRANB1-knockdown (blue) or EZH2-knockdown (pink) LM2 cells. Threshold values are as follows: corrected p value < 0.05 and absolute log2 fold change (log2FC) > 0.7. n = 3 samples per group. Additional information is available in Table S1.
See also Figures S1 and S2.
To corroborate the siRNA results, we used a CRISPR-Cas9 approach to knock out ZRANB1 in HEK293A cells that express abundant endogenous ZRANB1 protein (Figure 1E). Compared with cells transfected with a control guide RNA (gRNA), two independent clones derived from transfection with the ZRANB1 gRNA both showed depletion of ZRANB1 (gene editing in the targeted region was confirmed by sequencing the clones) and downregulation of EZH2 (Figure 1E). Consistent with a reported role of EZH2 in stabilizing the PRC2 (Kim et al., 2015), protein levels of other PRC2 components, SUZ12 and EED, were decreased in ZRANB1-knockout cells (Figure 1E). Moreover, ablation of ZRANB1 abrogated H3K27me3, but not H3K27me1 or H3K27me2 (Figure 1E). Similarly, GSK126 treatment has been shown to diminish H3K27me3 without affecting H3K27me1 or H3K27me2 at 25–500 nM (McCabe et al., 2012).
Furthermore, we observed the interaction of ZRANB1 with endogenous EZH2 and SUZ12 (Figure S2F) and, reciprocally, the interaction of either exogenous (Figure S2G) or endogenous (Figure 1F) EZH2 with endogenous ZRANB1, SUZ12, and EED, suggesting that ZRANB1 may regulate EZH2 that is present in the PRC2. Interestingly, knockdown of EZH2 impaired but did not abolish the interaction of ZRANB1 with SUZ12, RBBP4, and EED (Figure S2H), indicating that EZH2 partially mediates the association of ZRANB1 with the PRC2. We then examined the expression of 10 previously reported PRC2 target genes, and we found that ADRB2 and DAB2IP were significantly upregulated by knockdown of EZH2 in MDA-MB-231 cells (Figure S2I); consistently, knockdown of ZRANB1 in MDA-MB-231 cells also increased ADRB2 and DAB2IP mRNA levels (Figure S2J). Moreover, we performed RNA sequencing (RNA-seq) analysis to compare the effect of ZRANB1 and EZH2 on global gene expression. A total of 63 genes, including known EZH2 targets or downstream genes SERPINB2 (Kim et al., 2012), PTGS2 (encoding cyclooxygenase-2) (Coward et al., 2014), ABCA1 (Lv et al., 2016), and MMP9 (Delgado-Olguín et al., 2014), were differentially expressed (49 upregulated and 14 downregulated) by at least 1.6-fold (p < 0.05) upon knockdown of either ZRANB1 or EZH2 in LM2 cells (Figures 1G and 1H; Table S1). Taken together, our data suggest that ZRANB1 is a functional regulator of EZH2.
ZRANB1 Is an EZH2 Deubiquitinase
To determine whether ZRANB1 stabilizes EZH2 protein, we first examined the levels of ectopically expressed EZH2 protein in HEK293T cells in the presence of cycloheximide (CHX), an inhibitor of protein synthesis. As expected, overexpression of ZRANB1 led to a prominent increase in the basal level and half-life of EZH2 protein, whereas the level and stability of co-transfected GFP were not affected (Figures 2A and S3A). Conversely, knockdown of ZRANB1 in LM2 or MDA-MB-231 cells markedly shortened the half-life of endogenous EZH2, SUZ12, and EED (Figures 2B, S3B, and S3C). Unlike ZRANB1, USP22 overexpression did not affect EZH2 protein half-life (data not shown).
Figure 2. ZRANB1 Is an EZH2 Deubiquitinase.

(A) HEK293T cells were co-transfected with MYC-EZH2, MYC-GFP, and SFB-ZRANB1; treated with 50 μg mL−1 cycloheximide (CHX); harvested at different time points; and then immunoblotted with antibodies against MYC and FLAG. MYC-GFP serves as the control for transfection.
(B) LM2 cells were transfected with ZRANB1 siRNA, treated with 50 μg mL−1 CHX, harvested at different time points, and then immunoblotted with antibodies against EZH2 and β-actin.
(C) ZRANB1-knockout HEK293A cells were co-transfected with MYC-EZH2 and SFB-ZRANB1, followed by immunoprecipitation with anti-MYC beads and immunoblotting with antibodies against ubiquitin (Ub) and MYC. Cells were treated with 10 μM MG132 for 6 hr before collection. Before immunoprecipitation, lysates were heated at 95°C for 5 min in the presence of 1% SDS (for denaturing), followed by 10-fold dilution with lysis buffer and sonication.
(D) Ubiquitinated MYC-EZH2 was purified with anti-MYC beads from ZRANB1-knockout HEK293A cells co-transfected with MYC-EZH2 and HA-ubiquitin (Ub), and it was incubated with His-ZRANB1 purified from insect cells. After the in vitro deubiquitination reaction, the bound proteins were eluted by boiling in Laemmli sample buffer and immunoblotted with antibodies against HA and EZH2.
(E) Control and ZRANB1-knockout HEK293A cells were co-transfected with MYC-EZH2 and lysine-specific mutants of HA-ubiquitin (Ub), followed by immunoprecipitation with anti-HA beads and immunoblotting with antibodies against EZH2 and HA. Cells were treated with 10 μM MG132 for 6 hr before collection. Before immunoprecipitation, lysates were heated at 95°C for 5 min in the presence of 1% SDS (for denaturing), followed by 10-fold dilution with lysis buffer and sonication.
(F) K33 linkage-specific ubiquitinated MYC-EZH2 was purified with anti-MYC beads from ZRANB1-knockout HEK293A cells co-transfected with MYC-EZH2 and the K33-specific mutant of HA-ubiquitin (Ub), and it was incubated with His-ZRANB1 purified from insect cells. After the in vitro deubiquitination reaction, the bound proteins were eluted by boiling in Laemmli sample buffer and immunoblotted with antibodies against HA and EZH2.
See also Figure S3.
We reasoned that ZRANB1 stabilizes EZH2 through deubiquitination. Indeed, overexpression of ZRANB1 in ZRANB1-knockout HEK293A cells substantially reduced EZH2 polyubiquitination (Figure 2C, denaturing condition). It should be noted that ZRNAB1 overexpression also decreased the polyubiquitination of SUZ12, but not EED (Figure S3D). To further determine whether ZRANB1 directly deubiquitinates EZH2, we incubated purified ZRANB1 and ubiquitinated EZH2 purified from ZRANB1-knockout HEK293A cells in a cell-free system. ZRANB1 purified from insect cells markedly decreased EZH2 polyubiquitination in vitro (Figure 2D), suggesting that EZH2 is a substrate of ZRANB1. Consistent with this, knockout of ZRANB1 by gRNA increased EZH2 ubiquitination (Figure S3E). Previous studies have revealed that ZRANB1 preferentially cleaves K29-, K33-, and K63-linked poly-ubiquitin chains (Licchesi et al., 2011; Mevissen et al., 2013; Michel et al., 2015). Using all seven lysine-specific mutants of ubiquitin (e.g., the K33 mutant contains only a single lysine, K33, with all other six lysines mutated to arginine), we found that ZRANB1-knockout cells showed upregulation of K33-linked, but not other lysine-linked, polyubiquitination of EZH2 (Figure 2E, denaturing condition; Figure S3E, non-denaturing condition); moreover, purified ZRANB1 strongly deubiquitinated K33 linkage-specific ubiquitinated EZH2 in vitro (Figure 2F).
ZRANB1 consists of three N-terminal zinc-finger (NZF) domains, a central ankyrin repeat ubiquitin-binding domain (AnkUBD), and a C-terminal OTU domain (Michel et al., 2015). Deletion analysis using various truncation mutants (M1, NZFs; M2, NZFs + AnkUBD; M3, AnkUBD + OTU; Figure 3A) demonstrated that the OTU domain not only mediated the physical interaction of ZRANB1 with EZH2 (Figure 3B) but also was required for the stabilization of EZH2 by ZRANB1 (Figure 3C). Consistent with a previous report (Jin et al., 2016), we observed both nuclear and cytoplasmic expression of ZRANB1 (Figure 3D), as well as its co-localization with EZH2 in the nucleus (Figure S3F). It should be noted that all three truncation mutants of ZRANB1 were also localized in both the nucleus and the cytoplasm (Figure 3D), suggesting that the M1 and M2 mutants of ZRANB1 did not interact with EZH2 because they lack the OTU domain, not because they were not localized in the nucleus. Therefore, the OTU domain of ZRANB1 is a functional domain in regulating EZH2. Furthermore, binding-domain mapping analysis showed that both the N-terminal region (EID + D1) and the CXC domain of EZH2 could interact with ZRANB1 (Figures 3E, 3F, S3G, and S3H).
Figure 3. ZRANB1 Interacts with and Stabilizes EZH2 through Its OTU Domain.

(A) Schematic diagram of full-length ZRANB1 protein and its various deletion mutants.
(B) HEK293T cells were co-transfected with equal amounts of MYC-EZH2 and SFB-tagged full-length ZRANB1 or its deletion mutants, followed by pull-down with S-protein beads and immunoblotting with antibodies against MYC and FLAG.
(C) HEK293T cells were co-transfected with MYC-EZH2, MYC-GFP, and excessive amounts of SFB-tagged full-length ZRANB1 or its deletion mutants. 24 hr after transfection, cells were harvested and immunoblotted with antibodies against MYC and FLAG.
(D) Immunoblotting of FLAG-ZRANB1, Vinculin (cytoplasmic marker), and Lamin A/C (nuclear marker) in cytoplasmic and nuclear fractions of HEK293T cells transfected with SFB-tagged full-length ZRANB1 or its deletion mutants.
(E) Schematic diagram of full-length EZH2 protein and its various deletion mutants. EID, EED interaction domain; D1 and D2, homologous domains 1 and 2; CXC, cysteine-rich domain; SET, SU(var)3–9, E(z), and Trithorax histone methyltransferase domain.
(F) HEK293T cells were co-transfected with HA-ZRANB1 and FLAG-tagged full-length EZH2 or its deletion mutants, followed by immunoprecipitation with anti-HA beads and immunoblotting with antibodies against FLAG and HA.
See also Figure S3.
ZRANB1 Is a Cancer-Promoting Deubiquitinase and a Potential Therapeutic Target
Whereas EZH2 has been shown to promote mammary tumorigenesis and metastasis (Alford et al., 2012; Chang et al., 2011; Gonzalez et al., 2009, 2014; Kleer et al., 2003; Li et al., 2009; Moore et al., 2013), the function of ZRANB1 in cancer is unknown. We transfected two independent ZRANB1 siRNAs into MDA-MB-231 cells, and we found that both siRNAs drastically inhibited cell proliferation (Figure 4A) and migration (Figure 4B). Similarly, two independent ZRANB1 gRNA clones of HEK293A cells showed severe growth defect (Figure 4C). siRNA-mediated knockdown of ZRANB1 also led to EZH2 down-regulation and proliferative deficiency in nine additional TNBC cell lines, BT549, SUM149, SUM159, SUM229, MDA-MB-157, MDA-MB-436, MDA-MB-468, HCC1806, and Hs578T (Figures S4A–S4C). However, non-TNBC cell lines, T47D (ER+HER2−), ZR75-1 (ER+HER2+), SKBR3 (ER−HER2+), and MCF7 (ER+HER2−), showed variable responses: knockdown of ZRANB1 inhibited the proliferation of SKBR3 and MCF7 cells, but not T47D and ZR75-1 cells, even though EZH2 was downregulated (Figures S4D–S4F). Importantly, re-expression of ZRANB1 rescued the inhibitory effect of ZRANB1 siRNA (Figures S5A and S5B) and ZRANB1 gRNA (Figures 4D and 4E) on EZH2 protein level and cell proliferation. In addition, knockdown of ZRANB1 in MDA-MB-231 cells increased early apoptosis and late apoptosis (cell death) by 3-fold and 9-fold, respectively (Figure S5C).
Figure 4. ZRANB1 Is a Growth-Promoting Deubiquitinase.

(A) Growth curves of MDA-MB-231 cells transfected with ZRANB1 siRNA or a scramble control. n = 3 biological replicates.
(B) Transwell migration assays of MDA-MB-231 cells transfected with ZRANB1 siRNA or a scramble control. Cells were stained 18 hr after seeding.
(C) Growth curves of two independent clones of ZRANB1-knockout HEK293A cells generated by CRISPR-Cas9. n = 3 biological replicates.
(D) Immunoblotting of EZH2, ZRANB1, and HSP90 in control, ZRANB1-knockout, and ZRANB1-restored HEK293A cells.
(E) Growth curves of control, ZRANB1-knockout, and ZRANB1-restored HEK293A cells. n = 3 biological replicates.
(F and G) LM2 cells were transduced with EZH2 shRNA or a scramble control, and they were then transfected with SFB-ZRANB1. 24 hr after transfection, cells were harvested for immunoblotting with antibodies against EZH2, FLAG, and β-actin (F) or seeded in 24-well plates for growth curves (G). n = 3 biological replicates in (G).
(H) Immunoblotting of EZH2 and β-actin in control and MYC-EZH2-transduced LM2 cells with or without ZRANB1 siRNA transfection.
(I) Growth curves of control and MYC-EZH2-transduced LM2 cells with or without ZRANB1 siRNA transfection. n = 3 biological replicates.
(J and K) ZRANB1 siRNA-transfected MDA-MB-231 cells were transfected with SFB-tagged full-length EZH2 (FL) or its catalytically inactive mutant (ΔSET), and they were then harvested for immunoblotting with antibodies against EZH2, FLAG, and β-actin (J) or seeded in 6-well plates for growth curves (K). n = 3 biological replicates in (K).
Error bars in (A), (C), (E), (G), (I), and (K) are SEM; p values were calculated from a two-tailed t test. See also Figures S4 and S5.
We asked whether EZH2 is a functional effector of ZRANB1. Ectopic expression of ZRANB1 in both LM2 cells (Figures 4F and 4G) and HEK293T cells (Figures S5D and S5E) markedly upregulated endogenous EZH2 protein and increased cell proliferation, which could be reversed by knockdown of EZH2. Conversely, expression of either wild-type EZH2 or the catalytically inactive mutant (ΔSET) largely rescued ZRANB1 siRNA-induced growth defect in TNBC cells (Figures 4H–4K), underscoring the importance of EZH2’s non-catalytic function. Moreover, depletion of ZRANB1 in LM2 cells inhibited secondary mammosphere formation, a surrogate assay of breast tumor-initiating ability, and this inhibition was partially reversed by ectopic expression of EZH2 (Figure S5F). Taken together, ZRANB1 is a growth-promoting deubiquitinase, and its function is mediated, at least in part, by its substrate EZH2.
To determine whether the inhibition of ZRANB1 has anticancer effects in vivo, we incorporated ZRANB1 siRNA or a scramble control into the neutral nanoliposome 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC), which can remain in animal issues for at least 48 hr without detectable toxicity (Gutiérrez-Puente et al., 1999) and has been used to deliver siRNA or micro-RNA (miRNA) oligonucleotides to tumor-bearing mice (Wu et al., 2014; Zhang et al., 2014). We implanted luciferase-expressing LM2 cells into the mammary fat pads of NSG (non-obese diabetic, severe combined immunodeficiency, interleukin-2 receptor gamma chain-null) mice. Three days after tumor cell implantation, we confirmed the initial tumor establishment by bioluminescent imaging, excluded the outliers, and randomly divided the mice into four treatment groups: (1) vehicle (PBS), (2) scramble-DOPC, (3) si-ZRANB1#2-DOPC, and (4) si-ZRANB1#5-DOPC. The treatment was administered through the tail vein twice weekly at a dose of 250 μg kg−1 body weight. At 4 weeks, the average tumor volumes in the two ZRANB1 siRNA treatment groups were 276.9 mm3 and 231.8 mm3, respectively, which was approximately 60% less than those in the vehicle group (674 mm3) and the scramble-DOPC group (692.7 mm3; Figure 5A). Similarly, the two independent ZRANB1 siRNAs also reduced tumor weight by approximately 60% (Figure 5B). As anticipated, EZH2 protein in the tumor was markedly downregulated in the two ZRANB1 siRNA treatment groups compared with both the vehicle group and the scramble control group (Figure 5C), demonstrating that in vivo delivery of ZRANB1 siRNA was effective in destabilizing EZH2.
Figure 5. Depletion of ZRANB1 Leads to Antitumor and Antimetastatic Effects.

(A and B) Tumor growth curves (A) and tumor weight (B, at week 4) of NSG mice with mammary fat pad injection of LM2 cells. From day 3, mice received twice weekly intravenous injections of the vehicle, DOPC-encapsulated ZRANB1 siRNA, or scramble oligonucleotides. n = 3 (the vehicle group) or 5 (all other groups) mice per group.
(C) Immunoblotting of EZH2 and β-actin in tumor lysates from the mice described in (A) and (B). n = 3 mice per group.
(D) qPCR of human-specific ZRANB1 in lung tissues from NSG mice with tail vein injection of LM2 cells. From day 3, mice received twice weekly intravenous injections of the vehicle, DOPC-encapsulated ZRANB1 siRNA, or scramble oligonucleotides. n = 5 mice per group.
(E and F) Bioluminescent imaging (E) and quantification of photon flux (F) of the mice described in (D). n = 6 mice per group at the time of the first treatment; n = 5 or 6 mice per group at the endpoint.
(G) H&E staining of lungs isolated from the mice described in (D). Scale bar, 500 μm. The corresponding high-magnification images are presented in Figure S5G.
Error bars in (A), (B), (D), and (F) are SEM; p values were calculated from a two-tailed t test. See also Figure S5.
To assess the effect of ZRANB1 inhibition on lung metastatic colonization, we injected LM2 cells into NSG mice through the tail vein and then used the same dosage regimen as in the tumor growth study. Using a human ZRANB1-specific TaqMan assay, we found that systemic treatment with the two ZRANB1 siRNAs decreased human ZRANB1 mRNA levels in lung issues by 60% and 73%, respectively, compared with the scramble control group (Figure 5D). Bioluminescent imaging of live animals revealed the consistent reduction of lung metastases in mice treated with DOPC-encapsulated ZRANB1 siRNA, but not in mice treated with DOPC-encapsulated scramble RNA oligonucleotides (Figures 5E and 5F). At week 5, the si-ZRANB1#2-DOPC group and the si-ZRANB1#5-DOPC group exhibited 55% and 86% reductions, respectively, in lung metastases relative to the vehicle group, whereas the scramble-DOPC group showed no significant difference from the vehicle group (Figures 5F, 5G, and S5G). Collectively, delivery of nanoliposomal ZRANB1 siRNA led to marked antitumor and antimetastatic effects in this TNBC model.
A ZRANB1 Inhibitor Destabilizes EZH2 and Inhibits Cell Viability through ZRANB1
Although the function of ZRANB1 in cancer was not reported before, Wu and colleagues searched for compounds in the National Cancer Institute (NCI) database that potentially bind to ZRANB1’s catalytic site based on the crystal structure of the OTU domain (Shi et al., 2012). Then 125 compounds that stood out in the in silico screen were tested for their ability to inhibit the cleavage of K63-linked hexa-ubiquitin by ZRANB1 in vitro. Two of them, NSC112200 and NSC267309, blocked ZRANB1’s deubiquitinase activity at 10 μM, whereas a compound with the similar chemical structure, NSC60650, showed no inhibition even at 30 μM (Shi et al., 2012).
NSC112200 is a mixture of 2,5-dibromo-3,6-dimethyl-1,4-benzenediol and 2,5-dibromo-3,6-dimethyl-1,4-benzoquinone (Shi et al., 2012), which we confirmed by ultrahigh-pressure liquid chromatography-mass spectrometry (UPLC-MS) and nuclear magnetic resonance (NMR) analysis. In an in vitro deubiquitination assay using ubiquitin chains as substrates, we found that ZRANB1 cleaved K33-linked di-ubiquitin; this cleavage was blocked by NSC112200, but not by the control compound NSC60650 (1,5-dichloro-2,4-dihydroxybenzene) (Shi et al., 2012) (Figure 6A). We also found that NSC112200 inhibited the deubiquitinase activity of ZRANB1, but not another OTU family member, A20 (Figure S6A). These in vitro DUB activity assays suggest that NSC112200 can directly act on ZRANB1.
Figure 6. A ZRANB1 Inhibitor Destabilizes EZH2 and Inhibits Cell Viability through ZRANB1.

(A) Purified His-ZRANB1 was pretreated with NSC60650 or NSC112200 for 10 min and then incubated with K33-linked di-ubiquitin in the presence of the compound at 37°C for 1.5 hr. Samples were then analyzed by immunoblotting with a ubiquitin-specific antibody.
(B) Immunoblotting of EZH2 and β-actin in LM2 cells treated with NSC112200 at the indicated doses for 24 hr.
(C) LM2 cells were treated with NSC112200 at the indicated doses. 24 hr after treatment, viable cells were quantitated by an MTT assay. n = 4 biological replicates.
(D) Immunoblotting of EZH2 and β-actin in BT549 cells treated with NSC112200 at the indicated doses for 24 hr.
(E) BT549 cells were treated with NSC112200 at the indicated doses. 24 hr after treatment, viable cells were quantitated by an MTT assay. n = 4 biological replicates.
(F and G) Immunoblotting of EZH2, SUZ12, and β-actin in LM2 (F) and BT549 (G) cells treated with NSC60650 or NSC112200 at the indicated doses for 24 hr.
(H) Immunoblotting of EZH2 and β-actin in LM2 cells pretreated with 2 μM MG132 for 1 hr and then treated with 10 μM NSC112200 in the presence of MG132 overnight.
(I) HEK293T cells were co-transfected with MYC-EZH2 and HA-ubiquitin (Ub), pretreated with 2 μM MG132 for 1 hr, and then treated with 10 μM NSC112200 in the presence of MG132 overnight, followed by immunoprecipitation with anti-MYC beads and immunoblotting with antibodies against HA and EZH2. N.S., non-specific signal.
(J) Left panel: immunoblotting of EZH2 and β-actin in control and ZRANB1-knockout HEK293A cells treated with 10 μM NSC112200 overnight. Right panel: quantification of EZH2 protein levels (normalized to β-actin) is shown.
(K) Control, ZRANB1-knockout, and ZRANB1-restored HEK293A cells were treated with 10 μM NSC112200 for 24 hr, and viable cells were quantitated by an MTT assay. Data are normalized to vehicle-treated cells for each group. n = 4 biological replicates.
Error bars in (C), (E), and (K) are SEM; p values were calculated from a two-tailed t test. See also Figure S6.
We asked whether NSC112200 regulates EZH2 and cell viability. Indeed, treatment of LM2 cells (which are resistant to GSK126; Figure S1C) with NSC112200 downregulated EZH2 protein and inhibited cell viability in a dose-dependent manner (Figures 6B and 6C), without significantly altering mRNA levels of EZH2 and other PRC2 components (Figures S6B and S6C); similar effects were also observed in BT549 cells (Figures 6D and 6E). In contrast, treatment of LM2 and BT549 cells with NSC60650, which has the similar chemical structure but does not inhibit ZRANB1’s deubiquitinase activity (Shi et al., 2012) (Figure 6A), had little or no effect on EZH2 protein level and cell viability (Figures 6C and 6E–6G).
We further assessed the dependence of NSC112200-induced EZH2 downregulation and cell killing on ZRANB1 and the ubiquitin-proteasome system. Treatment with the proteasome inhibitor MG132 abolished the downregulation of EZH2 by NSC112200 (Figure 6H), suggesting that NSC112200 promotes proteasomal degradation of EZH2. Indeed, treatment with NSC112200 markedly increased the ubiquitination of EZH2 (Figure 6I) and decreased its half-life (Figure S6D), which recapitulated the effects of ZRANB1 genetic depletion. Importantly, NSC112200 treatment downregulated EZH2 protein in HEK293A cells transfected with control gRNA, but not in isogenic ZRANB1-knockout HEK293A cells (Figure 6J), indicating that this compound destabilizes EZH2 through ZRANB1. Moreover, we assessed the viability of NSC112200-treated ZRANB1-knockout HEK293A cells, and we found that they were much more resistant to NSC112200 than isogenic ZRANB1 wild-type cells; re-expression of ZRANB1 in these ZRANB1-knockout cells restored their sensitivity to NSC112200 (Figure 6K). Collectively, these data suggest that the inhibition of ZRANB1 may selectively kill cells with abundant ZRANB1 and EZH2 expression.
It should be noted that Zranb1-null mice had normal development and growth (Jin et al., 2016) and that tissues from these mutants showed a marginal decrease or no change in Ezh2 protein levels (Figure S6E), suggesting that normal cells are not dependent on ZRANB1. One possibility could be low expression of ZRANB1 in normal cells. Indeed, in the normal human mammary epithelial cell lines MCF10A and HMLE, which showed undetectable ZRANB1 protein, ZRANB1 siRNA or NSC112200 treatment had no effect on EZH2 and little effect on cell proliferation and viability (Figures S6F–S6K). Similarly, knockdown of Zranb1 by siRNA (5) did not affect mouse embryonic fibroblast (MEF) proliferation (Figures S6L and S6M). Furthermore, normal C57BL/6 mice treated with DOPC-encapsulated ZRANB1 siRNA (5) showed no significant changes in survival, behavior, body weight, or liver weight (Figures S6N and S6O). Livers from all three treatment groups (vehicle, scramble, and ZRANB1 siRNA) exhibited no necrosis, fibrosis, steatosis, or biliary changes (Figure S6P). Taken together, these data demonstrated the safety of ZRANB1 depletion or inhibition in normal tissues.
ZRANB1 Levels Correlate with EZH2 Levels and Poor Survival in Human Breast Cancer
To address whether the regulation of EZH2 by ZRANB1 is relevant in human cancer, we performed immunohistochemical staining of these two proteins on human breast tumor tissue microarrays (125 cases were TNBC and 83 cases were non-TNBC). Consistent with fractionation assays and immunofluorescent staining of cell lines (Figures 3D and S3F), ZRANB1 protein was localized in both the nucleus and the cytoplasm in primary tumor tissues, whereas EZH2 protein was found only in the nucleus (Figure 7A). Notably, 67% (106 of 158) of the tumors with low ZRANB1 expression exhibited low EZH2 expression, and 84% (42 of 50) of the tumors with high ZRANB1 expression showed high EZH2 expression (Spearman correlation R = 0.44, p < 1 × 10−6; Figure 7B). We also plotted the EZH2 protein score versus the ZRANB1 protein score for individual patients, which revealed a highly significant correlation (linear regression R2 = 0.49, p < 1 × 10−6; Figure 7C).
Figure 7. ZRANB1 Levels Correlate with EZH2 Levels and Poor Survival in Human Breast Cancer.

(A) Immunohistochemical staining of ZRANB1 and EZH2 in representative breast tumor specimens. Scale bar, 50 μm.
(B) Correlation between ZRANB1 and EZH2 protein levels in human breast tumors. The p value was calculated from a chi-square test. R is the Spearman correlation coefficient.
(C) ZRANB1 protein scores (x axis) in primary breast tumors positively correlate with EZH2 protein scores (y axis) in individual patients. The p value was calculated from a linear regression analysis. R is the correlation coefficient. Protein score = the percentage of immunopositive cells × immunostaining intensity.
(D) Kaplan-Meier curves of overall survival of breast cancer patients (n = 138 patients), stratified by ZRANB1 protein levels. Protein score = the percentage of immunopositive cells × immunostaining intensity. High or low protein expression was defined using the mean score of all samples as a cutoff point. The p value was calculated from a log rank test. HR, hazard ratio.
See also Figure S7.
To evaluate whether high ZRANB1 expression is associated with poor clinical outcome, we performed immunohistochemical staining of ZRANB1 on 138 tumor tissue specimens from breast cancer patients (23 cases were TNBC) with a long-term (~10 years) follow-up record (Figure S7A). Kaplan-Meier analysis showed that patients with high levels of ZRANB1 protein had shorter overall survival than patients with low levels of ZRANB1 (HR = 2.6, p = 0.01; Figure 7D). Finally, analysis of The Cancer Genome Atlas (TCGA) data revealed amplifications (at 10q26.13; Figure S7B) or missense mutations of the ZRANB1 gene in a subset of breast cancer and ovarian cancer. It is possible that additional mechanisms also contribute to ZRANB1 alternations in human tumors, which warrants future investigation.
DISCUSSION
Genetically engineered mouse models have provided strong evidence that EZH2 drives or accelerates oncogenesis in multiple cancer types, such as lymphoma (Béguelin et al., 2013; Berg et al., 2014; Souroullas et al., 2016), melanoma (Souroullas et al., 2016), breast cancer (Gonzalez et al., 2014; Li et al., 2009), and lung cancer (Zhang et al., 2016). Whereas previous studies attributed the cancer-promoting function of EZH2 to the repression of PRC2 target genes through H3K27me3-mediated epigenetic silencing (Bonasio et al., 2010), accumulating evidence has demonstrated that the non-catalytic activity of EZH2 contributes substantially to tumor formation and progression (Kim et al., 2015; Lee et al., 2011; Shi et al., 2007; Xu et al., 2012), which could explain the resistance of many cancers to EZH2 enzymatic inhibitors. On the other hand, genetic depletion of EZH2 has been shown to suppress tumorigenesis and metastasis.
Several ubiquitin ligases promote EZH2 ubiquitination and degradation (Jin et al., 2017; Sahasrabuddhe et al., 2015; Yu et al., 2013). Destabilization of EZH2 by targeting its deubiquitinase may offer an alternative therapeutic approach to treating EZH2-overexpressing tumors, such as TNBC and ovarian cancer. In this study, we identified ZRANB1 as the EZH2 deubiquitinase that regulates the polyubiquitination and protein stability of EZH2. Ablation of ZRANB1 is compatible with the viability of normal tissues, since Zranb1-null mice were viable and did not exhibit phenotypic differences compared with wild-type mice under normal physiological conditions, although they were resistant to the induction of experimental autoimmune encephalomyelitis (Jin et al., 2016). Similarly, our data revealed that ZRNAB1 siRNA treatment was not detrimental to normal human and mouse cells and was not toxic to mice. In contrast, depletion of ZRANB1 in TNBC cells markedly suppressed cell proliferation and migration and induced apoptosis, and systemic delivery of nanoliposome-encapsulated ZRANB1 siRNA led to pronounced anticancer effects in xenograft models of TNBC, suggesting that ZRANB1 could be a novel therapeutic target. Notably, ZRANB1 protein is abundantly expressed in TNBC cell lines, which are responsive to ZRANB1 siRNA or inhibitor, but it is undetectable in normal human mammary epithelial cell lines, which are resistant to knockdown or chemical inhibition of ZRANB1.
In patients with breast cancer, ZRANB1 gene amplification was found in a subset of cases, and ZRANB1 protein levels correlated with EZH2 protein levels and poor survival (both TNBC and non-TNBC). Moreover, unlike normal mammary epithelial cells, all human breast cancer cell lines examined (both TNBC and non-TNBC) showed downregulation of EZH2 protein upon ZRANB1 knockdown, suggesting that ZRANB1 is a general regulator of EZH2 in breast cancer cells. On the other hand, however, ZRANB1 may have higher functional importance and better therapeutic potential in TNBC than in non-TNBC, given that knockdown of ZRANB1 led to severe growth defect in all 11 TNBC cell lines, whereas ER+ or HER2+ breast cancer cell lines showed variable responses (either responsive or resistant) to ZRANB1 depletion. This suggests that some of the non-TNBC cells are not dependent on ZRANB1 and EZH2 for their proliferation.
In the present study, a small-molecule ZRANB1 inhibitor, NSC112200, destabilized EZH2 through ubiquitination and the proteasome, and it inhibited cell viability through, at least in part, ZRANB1. In contrast, it had no effect on EZH2 protein level and little effect on viability in normal mammary epithelial cells with no detectable ZRANB1 expression. It should be noted that NSC112200 is not water soluble (dissolved in DMSO or corn oil) and that it inhibits ZRANB1’s DUB activity, destabilizes EZH2, and kills TNBC cells at micromolar, but not nanomolar, concentrations. Moreover, some of the NSC112200-treated C57BL/6 mice exhibited acute responses (low body temperature, less active) in preliminary animal testing (data not shown), indicating some toxicity. Thus, NSC112200 can serve as a tool compound but may not behave in a drug-like manner in vivo. Nevertheless, here we provide a proof of principle that the EZH2 deubiquitinase identified in this study is amenable to inhibition by small molecules. This represents a starting point to target ZRANB1. Considering that NSC112200 was from in silico screening, further development of a clinical ZRANB1 inhibitor may involve lead compound identification via high-throughput chemical screening and the use of the structure-activity relationship (SAR) analysis to optimize the activity, selectivity, pharmacokinetics, safety profile, and physical properties of lead compounds, which will enable full evaluation in animal models.
EXPERIMENTAL PROCEDURES
Further details and an outline of resources used in this work can be found in the Supplemental Experimental Procedures.
Mice
All mice used in this study were supplied by and housed in the Research Animal Support Facility at MD Anderson Cancer Center. Six-week-old female NSG mice were used for mammary fat pad injection or intravenous injection of human breast cancer cells. Six-week-old C57BL/6J mice were used for toxicity assessment of ZRANB1 siRNA. All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of MD Anderson Cancer Center.
Human Tumor Samples
Two breast tumor tissue microarrays, BR487b (48 TNBC cases) and BR1921 (80 invasive ductal carcinoma cases and 80 invasive lobular carcinoma cases), were purchased from Biomax to determine the correlation between ZRANB1 and EZH2 protein levels. For survival analysis, 138 cases of human tumor tissue specimens were obtained from patients undergoing surgical resection of breast cancer as primary treatment at China Medical University Hospital (Taichung, Taiwan) between 2005 and 2008, under the guidelines approved by the Institutional Review Board, and written informed consent was obtained from patients in all cases at the time of enrollment.
Statistical Analysis
Unless otherwise noted, data are presented as mean ± SEM, and the Student’s t test (unpaired, two tailed) was used to compare two groups of independent samples. The chi-square test and linear regression analysis were used for statistical analysis of the correlation between ZRANB1 and EZH2. The log rank test was used to compare Kaplan-Meier survival curves. p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
ZRANB1 binds, deubiquitinates, and stabilizes EZH2 protein
Depletion or inhibition of ZRANB1 causes EZH2 destabilization and anticancer effect
ZRANB1 levels correlate with EZH2 levels and poor survival in human breast cancer
Acknowledgments
We thank Xiang Zhang for providing luciferase-expressing LM2 cells, Dihua Yu for providing SKBR3 cells, Jae-Il Park for providing EZH2 constructs, Qi Cao for thoughtful suggestions, and Hyemin Lee for assistance with flow cytometric analysis. We also thank MD Anderson’s shRNA and ORFeome Core, Small Animal Imaging Facility, Characterized Cell Line Core Facility, and Pharmaceutical Chemistry Facility for technical assistance. L.M. is supported by NIH grants R01CA166051 and R01CA181029, a Cancer Prevention and Research Institute of Texas (CPRIT) grant RP150319, and a Stand Up To Cancer Innovative Research Grant (403235). Y.H. is supported by a CPRIT grant RR140053, an Innovation Award from the American Heart Association (16IRG27250155), a John S. Dunn Foundation Collaborative Research Award, and the Texas A&M University Start-up Funds. X.Y. was supported by NIH grants GM086937 and GM100777. M.J.Y. was supported in part by NIH R01CA164346 and R01CA200703 and CPRIT RP140402. Z.X. is supported by a Rosalie B. Hite Graduate Fellowship. P.Z. is a scholar in the National 1000 Young Talents Program of China.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE104910.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.078.
DECLARATION OF INTERESTS
X.Y. is an employee of AM Biotechnologies. All other authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Y.S. and L.M. conceived the study. P.Z. and Z.X. designed, performed, and analyzed most of the experiments. S.W. initiated the EZH2 DUB screening. M.Z., M.L., J.L., Y.H., and D.S. performed RNA-seq and data analysis. Y. Wei and M.-C.H. performed tissue microarray staining and analysis. Q.H., J.K., F.Y., Z.Z., L.Z., X.H., S.E.L., X.S., J.C., and Y.S. contributed reagents and provided technical assistance. J.K., B.N.T., and A.N.S. maintained mice and performed xenograft implantation. C.R.-A., G.L.-B., and A.K.S. assisted with nanoliposomal siRNA delivery. Y. Wang and H.L. performed database analysis. X.X., X.C., and S.-C.S. provided Zranb1-null mice. L.-C.L., H.-W.C., S.-F.C., and M.-C.H. collected tissue samples. M.J.Y. performed histopathological analysis. X.Y. synthesized large-scale ZRANB1 siRNA for animal studies. P.Z., Z.X., and L.M. wrote the manuscript with input from all other authors. L.M. provided scientific directions, established collaborations, and allocated funding for this study.
References
- Alford SH, Toy K, Merajver SD, Kleer CG. Increased risk for distant metastasis in patients with familial early-stage breast cancer and high EZH2 expression. Breast Cancer Res Treat. 2012;132:429–437. doi: 10.1007/s10549-011-1591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Thoene S, Yap D, Wee T, Schoeler N, Rosten P, Lim E, Bilenky M, Mungall AJ, Oellerich T, et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood. 2014;123:3914–3924. doi: 10.1182/blood-2012-12-473439. [DOI] [PubMed] [Google Scholar]
- Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IeM, Conejo-Garcia JR, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell. 2011;19:86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Coward WR, Feghali-Bostwick CA, Jenkins G, Knox AJ, Pang L. A central role for G9a and EZH2 in the epigenetic silencing of cyclooxygenase-2 in idiopathic pulmonary fibrosis. FASEB J. 2014;28:3183–3196. doi: 10.1096/fj.13-241760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Delgado-Olguín P, Dang LT, He D, Thomas S, Chi L, Sukonnik T, Khyzha N, Dobenecker MW, Fish JE, Bruneau BG. Ezh2-mediated repression of a transcriptional pathway upstream of Mmp9 maintains integrity of the developing vasculature. Development. 2014;141:4610–4617. doi: 10.1242/dev.112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, Ljungman M, Merajver SD, Kleer CG. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene. 2009;28:843–853. doi: 10.1038/onc.2008.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, Kidwell KM, Kleer CG. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci USA. 2014;111:3098–3103. doi: 10.1073/pnas.1308953111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Puente Y, Tari AM, Stephens C, Rosenblum M, Guerra RT, Lopez-Berestein G. Safety, pharmacokinetics, and tissue distribution of liposomal P-ethoxy antisense oligonucleotides targeted to Bcl-2. J Pharmacol Exp Ther. 1999;291:865–869. [PubMed] [Google Scholar]
- Jin J, Xie X, Xiao Y, Hu H, Zou Q, Cheng X, Sun SC. Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase Trabid. Nat Immunol. 2016;17:259–268. doi: 10.1038/ni.3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, Liu T, Wang D, Wu H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem. 2017;292:6269–6280. doi: 10.1074/jbc.M116.764407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim KB, Son HJ, Chae YC, Oh ST, Kim DW, Pak JH, Seo SB. H3K27 methylation and H3S28 phosphorylation-dependent transcriptional regulation by INHAT subunit SET/TAF-Iβ. FEBS Lett. 2012;586:3159–3165. doi: 10.1016/j.febslet.2012.06.026. [DOI] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491–1496. doi: 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798–810. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- Li X, Gonzalez ME, Toy K, Filzen T, Merajver SD, Kleer CG. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol. 2009;175:1246–1254. doi: 10.2353/ajpath.2009.090042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licchesi JD, Mieszczanek J, Mevissen TE, Rutherford TJ, Akutsu M, Virdee S, El Oualid F, Chin JW, Ovaa H, Bienz M, Komander D. An ankyrin-repeat ubiquitin-binding domain determines TRABID’s specificity for atypical ubiquitin chains. Nat Struct Mol Biol. 2011;19:62–71. doi: 10.1038/nsmb.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, Armaiz-Pena GN, Hu W, Stone RL, Munkarah A, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18:185–197. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv YC, Tang YY, Zhang P, Wan W, Yao F, He PP, Xie W, Mo ZC, Shi JF, Wu JF, et al. Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. PLoS ONE. 2016;11:e0157265. doi: 10.1371/journal.pone.0157265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SM, Komander D. Assembly and specific recognition of k29- and k33-linked polyubiquitin. Mol Cell. 2015;58:95–109. doi: 10.1016/j.molcel.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore HM, Gonzalez ME, Toy KA, Cimino-Mathews A, Argani P, Kleer CG. EZH2 inhibition decreases p38 signaling and suppresses breast cancer motility and metastasis. Breast Cancer Res Treat. 2013;138:741–752. doi: 10.1007/s10549-013-2498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyl-transferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KS. Oncogenic Y641 mutations in EZH2 prevent Jak2/β-TrCP-mediated degradation. Oncogene. 2015;34:445–454. doi: 10.1038/onc.2013.571. [DOI] [PubMed] [Google Scholar]
- Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27:5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Bao J, Wang NX, Zheng J, Wu D. Identification Of Small Molecule TRABID Deubiquitinase Inhibitors By Computation-Based Virtual Screen. BMC Chem Biol. 2012;12:4. doi: 10.1186/1472-6769-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu JY, Paulk J, Xiong J, Clark KS, Fedoriw Y, Qi J, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med. 2016;22:632–640. doi: 10.1038/nm.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- Wu SY, Yang X, Gharpure KM, Hatakeyama H, Egli M, McGuire MH, Nagaraja AS, Miyake TM, Rupaimoole R, Pecot CV, et al. 2′-OMe-phosphorodithioate-modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat Commun. 2014;5:3459. doi: 10.1038/ncomms4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zhang P, Ma L. The role of deubiquitinases in breast cancer. Cancer Metastasis Rev. 2016;35:589–600. doi: 10.1007/s10555-016-9640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YL, Chou RH, Shyu WC, Hsieh SC, Wu CS, Chiang SY, Chang WJ, Chen JN, Tseng YJ, Lin YH, et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med. 2013;5:531–547. doi: 10.1002/emmm.201201783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, Wang M, Chen D, Sun Y, Hung MC, et al. Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol. 2013;15:1486–1494. doi: 10.1038/ncb2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wang L, Rodriguez-Aguayo C, Yuan Y, Debeb BG, Chen D, Sun Y, You MJ, Liu Y, Dean DC, et al. miR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat Commun. 2014;5:5671. doi: 10.1038/ncomms6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qi J, Reyes JM, Li L, Rao PK, Li F, Lin CY, Perry JA, Lawlor MA, Federation A, et al. Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer Discov. 2016;6:1006–1021. doi: 10.1158/2159-8290.CD-16-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.