

Abstract
FMR1 premutation carriers (55–200 CGG repeats) are at risk for developing Fragile X-associated Tremor/Ataxia Syndrome (FXTAS), an adult onset neurodegenerative disorder. Approximately 20% of female carriers will develop Fragile X-associated Primary Ovarian Insufficiency (FXPOI), in addition to a number of clinical problems affecting premutation carriers throughout their life span. Marked elevation in FMR1 mRNA levels have been observed with premutation alleles resulting in RNA toxicity, the leading molecular mechanism proposed for the FMR1 associated disorders observed in premutation carriers.
The FMR1 gene undergoes alternative splicing and we have recently reported that the relative abundance of all FMR1 mRNA isoforms is significantly increased in premutation carriers.
In this study, we characterized the transcriptional FMR1 isoforms distribution pattern in different tissues and identified a total of 49 isoforms, some of which observed only in premutation carriers and which might play a role in the pathogenesis of FXTAS. Further, we investigated the distribution pattern and expression levels of the FMR1 isoforms in asymptomatic premutation carriers and in those with FXTAS and found no significant differences between the two groups.
Our findings suggest that the characterization of the expression levels of the different FMR1 isoforms is fundamental for understanding the regulation of the FMR1 gene as imbalance in their expression could lead to an altered functional diversity with neurotoxic consequences. Their characterization will also help to elucidating the mechanism(s) by which “toxic gain of function” of the FMR1 mRNA may play a role in FXTAS and/or in the other FMR1-associated conditions.
Keywords: Alternative splicing, FMR1 gene, FXTAS, RNA toxicity, SMRT sequencing, Isoforms
1. Introduction
The human genome harbors approximately 22,000 coding genes that contain a variable number of exons which can be alternatively spliced in numerous mRNA isoforms generating two or more distinct proteins with multiple biological functions [1].
Thus, alternative splicing is a mechanism that encourages proteomic diversity and plays a vital role in regulating gene expression and disease etiology. It was shown that alternative splicing determines intracellular localization, binding properties, protein stability, enzymatic activity and post- translational modifications of a vast number of proteins [1,2].
There is growing interest in the alternative splicing phenomenon due to the evidence of its clinical relevance and its promising therapeutic potential. In fact, it has been shown that the alteration in the splicing pattern can result in aberrant splice variants, which are involved in > 50% of genetic disorders [2] including autism spectrum disorders [3,4]. Alternative splicing is most common in the nervous system, and changes in alternative splicing have been observed in different neurodevelopmental stages [5,6]. Hence, alternative splicing is a major player in the normal function of the nervous system and indeed impairments in the alternative splicing process has been demonstrated for a number of neurologic diseases [7–9]. In addition to mutations within splice sites, mutations in genes encoding splicing factors that are involved in regulating and directing splicing of target genes have been shown to change splicing patterns leading to neurological disorders [10,11]. Different types of neuronal cells express various combinations of splice regulators and show a distinct susceptibility to their mutation [12].
The FMR1 gene is a highly conserved gene that consists of 17 exons spanning approximately 38 kb of genomic DNA. The lack of expression of fragile X mental retardation protein (FMRP) due to an expansion of > 200 CGG repeats and consequent methylation in the 5′UTR, leads to the pathogenesis of fragile X syndrome (FXS). The gene undergoes extensive alternative splicing yielding different FMR1 mRNA isoforms, resulting in several FMRP isoforms that were demonstrated to exist in both human and mouse [13–15]. On the contrary, the FMR1 pre- mutation expansion (55–200 CGG repeats) is characterized by an in- crease in the levels of mRNA expression causing RNA toxicity and leading to a higher risk of developing FMR1 associated disorders [16]. Previously, we reported on the presence of 16 different isoforms, out of the 24 predicted ones, in both normal and premutation samples [17]. Interestingly we also detected a novel splicing isoform involving exon 3. All detected isoforms were overexpressed in premutation carriers who showed elevated levels of FMR1 mRNA. However, the isoforms 10 (Iso10) and 10b (Iso10b), lacking the C-terminal functional sites of FMRP, showed the highest levels of expression in premutation samples [17]. The role of these isoforms is yet unknown; however, their presence suggests a potential functional relevance in the pathology of FMR1-associated disorders.
In this study, we have further characterized the FMR1 transcriptional landscape in different tissues including muscle, brain, heart and testes from three individuals with a premutation allele and compared them to the isoform expression profiles of three age-matched controls. A total of 49 isoforms were identified, including the 16 previously identified isoforms. Among the novel isoforms with elevated expression levels in premutation samples, there were transcripts including new splicing sites, skipping of multiple exons and retention of partial intron and exon sequences. We did not find strong evidence for tissue-specific isoform expression profiles. In addition, we compared the relative expression of the novel isoforms between premutation carriers affected by FXTAS, asymptomatic premutation carriers (having premutation without symptoms of FXTAS) and age-matched controls.
2. Materials and methods
2.1. Subjects
Tissue samples from six subjects including three premutation male carriers and three age-matched male controls were used in this study. Cerebellum brain tissue in addition to testis, muscle and heart were used (Supplementary Table 1). Post Mortem Interval (PMI), age at death and the number of sequencing reads are shown in Supplementary Table 1.
Peripheral blood samples from 45 male participants including 15 asymptomatic premutation carriers (FXTAS stage 0 or 1), 15 premutation carriers with FXTAS (FXTAS stage 4 or 5) and 15 non-carrier controls were included in the study. Participants were matched by age and within the premutation groups by CGG repeat number. Demographic, molecular and clinical data are shown for all participants in Supplementary Table 2.
The three premutation male carriers and three age-matched male controls had an age mean/SD of (68.3 ± 10.5) and (53 ± 11.3) respectively. The CGG number mean/SD was (92 ± 5.3) in premutation carriers, and (30.3 ± 10.5) in controls (Supplementary Table 1). In addition, the 45 male participants including 15 asymptomatic pre- mutation carriers, 15 premutation carriers with FXTAS and 15 non- carrier controls were all age matched with a mean/SD of (62.3 ± 8), (64.8 ± 8), and (63.3 ± 8.9) respectively. They were also matched within the premutation groups by CGG repeat number: asymptomatic premutation carriers (90.3 ± 22.8 CGG repeats), FXTAS premutation carriers (98.5 ± 15.8 CGG repeats) and controls (29.4 ± 4.7 CGG repeats) (Supplementary Table 2).
Clinical severity of FXTAS as an indication of the impact of motor aspects of the disease on activities of daily living (ADLs) was assessed by stages. The stages range from 0 to 6, where stage 0 describes normal functions; stage 1 indicates subtle tremor or mild balance problems with no interference with ADLs; stage 2 indicates clear tremor and/or balance problems with minor interference with ADLs; stage 3 indicates moderate tremor and/or balance problems and occasional falls with significant interference with ADLs; stage 4 indicates severe tremor and/or balance problems with at least intermittent use of a cane or a walker; stage 5 involves the use of a wheelchair on a daily basis; and in stage 6, subjects are bedridden [18]. Samples from all subjects were collected under approved UC Davis Institutional Review Board (IRB) protocols. Postmortem tissues were obtained from University of California Davis FXTAS/FXS Repository and from the NIH NeuroBiobank.
2.2. Assigning isoforms by splicing patter
Our previous study identified 16 out of 24 predicted isoforms in group A to D [17] in both premutation carriers and controls (Supple- mentary Fig. 1). In this study we detected five additional isoforms, one in group A, three in group B and one in group D.
We also added two new groups (E and F) with a total of 28 new isoforms. Group E contained 10 isoforms missing exons 11,12,13 and 14. The remaining 18 isoforms were all included in group F (Fig. 1 and Supplementary Table 3). The final number of isoforms within group A through group F was 49. The exon coordinates of these isoforms are shown in Supplementary Table 4. The list of coordinates of the alternative splice sites’ donors and acceptors observed in the novel isoforms are shown in Supplementary Tables 5. The frequency of each donor-acceptor motif is shown in Supplementary Table 6.
Figure 1.
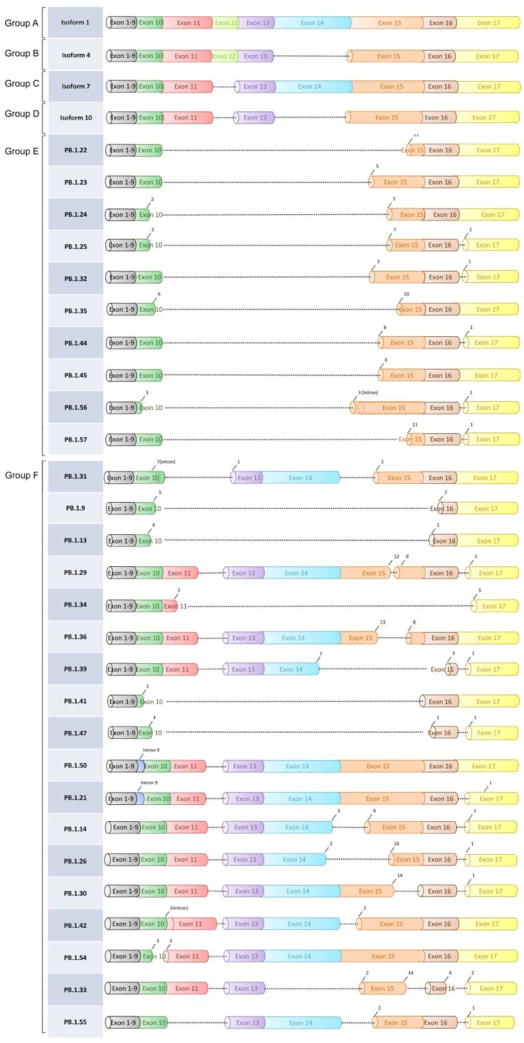
One isoform (containing the entire exon 15) representative of Group A to D is depicted at the top of the figure. The new FMR1 splicing groups identified in this study are also shown. Group E skips exon 11 through 14. Group F includes all additional isoforms not belonging to patterns from group A-E. For full definition of exonic coordinates and splicing patterns, see Supplementary Table 3 and 4.
2.3. Data availability
PacBio sequencing raw data have been deposited at NCBI SRA (accession SRP093166). Intermediate FLNC and nFL sequences are deposited in Zenodo (http://dx.doi.org/10.5281/zenodo.185011).
2.4. CGG sizing
Genomic DNA (gDNA) was isolated from either 3 ml of peripheral blood or from 100 to 200 mg of human postmortem tissues using Gentra Puregene Blood Kit (Qiagen, Valencia, CA) following standard procedure. A combination of PCR and Southern blot analysis was used to size FMR1 allele and determine methylation status, as previously described [19, 20].
2.5. RNA isolation
Total RNA from whole blood was isolated using Tempus tubes and/or PAX gene tubes according to manufacturer’s instructions (Applied Biosystems, Foster City, California, USA; Qiagen, Valencia, CA, USA). Total RNA from postmortem tissues was isolated using Trizol also ac- cording to manufacturer’s instructions (Life Technologies, Carlsbad, California, USA). All tissues were stored at −80 °C and 100-200 mg and were pulverized in liquid nitrogen for RNA isolation. Total RNA quantification was performed using Qubit fluorometer (Invitrogen, Waltham, Massachusetts, USA) and quality control of total RNA was performed using the Agilent 2100 BioAnalyzer system. RNA, quantification was performed using Nanodrop.
2.6. qRT-PCR
cDNA synthesis was performed as described in (Tassone 2000). PCR was carried out using specific primers for the different group of iso- forms. Their sequences are as follow: for isoforms 4 and 4b of group B (Forward: 5′- TGGCTTCATCAGTTGTAGCAGG -3′. Reverse: 5′- CAGAATTAGTTCCTTTAAATAGTTCAGG -3′. Probe: 5′ 6 FAM- CAG CAT CGC TAA TGC- 3′ MGBNFQ). For group E (Forward; 5′- GTTCCACAAGAAGAGGAACTAATTCTG -3′, Reverse; 5′- TAGCCTCTC TTGGATTACGTGG -3′). For group F (Forward; 5′- GTTGTGAGGGTG AGGATTGAGGCT -3′, Reverse; 5′- TCCAATCTGTCGCAACTGCTCATC-3′), and for the isoform containing the fragment from intron 9 (Forward 5′- TTCTCCATTTTGACTTGAGTAGTGTTTT -3′. Reverse: 5′- TCCTGAATCAGCTTTCCATTTTTT -3′. Probe: 5′- CACCATCACCATC| GGCAAAGTAATAGG-3′). Finally, as for the isoform missing exon 3 (Forward 5′- AGGCATTTGTAAAGGATGTTCATG -3′. Reverse 5′- GCAAGGCTCTTTTTCATTTGCT -3′. Probe 5′ 6 FAM AACAGTTG CATTTGAAAACAAGTGTATTCCA 3′ TAMRA).
2.7. Generation of SMRT libraries
cDNA library for each patient was generated by synthesizing first the single-strand cDNA from total RNA of each specimen tissues using SMARTer™ cDNA Synthesis Kit (Clontech Laboratories, Mountain View, California, USA), followed by synthesis of the whole transcriptome cDNA using KAPA HiFi PCR Kit (Wilmington, Massachusetts, USA) according to manufactures’ instructions. A barcoded library was generated for each specimen tissue from their whole transcriptome cDNA using barcoded FMR1 gene-specific PCR primers (Supplementary Table 7) to amplify FMR1-specific amplicons (including all the ex- pressed isoforms of different size depending on the inclusion/exclusion of exons and location of the splice sites) of 847 bp between the exon 8/9 and 3′ UTR boundary of the FMR1 gene as previously described [17]. Moreover, four unique pairs of barcode sequences assigned to identify the five different specimen tissues were attached to pairs of forward and reverse primers. The SMRT bell libraries were assembled by pulling together an equal amount of their corresponding barcoded libraries, followed by DNA damage repair and SMRTbell ligation using the SMRTbell Template Prep Kit 1.0 (Pacific Biosciences, Menlo Park, California, USA) as by manufacturer’s protocol.
2.8. Bioinformatics analysis
Each sample (pooled barcoded library of each individual) was run through the IsoeSeq bioinformatics pipeline to obtain full-length, high- quality isoform consensus sequences [21]. Briefly, for each sequencing molecule, an intra-molecular circular consensus (CCS) read was generated. The CCS reads were then classified as full-length if a matching pair of barcode sequences was observed on the two ends of the read (Supplementary Table 8). The full-length (FL) reads were then run through an iterative clustering algorithm (ICE), where each cluster contains FL reads that belong to the same isoform. Each FL read can belong to exactly one cluster. After ICE, non-full-length reads were mapped to the ICE consensus sequences; non-FL reads were allowed to map to more than one cluster. Finally, Quiver was used to create a polished consensus sequence for each cluster. Based on the Quiver consensus predicted accuracy, only sequences that have a predicted accuracy of > 99% were deemed high-quality (HQ) and were used for the next part of the analysis.
The HQ Quiver sequences were mapped back to the hg19 genome using GMAP. We kept only sequences that satisfied the following criteria: (a) alignment coverage > = 99%; (b) alignment identity >= 95%; (c) has at least 5 supporting FL reads; (d) alignment start is between chrX:147,014,079–147,014,470 and alignment end is between chrX:147030158-147030505, where the targeted primers were de- signed. Redundant sequences, that is, sequences that have the same exon structures, were collapsed into unique isoforms. This resulted in 57 unique isoforms. We further manually inspected the evidence for each isoform by checking the FL alignment to each isoform consensus, and as result, we removed 8 out of 57 that we deemed to have lower level of alignments and consensus, generating a final total of 49 unique isoforms. For each isoform, the Iso-Seq output maintained the list of FL and non-FL reads that were associated with it. All FL reads had identifiable tissue-specific barcodes. We extracted the barcode-identifiable FL reads associated with each isoform and normalized it by the total number of FL reads in that sample. The normalized quantity is shown as a percentage in Supplementary Table 9.
2.9. Open reading frame prediction
Since our isoform sequences began from exon 9, to predict the full open reading frame, we inserted the first eight exons of FMR1 mRNA based on canonical exon annotation (Supplementary Table 4) using hg19 reference genome bases. To further eliminate any remaining base errors and exclude individual-specific SNPs, we again used the hg19 reference genome bases to create a “genome-corrected” version of the final 49 isoforms. We predicted the open reading frames using the ANGEL software (https://github.com/pacificbiosciences/ANGEL) to identify the first open reading frame that is longer than 100aa. The predicted open reading frames ORFs were then aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
3. Results
In this study, we have used the single-molecule long-read sequencing methodology, (SMRT sequencing), to investigate the distribution of the FMR1 splicing isoforms and to determine aberrations in the splicing pattern in different tissues derived from premutation male carriers and male controls. We identified a total of 49 isoforms (including both previously known isoforms and novel ones) generated by the usage of new splicing sites, skipping of multiple exons or retention of intron and exon sequences. Several isoforms were expressed a low level and detected only in pre- mutation carriers. As we have previously demonstrated [17], differently from qRT-PCR, the SMRT sequencing approach allows us to build a transcriptional map of all of the possible splice combinations within a single FMR1 mRNA molecule and therefore it represents a powerful tool for this type of analysis. We mainly focused on the alternative splicing occurring between exon 9 and 17 as, with the exception of an alternative splicing we previously identified in exon 3 [17], no additional splicing events appear to occur in the first half region of the FMR1 gene.
However, we detected de- creased expression levels of the isoform missing exon 3 in male carriers confirming our previous results [17]. Below, we describe the main findings of this study.
3.1. Identification of novel splicing variants of the FMR1 gene
We obtained between 23,000 and 28,000 full-length non-chimeric (FLNC) reads in all tissue samples derived from premutation carriers and between 10,000 and 12,000 FLNC reads for samples derived from age and gender matched controls (Supplementary Table 8). After de- multiplexing, we obtained between 3000 and 11,000 FLNC reads per tissue per sample (Supplementary Table 1). To obtain unique isoforms, each sample (from pooled tissues but with barcode information re- corded) was run through the ToFU bioinformatics pipeline [21] to get high-quality consensus isoform sequences, which were then mapped to the hg19 reference genome. Isoforms were further filtered to start and end at the primer targeted region and included if at least 5 FLNC read support were observed (see Methods). We identified a total of 49 unique isoforms, categorized by their splicing patterns into six different groups (A, B, C, D, E, and F) (Fig. 1, Supplementary Fig. 1). Within group A-D, we identified 21 out of a total of 24 predicted isoforms. The majority of the newly identified isoforms within group A-D came from group B, which were detected at low levels in select tissues derived from premutation carriers but were below the detection level in the control samples.
Specifically, among the 21 isoforms identified, we detected three isoforms in group B (Iso4b, Iso6, Iso16) out of the six predicted isoforms; the remaining three within group B (Iso4, Iso5, and Iso5b) were not detected (Supplementary Fig 1).
3.2. Identification of a novel isoform containing a portion of intron 9 between exon 9 and 10
We identified two novel isoforms retaining a portion of intronic sequence, between exons 9 and 10 (PB1.21 and PB1.50, Fig. 1 and Fig. 2a) differing only in the usage of the splicing acceptor in exon 17. The retained sequence was 140 bp long, (from chrX:147014920147015059; hg19 coordinates), and had a canonical donor/receptor site of GT/AG. Specifically, PB1.21 had the canonical acceptor site while isoform PB1.50 had the AG-acceptor site downstream. Based on ORF analysis (see section below), the addition of the 140 bp intronic sequence disrupts translation, creating a premature stop codon and resulting in truncated FMRP isoforms. The two isoforms containing this novel “exon” were found in low abundance (0.02%–1.6%) in two pre- mutation individuals.
Figure 2.
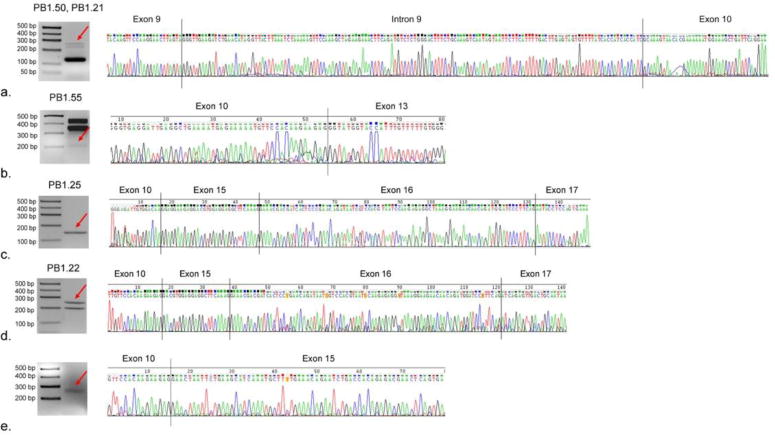
RT-PCR and Sanger sequencing confirmed the existence of several novel splicing isoforms. From top to bottom, the electrophoresis showing the sequence of (a) Group F isoform retaining a portion of intronic fragment 9 (PB1.50/PB1.21) different only in acceptor site at exon 17, (b) Group F isoforms missing exons 11 and 12 ((PB1.55), (c & d) Group E isoforms missing exons 11 through 14 and containing different portions of exon 15; and (e) Group E isoforms (missing the entire region of Exon 11 through 14) detected by qRT-PCR but below the threshold for SMRT sequencing.
3.3. FMR1 Isoforms distribution by group
We determined the representation from all isoforms within each isoform group in a specific tissue sample and the collective percentages are shown in Table 1. A detailed percentage for each isoform is shown in the Supplementary Table 9. We calculated the percentage of FL reads supporting each isoform in each sample-tissue. While this is not a direct abundance measure of the RNA level and is subject to bias in PCR amplification and loading bias in sequencing, two factors still make the normalized FL counts a useful information: the library was constructed without size selection and all isoforms have relatively similar lengths, further mitigating the effects of length differences in amplification and loading bias.
Table 1.
Aggregated normalized full-length read counts by different splice groups. Each cell denotes the percentage of FL reads coming from all isoforms in that group for that sample-tissue. Those shown with 0.0% are either truly undetected or less than 0.1%. For a full table showing absolute and normalized read count for each isoform, see Supplementary Table 9.
| Sample # | Carrier - 1 | Carrier - 2 | Carrier - 3 | Normal - 1 | Normal - 2 | Normal - 3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample Type | testes | brain | muscle | testes | brain | heart | muscle | testes | heart | muscle | testes | brain | testes | brain | muscle | brain | heart | muscle |
| Group A % | 1.5% | 1.8% | 2.0% | 3.0% | 0.7% | 0.0% | 1.4% | 5.5% | 2.4% | 1.0% | 0.0% | 0.0% | 0.4% | 1.1% | 0.0% | 0.3% | 5.2% | 0.6% |
| Group B % | 0.0% | 0.4% | 0.8% | 0.0% | 0.0% | 0.0% | 3.3% | 0.1% | 1.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| Group C % | 95.4% | 96.3% | 89.2% | 55.1% | 90.6% | 95.9% | 85.7% | 86.8% | 73.8% | 61.7% | 93.5% | 95.0% | 93.1% | 95.8% | 73.5% | 97.3% | 93.3% | 87.1% |
| Group D % | 2.9% | 0.8% | 6.8% | 0.0% | 2.5% | 0.0% | 5.6% | 7.1% | 2.6% | 11.2% | 3.0% | 2.7% | 2.3% | 2.1% | 23.8% | 0.2% | 0.1% | 12.2% |
| Group E % | 0.2% | 0.1% | 0.1% | 36.8% | 6.2% | 0.9% | 0.7% | 0.4% | 17.4% | 25.1% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.6% | 0.0% | 0.0% |
| Group F % | 0.0% | 0.6% | 1.1% | 5.1% | 0.1% | 3.2% | 3.3% | 0.1% | 2.4% | 1.0% | 3.5% | 2.3% | 4.2% | 1.1% | 2.6% | 1.6% | 1.4% | 0.0% |
In concordance with our previous study, Iso7 + Iso17 were found to be the predominant isoforms among all the isoforms and among those within group C, in both premutation and control groups. Also in agreement with our previous study we observed that Iso10 and Iso10b within group D were more expressed in the premutation than the control group. Our new findings include the detection of three of the predicted group B isoforms, Iso4b, Iso6, and Iso16, which were present in low abundances (0.02%–3.25%) in the premutation group but were undetected in the control group. These isoform were not detected in our previous study [17].
Interestingly, group E (missing exon 11 through 14) was highly detected in the premutation group but largely absent in the control group. Group F contains isoforms that could not be easily categorized into group A-E as they either contain an additional novel exon, a missing non-consecutive exon or were spliced within exons, and they were expressed at very low levels in our samples.
Additionally, we discovered, while previously using a lower cutoff of full-length read support (instead of 5 full-length read support, see Methods), that there was an isoform missing exon 11 through 14 detected only in one of the premutation sample (carrier 1) with only 3 full-length sequenced transcript. qRT-PCR experiment confirmed the presence of this isoform (Fig. 2e) suggesting that there could be more novel isoforms or splicing groups expressed at very low levels.
3.4. qRT-PCR confirmed the existence of novel FMR1 mRNA isoforms
In total, we identified 49 unique isoforms that were present either in the premutation group or the control group, or both (Table 2). The total number of isoforms in carriers was 46 compared to 19 in the control group (Table 2).
Table 2.
Number of isoforms detected in each isoform group in the premutation carrier and in the normal control samples. For group A through E, the isoforms detected in the control group were also detected in the premutation group, while some isoforms, within group F, were mutually exclusive to either the premutation (n=15) or control group (n=3).
| Group | Premutation | Control | Premutation + Control | Isoforms in Premutation only | Isoforms in controls only | Isoforms Common in Both Groups |
|---|---|---|---|---|---|---|
| A | 6 | 4 | 6 | 2 | 0 | 4 |
| B | 3 | 0 | 3 | 3 | 0 | 0 |
| C | 6 | 6 | 6 | 0 | 0 | 6 |
| D | 6 | 5 | 6 | 1 | 0 | 5 |
| E | 10 | 1 | 10 | 9 | 0 | 1 |
| F | 15 | 3 | 18 | 15 | 3 | 0 |
| Total | 46 | 19 | 49 | 30 | 3 | 16 |
In all groups except for group F, all novel isoforms observed in the control group were also observed in the premutation group. In group F, the isoforms were mutually exclusive; three isoforms (PB.1.9, PB.1.41, PB.1.47) were found only in the control group, while the remaining 15 are found only in the premutation group. In total, 30 isoforms were found to be present in the premutation group but not in the control group, with group E having the highest difference accounting for 9 isoforms detected only in premutation carriers. We confirmed the existence of several novel isoforms by qRT- PCR and Sanger sequencing: three isoforms within group F (including two retaining a portion of intron 9 and one missing exon 11 and 12) as illustrated in Fig. 2a and b respectively; two within group E (missing exon 11 through 14 but containing different portion of exon 15), as shown in Fig. 2c and d respectively. In addition, the existence of an isoform within group E, which was identified by SMRT sequencing but present below our inclusion criteria, was confirmed by qRT-PCR and Sanger sequencing (Fig. 2e).
3.5. Expression levels of the novel FMR1 mRNA isoforms are elevated in premutation carriers
RT-PCR analysis showed significant increases in expression levels of the novel FMR1 isoforms, including those in group B and the isoforms PB1.21/PB1.50 in group F, in premutation carriers compared to controls. For these isoforms, while a significant difference was observed between the premutation group and the control group (p < 0.001 for group B and p = 0.002 for group F), no significant differences were observed between the asymptomatic premutation carriers and the premutation carriers with FXTAS groups (p = 0.507 and p = 0.135 respectively) (Fig. 3a, b). Interestingly, we previously reported on a novel alternative splicing event that excludes exon 3, which was identified in both premutation carriers and controls [17]. Although a splice variant lacking exon 3 was detected at very low levels, there was a statistical significant difference in expression levels between premutation and control groups (p = 0.001) (Fig. 3c). No difference between the premutation asymptomatic group and the group with FXTAS was observed for any of the isoforms identified.
Figure 3.
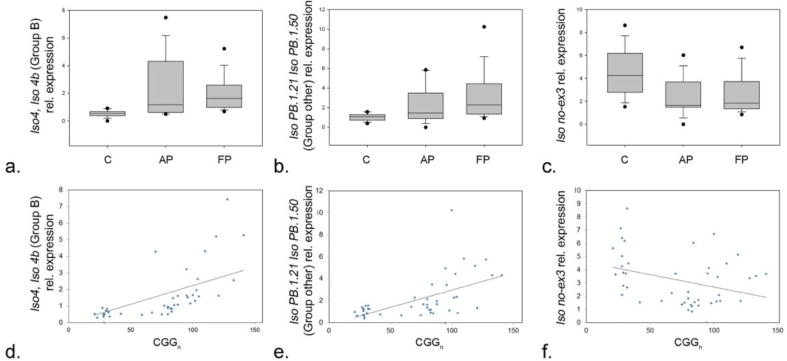
Box plot charts comparing the level of expression of (a) Group B, (b) intronic fragment 9 and (c) exon 3, in controls, asymptomatic premutation carriers and premutation carriers with FXTAS. Correlation charts showing the expression pattern of the Iso4 and Iso4b isoforms within (d) Group B, (e) of IsoPB1.21 and IsoPB 1.50 within group F, and of the isoform missing exon 3 (f) as function of CGG repeat number.
Finally, increased expression levels were demonstrated for the Iso4 and Iso4b isoforms within group B (Fig. 3d) and of IsoPB1.21 and IsoPB 1.50 within group F (Fig. 3e) as function of CGG repeats number. De- creased expression of the isoform missing exon 3 with increased CGG repeat number is shown in Fig. 3f.
3.6. Characteristics of newly identified alternative acceptor and donor sites
We observed many alternative acceptor and donor sites in all but exon 12. Exon 15 had by far the most diverse alternative acceptor sites (Supplementary Table 5). While many of them used canonical acceptor site (AG) and canonical donor site (GT), by far the most common alternative acceptor-donor site pattern was GA-AG (Supplementary Table 6).
All of the other donor-acceptor sites, while rare, have been observed in previous Genbank/EST data [22]. Manual inspection excludes this to be an aligner/mapper artifact, as the PacBio isoforms are full-length and span the whole targeted region. The genomic region immediately upstream of the exon 15 acceptor site has a high incidence of AGs that became the new acceptor sites for many group F isoforms.
Further work would need to be done to discern whether these novel AG- acceptor sites are a result of aberrant splicing, have any biological relevance, or could be an artifact of the retrotranscription process.
3.7. ORF prediction for all isoforms
Since the first 8 exons are based on reference, all predicted ORFs are identical for the 49 PacBio isoforms for exon 1 through 8 (see Methods). The last coding exon varied and is not always the same within each group (Supplementary Table 10). The peptide sequences alignments are shown in Supplementary Text 1. For group A isoforms (includes exon 9–17), the differences came from the use of alternative acceptor site in exon 15 and alternative acceptor site in exon 17, all of which were in-frame, and thus the predicted ORFs were only different in missing or including a few extra peptides but did not change the overall sequence. For group B, differences were also only in exon 15 and 17, but this generated slightly different ORF sequences, all of which have been previously predicted and annotated. Group C, with the most number of splice forms, had various acceptor sites in exon 15, 16, 17, which for the most part resulted in missing a few amino acids but largely remaining in-frame and identical to the canonical ORF, with only a few having premature stop codons. Group D (missing exon 12 and 14) isoforms consists of alternative junctions in exon 15, 16, 17, which creates either a premature stop codon or different peptides towards the end of the ORF. Group E (missing exon 11 through 14) has the most diversity in acceptor site in exon 15, contributing to premature stop codons and inclusions of novel amino acid sequences that were not seen in other groups. The rest of the isoforms within group F have a variety of alter- native junctions, resulting in premature stop codons and inclusion of small stretches of unique peptides as in case of isoforms PB 1.21 and PB 1.50.
Finally, despite having 49 unique isoforms based on splice patterns, ORF prediction showed only 44 unique predicted sequences, with pairs of isoforms from the same group sharing the same predicted ORFs. Their differences in splice patterns are often downstream of a pre- mature stop codon. For example, Isoform PB 1.21 and Isoform PB1.50 are the two isoforms that retain a portion of intron 9, which introduces a premature stop codon; however, the difference in exon 17 did not affect ORF prediction.
4. Discussion
Many transcripts in the central nervous system including those of the FMR1 gene are alternatively spliced potentially resulting in multiple proteins with different biochemical properties. Using qRT-PCR, several studies have shown different alternative splicing events taking place at FMR1 locus and involving multiple exons [13, 14, 23–25]. The expression profile of the resulting isoforms and their distribution was found to have an impact on the FMRP expression and function. Indeed, it was found that alternative protein isoforms might have different functions, hence, having a profound influence on the corresponding biological pathways [14, 26, 27]. Until recently, no data was available on the FMR1 mRNA isoforms differential expression and distribution as function of the CGG repeat number in premutation carriers. Although evidence suggests a strong role for regulation of the FMR1 gene expression in clinical outcomes, there have been no detailed molecular characterizations on the role of alternative splicing in the development of FMR1 premutation associated disorders.
Thus, we addressed these questions using SMRT sequencing technology and qRT-PCR on peripheral blood mononuclear cells, fibroblasts and brain tissue samples derived from premutation carriers and controls [17]. This previous study demonstrated the existence of 16 isoforms out of 24 predicted variants and of a significant increase in the abundance of all isoforms in the premutation group, although Iso10 and Iso10b mRNA isoforms showed the highest expression. These findings suggested their potential functional relevance in premutation-associated disorders [17]. Using a single-molecule long-read sequencing approach, in the current study, we further investigated the transcriptional FMR1 isoforms distribution pattern, in several human tissues from permutation carriers and age-matched controls and unravel a wide isoform diversity, although no tissue specific expression pattern was observed for any of the isoforms identified. We detected five additional predicted isoforms, within group A to D, bringing the total to 21 out of 24 predicted isoforms. The additional isoforms mostly belonged to group B and included Iso4b, Iso6, and Iso16. The remaining two isoforms were in Group A (Iso2) and Group D (Iso11).
A total final number of 49 isoforms were identified where 30 of them were exclusively detected in premutation carriers based on sequencing results and is mostly included in Groups E and F (Table 2). The six isoforms within group C were the only ones in both the pre- mutation and the control groups. The only isoforms that were uniquely detected in the controls were three isoforms that fell into the splice group we classified as Group F. We cannot exclude the possibility that some of the isoforms uniquely detected in the premutation group may also be present in the control group. Possible reasons for not having detected more isoforms in the control group could be due a much lower abundance of such isoforms in the control group compared to the premutation group or to a lower sequencing depth of the controls; however, we have performed rarefaction curves (subsampling analysis) and found that even the control samples were sufficiently sampled after 2500 full-length reads (Fig. 4).
Figure 4.
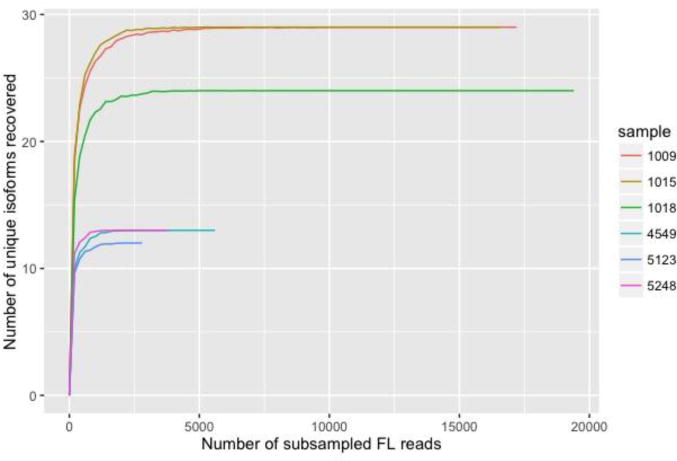
Rarefaction curve (subsampling analysis) for each sample shows that the sequencing has reached sufficient depth. We subsampled, for 100 iterations at each subsample size, a subset of FL reads and recorded the number of recovered isoforms at that sampling depth. We then show the average recovered isoforms for the 100 iterations at each subsampled point.
The discovery of an isoform detected below our cutoff and only present in a single patient-tissue sample but nevertheless validated later through RT-PCR suggests that there could be even more novel isoforms present at very low abundance and that the FMR1 gene may expressed more diverse transcripts that originally thought. In line with our previous study [17], Iso10 and Iso10b within group D were found to be the highest expressed in the premutation carrier group compared to the control group. These isoforms lead to truncated isoforms lacking both the function of the C-terminal RGG box and the nuclear export signal (NES).
Isoforms within Group E were also highly abundant in the pre- mutation carrier group compared to the control group (Table 1). Two isoforms, retaining a portion of an intronic sequence between exon 9 and 10, were identified and found to disrupt translation, leading to a premature stop codon and consequently encoding to truncated proteins. We observed many alternative acceptor and donor sites in all but exon 12.
Furthermore, exon 15 was found to have the most diverse alternative acceptor sites (Supplementary Table 5). Despite having 49 unique isoforms based on splice patterns, ORF prediction showed only 44 unique predicted sequences. The remaining sequences were mostly from truncated proteins, which showed the same ORF, as the differences in splice patterns were downstream of a premature stop codon. However, the question is if all the FMR1 mRNA isoforms potentially generating truncated proteins undergo RNA processing such that the RNA harboring premature termination codons are degraded through non-sense mediated decay [28], a post-transcriptional surveillance mechanisms developed by the cells to target messenger RNAs (mRNAs) carrying a mutation. Thus, whether all these identified truncated FMRP isoforms in premutation carriers have a functional role and could therefore contribute to the pathology of the FMR1 associate disorders is currently unknown.
Several studies have reported on the potential impact of the different splice events as result of alternative splicing of the FMR1 gene. It was shown that FMRP isoforms might: (i) have different binding affinities to RNA structures [29], (ii) might have altered posttranslational modification regions or (iii) lack the nuclear export sequence [15,30,31]. Some exons or group of exons out of the 17 within the FMR1 gene encode for functional domains that are essential for FMRP function. For example, exon 14 is responsible for the expression of the nuclear export signal (NES) domain which is important for FMRP functioning as a nucleocytoplasmic shuttling protein [32]. Exon 12 and 13 beside exons 8 to 10 encode for the K homology domain 2 (KH2) whereas exons 8 to 10 are also important for the expression of K homology domain 1 (KH1); both KH1 and KH2 are RNA binding do- mains. Thus, altered sequence of these regions is likely affecting the binding of FMRP to its mRNA targets.
A study done by Xie et al. showed that the presence or the absence of exon 12 can affect the ability of FMRP (specifically KH2) to recognize the loop-loop pseudoknot termed “kissing complex” structure that is included in some of the RNA targets [25] rendering those targets among the top candidates that might contribute to the pathogenesis of FXTAS when dysregulated (are no longer recognized by FMRP when KH2 region is missing or is mutated).
Alternative splicing events involving exons 15 and/or 16 are important for the expression of arginine-glycine-glycine domain (RGG). A study showed that the RGG region binds specifically to guanine-quadruplexes which are functionally implicated in an array of processes such as translational repression, alternative splicing and mRNA stability, supporting the role of FMRP as translational regulator [33].
FMRP has been shown to undergo post-translational modifications represented by serine phosphorylation in a region directly upstream of the RGG box domain and by arginine methylation within the RGG box domain [34,35]. The phosphorylation/dephosphorylation events of FMRP plays a significant role in the FMRP: miRNA interactions [36].
FMRP isoforms 2 and 3, which include all the 17 exons but lack specifically the phosphorylation site “Serine 500 residue” in exon 15 were suggested to fine-tune the FMRP translation regulatory function by controlling the translation process undergone by different mRNA tar- gets. As for methylation, it was found that one of the spliced variant of exon 15 in FMR1 reduced methylation by > 85% suggesting that differential FMRP exon 15 variants expression and methylation patterns play an important role in regulation of the translation of target mRNA [30].
Xie et al. [25] showed that alternative splicing at exon 15 produces FMRPs isoforms which differ in RNA binding ability and each is distinguished by unique post-translational modifications. Interestingly the isoforms containing the entire exon 15 are the most abundant within each isoform group [17] and Supplementary Fig. 9).
As shown in Table 2, we detected 9 isoforms in group E and 15 isoforms in group F in premutation carriers but not in controls. Isoforms within Groups E and F lead to a set of splice isoforms missing a combination of different exons (Fig. 1). However, the large majority of these isoforms either lacked exons 15 and 16 or portion of them potentially resulting in altered expression of the RGG domain, which consequently could affect the specificity of interaction between FMRP and target mRNAs contributing to a more global mRNA toxicity.
Isoforms within Group E, were only found in premutation carriers (i.e. PB.1.22, PB.1.25, PB.1.32, PB.1.35; Supplementary Table 9 and Supplementary Fig. 1), where the missing exons might lead to altered KH1 and KH2 domains (RNA binding) possibly influencing FMRP RNA binding specificity or affinity, in addition to the NES possibly altering FMRP shuttling between the nuclei and the cytoplasm. The KH2 domain and the NES region can promote homo- and heterodimerization and can participate in self-association [37–41]. Hence, there might be different aggregation and dimerization properties for the different FMRP isoforms based on the different splicing patterns and inclusion/exclusion of the exons that correspond to KH2 and NES.
Finally, this study has identified a number of isoforms leading to truncated proteins. Truncated proteins can have a significant role in the pathogenesis of different neurodegenerative disorders. Indeed, truncated forms of Alpha-Synuclein, play an important role in the pathology of Parkinson’s disease (PD) and can be found in PD brain extracts [42]. Furthermore, the expression of truncated human tau in mice leads to clumping of mitochondria, failure of axonal transport, mis-sorting of synaptic proteins and disruption of the Golgi apparatus [43].
On another note, the differential isoforms distribution pattern observed in this study, which indicates a dysregulation of the mRNA splicing process, could be attributed to the splicing regulator Muscle blind Like Splicing Regulator 1 (MBNL1) [44], found within the intra- nuclear inclusion in FXTAS brain [45]. Alternatively, altered splicing process could arise from the sequence-specific RNA binding protein (Sam68), which regulates the alternative splicing process during the cell cycle and apoptosis or from a number of pathways involved in splicing [44]. The authors also demonstrated that the expanded CGG repeats sequester Sam68 altering its splicing regulatory functions in FXTAS. They also found that when SAM68 is sequestered, it results in the expression of Bcl-xL, the inclusion of Exon-7 in SMN2 and the repression of Exon-28B in human ATP11B indicating altered splicing processing take place in premutation carriers affected by FXTAS [44].
Thus, it is possible that the translation of some of the “FMRP isoforms”, (whether truncated or not) resulting from an altered alternative splicing process of the FMR1 mRNA, might contribute to the pathogenesis of the premutation disorders. Such large isoform diversity in the FMR1 gene, as the one uncovered in this study, could reflect a functional diversity, which ultimately could have cellular and neurotoxic consequences. FMRP is an RNA binding protein, which acts as translator repressor at the synapses playing a key role in learning and memory [46]. Thus, FMRP is involved in many functions at the synapses, likely driven by different motifs regulated by alternative splicing; it would be therefore important to determine the functional significance of this large FMR1 isoform diversity and also their localization within the cells and at the synapses [47].
In conclusion, our findings suggest that an abnormal alternative splicing process is present in individuals with premutation alleles. The characterization of expression levels of the repertoire of FMR1 isoforms is fundamental for understanding the regulation of the FMR1 gene as well as for elucidating the mechanism(s) by which “toxic gain of function” of the FMR1 mRNA may play a role in FXTAS and/or in the other FMR1-associated conditions. In addition to the elevated levels of FMR1 isoforms, the altered abundance/ratio of the corresponding FMRP isomers may affect the overall function of FMRP and play a role in the FMR1- associated disorders observed in premutation carriers.
Finally, further studies are needed to characterize their specific roles played in the pathogenesis of FXTAS and in other FMR1 associated disorders.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health through grant number GM113929. It was also supported by Award Number T32MH073124 from the National Institute of Mental Health. This work is dedicated to the memory of Matteo.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbagrm.2017.08.007.
Footnotes
Transparency document
The http://dx.doi.org/10.1016/j.bbagrm.2017.08.007 associated with this article can be found, in online version.
Author disclosure statement
Authors have no competing financial interests to disclosure.
References
- 1.Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim Biophys Acta Mol basis Dis. 2009;1792(1):14–26. doi: 10.1016/j.bbadis.2008.09.017. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravindra T, Lakshmi N, Chaitanya K, Surender V, Ahuja Y. Clinical relevance of alternative splicing. Indian J Hum Genet. 2010;12(2):45. (Internet) [Google Scholar]
- 3.Smith RM, Sadee W. Synaptic signaling and aberrant RNA splicing in autism spectrum disorders. Front Synaptic Neurosci. 2011 Jan;3:1–8. doi: 10.3389/fnsyn.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stamova BS, Tian Y, Nordahl CW, Shen MD, Rogers S, Amaral DG, et al. Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders. Mol Autism. 2013;4(1):30. doi: 10.1186/2040-2392-4-30. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazin P, Xiong J, Liu X, Yan Z, Zhang X, Li M, et al. Widespread splicing changes in human brain development and aging. Mol Syst Biol. 2013;9(633):633. doi: 10.1038/msb.2012.67. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castle JC. Differential expression of 24,426 human alternative splicing events and predicted cis‐regulation in 48 tissues and cell lines. 2011 Oct;40(12):1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52(1):93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 8.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327–1332. doi: 10.1086/320609. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosewich H, Thiele H, Ohlenbusch A, Maschke U, Altmüller J, Frommolt P, et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. 2012;11(9):764–773. doi: 10.1016/S1474-4422(12)70182-5. (Internet) [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korir PK, Roberts L, Ramesar R, Seoighe C. A mutation in a splicing factor that causes retinitis pigmentosa has a transcriptome-wide effect on mRNA splicing. BMC Res Notes. 2014;7(1):401. doi: 10.1186/1756-0500-7-401. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vuong CK, Black DL, Zheng S. The neurogenetics of alternative splicing. Nat Rev Neurosci. 2016;17(5):265–281. doi: 10.1038/nrn.2016.27. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang T, Li LY, Shen Y, Qin XB, Pang ZL, Wu GY. Alternative splicing of the FMR1 gene in human fetal brain neurons. Am J Med Genet. 1996;64(2):252–255. doi: 10.1002/(SICI)1096-8628(19960809)64:2<252::AID-AJMG3>3.0.CO;2-W. (Internet) [DOI] [PubMed] [Google Scholar]
- 14.Brackett DM, Qing F, Amieux PS, Sellers DL, Horner PJ, Morris DR. Fmr1 transcript isoforms: association with polyribosomes; regional and developmental expression in mouse brain. PLoS One. 2013;8(3):1–12. doi: 10.1371/journal.pone.0058296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sittler A, Devys D, Weber C, Mandel JL. Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of FMR1 protein isoforms. Hum Mol Genet. 1996;5(1):95–102. doi: 10.1093/hmg/5.1.95. [DOI] [PubMed] [Google Scholar]
- 16.Hagerman PJ, Hagerman RJ. Fragile X-associated Tremor/Ataxia Syndrome. 2015;1338(1):58–70. doi: 10.1111/nyas.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pretto DI, Eid JS, Yrigollen CM, Tang HT, Loomis EW, Raske C, et al. Differential increases of specific FMR1 mRNA isoforms in premutation carriers. J Med Genet. 2014:1–11. doi: 10.1136/jmedgenet-2014-102593. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tassone F, Hall DA, editors. FXTAS, FXPOI, and Other Premutation Disorders [Internet] Springer International Publishing; 2016. [Google Scholar]
- 19.Chen L, Hadd A, Sah S, Filipovic-Sadic S, Krosting J, Sekinger E, et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn. 2010;12(5):589–600. doi: 10.2353/jmoldx.2010.090227. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10(1):43–49. doi: 10.2353/jmoldx.2008.070073. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon SP, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One. 2015;10(7):1–15. doi: 10.1371/journal.pone.0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burset M. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28(21):4364–4375. doi: 10.1093/nar/28.21.4364. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashley CT, Sutcliffe JS, Kunst CB, Leiner HA, Eichler EE, Nelson DL, et al. Human and murine FMR-1: alternative splicing and translational initiation down- stream of the CGG-repeat. Nat Genet. 1993;4:244–251. doi: 10.1038/ng0793-244. [DOI] [PubMed] [Google Scholar]
- 24.Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, et al. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363(6431):722–724. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- 25.Xie W, Dolzhanskaya N, LaFauci G, Dobkin C, Denman RB. Tissue and devel- opmental regulation of fragile X mental retardation 1 exon 12 and 15 isoforms. Neurobiol Dis. 2009;35(1):52–62. doi: 10.1016/j.nbd.2009.03.015. (Internet) [DOI] [PubMed] [Google Scholar]
- 26.Banerjee P, Schoenfeld BP, Bell AJ, Choi CH, Bradley MP, Hinchey P, et al. Short- and long-term Memory Are Modulated by Multiple Isoforms of the Fragile X Mental Retardation Protein. 2010;30(19):6782–6792. doi: 10.1523/JNEUROSCI.6369-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blackwell E, Ceman S. A New Regulatory Function of the Region Proximal to the RGG Box in the Fragile X Mental Retardation Protein. 2011:3060–3065. doi: 10.1242/jcs.086751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2015;44(4):1483–1495. doi: 10.1093/nar/gkw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD, et al. Purified Recombinant Fmrp Exhibits Selective RNA Binding as an Intrinsic Property of the Fragile X Mental Retardation Protein*. 1998;273(25):15521–15527. doi: 10.1074/jbc.273.25.15521. [DOI] [PubMed] [Google Scholar]
- 30.Dolzhanskaya N, Merz G, Denman RB. Alternative Splicing Modulates Protein Arginine Methyltransferase-Dependent Methylation of Fragile X Syndrome Mental Retardation Protein †. 2006:10385–10393. doi: 10.1021/bi0525019. [DOI] [PubMed] [Google Scholar]
- 31.Didiot MC, Tian Z, Schaeffer C, Subramanian M, Mandel JL, Moine H. The G-quartet Containing FMRP Binding Site in FMR1 mRNA is a Potent Exonic Splicing Enhancer. 2008;36(15):4902–4912. doi: 10.1093/nar/gkn472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardoni B, Sittler A, Shen Y, Mandel JL. Analysis of Domains Affecting Intracellular Localization of the FMRP Protein. 1997;336:329–336. doi: 10.1006/nbdi.1997.0142. [DOI] [PubMed] [Google Scholar]
- 33.Vasilyev N, Polonskaia A, Darnell JC, Darnell RB, Patel DJ. Crystal Structure Reveals Specific Recognition of a G-Quadruplex RNA by a β -turn in the RGG Motif of FMRP. 2015 doi: 10.1073/pnas.1515737112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siomi MC, Higashijima K, Ishizuka A, Siomi H, II, Kinase Casein. Phosphorylates the Fragile X Mental Retardation Protein and Modulates its Biological Properties. 2002;22(24):8438–8447. doi: 10.1128/MCB.22.24.8438-8447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stetler A, Winograd C, Sayegh J, Cheever A, Patton E, Zhang X, et al. Identification and Characterization of the Methyl Arginines in the Fragile X Mental Retardation Protein Fmrp. 2006;15(1):87–96. doi: 10.1093/hmg/ddi429. [DOI] [PubMed] [Google Scholar]
- 36.Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, et al. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphor- ylation, and mGluR signaling Ravi. Mol Cell. 2011;42(5):673–688. doi: 10.1016/j.molcel.2011.05.006. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siomi MC, Zhang YAN, Siomi H, Dreyfuss G. Specific Sequences in the Fragile X Syndrome Protein FMR1 and the FXR Proteins Mediate Their Binding to 60S Ribosomal Subunits and the Interactions Among Them. 1996;16(7):3825–3832. doi: 10.1128/mcb.16.7.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fatimy R, El Tremblay S, Dury AY, Solomon S, De Koninck P, Schrader JW, et al. Fragile Mental Retardation Protein Interacts with the RNA-Binding Protein Caprin1 in. Neuronal RiboNucleoProtein Complexes. 2012;7:6. doi: 10.1371/journal.pone.0039338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramos A, Hollingworth D, Pastore A. The Role of a Clinically Important Mutation in the Fold and RNA-binding Properties of KH Motifs. 2003:293–298. doi: 10.1261/rna.2168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valverde R, Pozdnyakova I, Kajander T, Venkatraman J, Regan L, Fragile X. Mental Retardation Syndrome: Structure of the KH1-KH2 Domains of Fragile X Mental Retardation Protein. 2007 Sep;:1090–1098. doi: 10.1016/j.str.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 41.Zalfa F, Adinolfi S, Napoli I, Ku E, Pastore A, Bagni C. Fragile X Mental Retardation Protein (FMRP) Binds Specifically to the Brain Cytoplasmic RNAs BC1/BC200 via a Novel RNA-binding Motif *. 2005;280(39):33403–33410. doi: 10.1074/jbc.M504286200. [DOI] [PubMed] [Google Scholar]
- 42.Lööv C, Scherzer CR, Hyman BT, Breakefield XO, Ingelsson M. A-synuclein in extracellular vesicles: functional implications and diagnostic opportunities. Cell Mol Neurobiol. 2016;36(3):437–448. doi: 10.1007/s10571-015-0317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ozcelik S, Sprenger F, Skachokova Z, Fraser G, Abramowski D, Clavaguera F, et al. Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol Psychiatry. 2015 Sep;21(2016):1–9. doi: 10.1038/mp.2015.228. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, et al. Sam68 Sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29(7):1248–1261. doi: 10.1038/emboj.2010.21. (Internet) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129(1):256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 46.Sidorov MS, Auerbach BD, Bear MF. Fragile X mental retardation protein and synaptic plasticity. Mol Brain. 2013;6(1):15. doi: 10.1186/1756-6606-6-15. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joel R, Bassell GJ, Eric K. Dysregulation and restoration of translational home- ostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16(10):595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.